
Abstract
A large fraction of genetic risk factors for Alzheimer’s Disease (AD) is still not identified, limiting the understanding of AD pathology and study of therapeutic targets. We conducted a genome-wide association study (GWAS) of AD cases and controls of European descent from the multi-center DemGene network across Norway and two independent European cohorts. In a two-stage process, we first performed a meta-analysis using GWAS results from 2,893 AD cases and 6,858 cognitively normal controls from Norway and 25,580 cases and 48,466 controls from the International Genomics of Alzheimer’s Project (IGAP), denoted the discovery sample. Second, we selected the top hits (p < 1 × 10−6) from the discovery analysis for replication in an Icelandic cohort consisting of 5,341 cases and 110,008 controls. We identified a novel genomic region with genome-wide significant association with AD on chromosome 4 (combined analysis OR = 1.07, p = 2.48 x 10-8). This finding implicated HS3ST1, a gene expressed throughout the brain particularly in the cerebellar cortex. In addition, we identified IGHV1-68 in the discovery sample, previously not associated with AD. We also associated USP6NL/ECHDC3 and BZRAP1-AS1 to AD, confirming findings from a follow-up transethnic study. These new gene loci provide further evidence for AD as a polygenic disorder, and suggest new mechanistic pathways that warrant further investigation.
Similar content being viewed by others
Introduction
Alzheimer’s disease (AD), the most common cause of dementia1,2, places a large personal and economic burden on families and society3,4. There are nearly 200,000 individuals suffering from AD in the Nordic countries alone (http://www.alzheimer-europe.org). In the absence of disease-modifying therapies, identifying AD prevention strategies is of importance. Converging evidence indicates that AD-associated pathological changes5 begin years, if not decades, before the onset of clinical symptoms6,7,8. In order for targeted prevention to be most effective, there is a need to better identify the genetic AD architecture to determine pre-symptomatic disease risk, as well as to provide new insight into disease mechanisms.
Multiple genes in combination with environmental risk factors affect AD neurodegeneration9,10. Apart from the major genetic risk factor of the APOE gene11,12,13, late onset AD does not exhibit a clear-cut pattern of inheritance and is probably caused by many common variants, each with a small effect9,14,15,16,17,18,19 (‘polygenic’), together with few rare variants with large effects9,19,20,21,22. In a genome-wide association study (GWAS), SNPs tagging these causal variants may be identified as association loci. In a large, two-stage GWAS meta-analysis15, 19 loci were associated with AD in addition to the previously reported APOE locus. Yet these sequence variants account only for a small portion of the disease heritability23,24,25,26. This “missing heritability” has been attributed to a number of potential causes, such as lack of typing of rare variants26, and lack of proper statistical methods for analyzing the polygenic architecture of AD. A concerted effort to increase sample sizes is required to reveal the small-effect genetic risk variants which are common in the population.
We conducted a GWAS of 2,893 AD cases and 6,858 controls from a Norwegian cohort and meta-analyzed this cohort with the publicly available IGAP GWAS data15. The most promising association signals were tested in an independent replication sample from Iceland. A novel locus was uncovered on chromosome 4 (rs6448807; hg19 chr4:11676144, OR = 1.07, p = 2.48 × 10−8). This novel locus is close to the HS3ST1 gene, suggesting the involvement of a new biochemical pathway in AD pathology.
Methods
Samples
Norwegian cohorts
We collected genotype data from the Norwegian DemGene network consisting of 2,893 cases and 1,660 healthy controls. A total of 4,553 Caucasian (1000 Genome population code: CEU) individuals were recruited and successfully genotyped. DemGene is a Norwegian network of clinical sites collecting cases from Memory Clinics based on standardized examination of cognitive, functional and behavioral measures and data on progression of most patients. We diagnosed 2,135 cases of AD dementia as well as 758 cases of Mild Cognitive Impairment (MCI) as a proxy of prodromal AD, from 7 studies (mean age = 73.2 ± 9.9): the Norwegian Register of persons with Cognitive Symptoms (NorCog), the Progression of Alzheimer’s Disease and Resource use (PADR), the Dementia Study of Western Norway (DemVest), the AHUS study, the Dementia Study in Rural Northern Norway (NordNorge), HUNT Dementia Study and the Nursing Home study, the TrønderBrain study, and the Dementia Disease Initiation study (DDI). Cases were diagnosed with dementia due to Alzheimer’s disease according to the recommendations from the National Institute on Aging–Alzheimer’s Association (NIA/AA) (AHUS), the NINCDS-ADRDA criteria (DemVest and TrønderBrain) or the ICD-10 research criteria (NorCog, PADR, NordNorge and HUNT). MCI was diagnosed according to the NIA-AA criteria (AHUS and DDI) or the Winblad criteria (NorCog, HUNT, PADR, DemVest, Trønderbrain). The controls from Norway were obtained through AHUS, NordNorge, HUNT and TrønderBrain studies. Controls were screened with standardized interview and cognitive tests from the population. To increase the statistical power of our association analysis, the controls were combined with additional 4,475 population controls from Norwegian blood donor samples (Oslo University Hospital, Ullevål Hospital, between 18–60 years) and 723 controls from the Thematically Organized Psychosis Research (TOP) Study27 (between 25–65 years). Control subjects from the TOP Study were of Caucasian origin without history of moderate/severe head injury, neurological disorder, mental retardation and were excluded if they or any of their close relatives had a lifetime history of a severe psychiatric disorder, a history of medical problems thought to interfere with brain function or significant illicit drug use27. The inclusion of population controls were corrected for population stratification28. For a complete description of the samples, please refer to Supplementary Text 1 and Supplementary Table 1.
Genotyping
The genotypes for the DemGene Study were obtained with Human Omni Express-24 v.1.1 (Illumina Inc., San Diego, CA, USA) at deCODE Genetics (Reykjavik, Iceland).
IGAP Study
We obtained summary statistic GWAS data from the IGAP Study, a large two-stage meta-analysis of GWAS in individuals of European ancestry. In stage 1, genotyped data were imputed into 7,055,881 SNPs to perform meta-analysis on 4 previously published GWAS data sets from four consortia: the Alzheimer’s Disease Genetic Consortium (ADGC), the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium, the European Alzheimer’s Disease Initiative (EADI) and the Genetic and Environmental Risk in Alzheimer’s Disease (GERAD) Consortium, consisting of 17,008 AD cases and 37,154 controls. In stage 2, 11,632 SNPs were genotyped and tested for association in an independent set of 8,572 AD cases and 11,312 controls.
Sample from Iceland
Patients from Iceland were diagnosed with definite, probable or possible AD dementia on the basis of the NINCDS-ADRDA criteria or according to guidelines for ICD-10 F00 and were compared to population controls17. Association testing was carried out using information from 5,341 AD patients and 110,008 population controls, adjusting for age, age-squared, sex and county of birth.
The case-control studies are summarized in Table 1.
Analysis
Before imputation, the genotype data underwent basic quality control including removal of SNPs with minor allele frequency (MAF) lower than 0.01, genotype call rate lower than 0.95, less than one in a million probability of being in Hardy-Weinberg equilibrium (for both cases and controls) and ambiguous strand assignment (A/T, C/G SNPs). The quality-controlled genotypes were used to compute genetic principal components analysis (PCA) with which to adjust for potential population stratification effects. PCA was performed using PLINK 1.9 based on the variance-standardized relationship matrix29.
MaCH software30 was used to impute the genotypes of all participants onto reference haplotypes derived from samples of European ancestry in the 1000 Genome Project (Build 37, Assembly Hg19)31. SNPs with MAF lower than 0.005 or a ratio of observed versus expected variance lower than 0.1 were excluded. The association of LOAD with genotype dosage was analyzed in PLINK 1.9 with a logistic regression model using gender and 20 top principal components as covariates. Sex chromosomes were not included from the analyses29. After filtering for SNPs with information value greater than 0.5, we obtained 9,155,276 association p-values. The inflation factor of the DemGene sample was calculated at 1.022 (Supplementary Fig. 1).
We analyzed 6,564,314 SNPs that overlap between DemGene and IGAP stage 1 data and 10,092 SNPs with IGAP stage 2. A meta-analysis with IGAP data was performed using PLINK 1.9 with fixed effects inverse-variance weighted effect sizes29.
Functional Annotation (FUMA)
We utilized FUMA for post-processing of our Stage 1 results32. FUMA incorporates 18 biological data repositories such as the Genotype-Tissue Expression (GTEx), Encyclopedia of DNA Elements (ENCODE), Roadmap Epigenomics Project and chromatin interaction information. FUMA requires GWAS summary statistics and outputs multiple tables and figures containing extensive information on, e.g., functionality of SNPs in genomic risk loci, including protein-altering consequences, gene-expression influences, open-chromatin states as well as three-dimensional (3D) chromatin interactions. Functionally annotated SNPs are subsequently mapped to prioritized genes based on (i) physical position mapping on the genome, (ii) expression quantitative trait loci (eQTL) mapping and (iii) 3D chromatin interactions (chromatin interaction mapping)32. Biological information for each prioritized gene is provided to gain insight into previously associated diseases. Beside the single gene level analyses, FUMA uses GTEx to identify 53 tissue specific expression levels of prioritized genes. We refer to the original publication for details on the methods and repositories of FUMA32.
Results
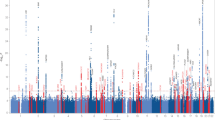
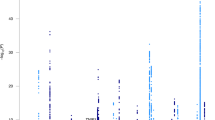
We performed GWAS in two stages33,34. First, we meta-analyzed the discovery samples using 10,092 SNPs imputed for both the DemGene and IGAP stage 1 + 2 samples (Manhattan plots in Fig. 1). From this discovery sample result, we narrowed our analysis to 1,035 SNPs with genome-wide or suggestive association (meta p < 1 × 10−6) and tested these SNPs in an Icelandic replication sample. Finally, we meta-analysed the discovery and replication samples into a combined analysis.

Manhattan plots of discovery sample from meta-analysis of DemGene data with IGAP 1 + 2. Note that color alternate between chromosomes for visualization only. Out of the loci passing genome-wide threshold p < 5 × 10−8, we found novel hits on Chromosomes 4, 10, 14 and 17.
We identified 20 genomic risk loci from the discovery sample using FUMA (r2 > 0.1 using Linkage Disequilibrium from 1000 Genomes hg1931), listed in Supplementary Table 2. Four out of the 20 loci had not reached genome-wide significance in IGAP at p < 5 × 10−8. We list the lead SNPs of the discovery sample in Table 2 and the corresponding lead SNPs of the combined analysis in Supplementary Table 3.
We highlight a genomic region on chromosome 4 spanning 11.55 Mb–11.76 Mb (best SNP: rs6448807, discovery OR = 1.08, p = 2.23 × 10−8) in Fig. 2. The lead SNP is located at position chr4:11676144 (minor/major allele: C/T), an intron of gene RP11-281P23.2, a non-coding RNA (Ensembl: ENSG00000249631). Its nearest protein-coding gene is HS3ST1 located 240 kb away. We found the same direction of effects within the replication sample (replication OR = 1.03, p = 0.17), and in the combined analysis the SNP is associated with AD at p = 2.48 × 10−8 (Table 2). Although this lead SNP of the discovery sample was not replicated at p < 0.05 in the replication sample, we found rs13133131 within the same genomic locus as a proxy SNP (r2 = 0.94) with better replication (replication p = 0.00319) and a stronger association in the combined analysis (combined OR = 1.07, p = 8.16 × 10−9), see Supplementary Table 3 for genomic risk loci based on the combined analysis.

Regional association plots of rs6448807 (chr 4) and rs79452530 (chr 14) in the discovery sample. Mapping of the genes is strictly based on position of the genomic risk loci.
Additionally we found a genomic region on chromosome 14 spanning 106.47 Mb–107.26 Mb with a novel risk associated locus at rs79452530 (chr14:107156009, OR = 0.89, p = 2.36 × 10−8), see Fig. 2. This is an intergenic SNP 3,860 kb away from the nearest gene IGHV1-68 (Ensembl: ENSG0000253703). The locus has not previously been associated with AD, but was mentioned in a study in rheumatic heart disease35. This genomic region was genome-wide significant in the discovery sample, but its effect was not observed in the replication sample (Table 2) and none of its proxies passed the significance threshold in the combined analysis (best SNP: rs78631692 at p = 6.57 × 10−8, Supplementary Table 3). The directions of effects, however, were consistent in all samples (Table 2).
We also found association regions at genome-wide significance at two additional loci on Chromosome 10 and 17, both of which had not passed significance threshold in the IGAP study. The first locus, rs7920721 (chr10:11720308, OR = 1.07, p = 1.82 × 10−8), is an intergenic SNP with nearest gene RP11-138I18.2 (Ensembl: ENSG00000271046) at 1366 kb away and in between protein-coding genes USP6NL/ECHDC3 (see Supplementary Fig. 2). This SNP is replicated at p = 0.0072 and has a strong association signal in the combined analysis (OR = 1.07, p = 4.84 × 10−10). The second locus, SNP rs2526378 (chr17:56404349, OR = 0.93, p = 3.64 × 10−9), is an intron of non-coding RNA BZRAP1-AS1 (Ensembl: ENSG00000265148) (Supplementary Fig. 2). A proxy of this SNP (r2 = 0.96) is significant in the combined analysis at p = 3.58 × 10−8, although it is mainly driven by the discovery sample (Supplementary Table 3). For both rs7920721 and rs2526378, we found agreement of its significance in a following transethnic AD study36, confirming our results.
We focused our FUMA analyses on the two novel loci in our study, the two genomic regions on chromosomes 4 and 14, represented by lead SNPs rs6448807 and rs79452530. Out of the functional consequences of 1,128 SNPs represented by the two loci on genes, we found the most SNPs being intergenic (n = 511 SNPs) and non-coding RNA-intronic (n = 340), see Supplementary Fig. 3. For the chromosome 4 locus, we mapped the associated variants through eQTL or 3-D chromatin interaction to 15 genes, eight of which are protein-coding: BOD1L1, CLNK, HS3ST1, NKX3-2, RAB28, SLC2A9, WDR1 and ZNF518B (Fig. 3). Notably, HS3ST1 was mapping using both eQTL and chromatin interaction. For the chromosome 14 locus, we mapped the associated variants to 117 genes (Supplementary Figs 3 and 4). Despite the large number of genes located in the 780 kb-span of the genomic risk region, none of them was identified as a protein-coding gene by FUMA.

Circos plots of mapped gene on chromosome 4 locus. Genomic risk loci are highlighted in blue. Genes are mapped by 3-D chromatin interaction (orange) or eQTLs (green), or both (red) notably gene HS3ST1.
Furthermore, we looked at the expression and enrichment tests of the protein-coding genes prioritized by FUMA. We found WDR1 to be highly expressed in comparison to other genes in all tissues except brain tissue, where it is only moderately highly expressed (Supplementary Fig. 5). We also found higher expression in RAB28 and BOD1L1 in all tissues. Although HS3ST1 is not highly expressed compared to other genes, its expression is specifically higher in the cerebellar hemisphere and the cerebellum. Lastly, we found IGHV-genes to have higher expression in the spleen, stomach, intestine and lung tissues, and lower expression specific to brain tissues.
Discussion
Our study presents the largest GWAS of AD cases and controls from multi-center network across Norway to date and its meta-analysis with two independent European cohorts. Our main finding in the present study is a novel locus associated with AD on chromosome 4, represented by rs6448807. Its closest protein-coding gene, HS3ST1, has been previously suggested as an AD susceptibility locus using conditional False Discovery Rate methods associating AD with dyslipidaemia37, and here we discover the same locus using standard GWAS analysis. According to brain expression data from the BrainSpan Atlas of the Developing Human Brain38,39, HS3ST1 (Heparan sulfate D-glucosaminyl 3-O-sulfotransferase 1) is expressed in the brain, with strong expressions in the cerebellar cortex, followed by the primary visual cortex. Similar heparan sulphate enzymes (HST3ST1, HS3ST2, HST3ST3A, HS3ST3B, HS3ST4 and HS3ST5) are expressed in human elderly hippocampus40, with HS3ST2 already proven to be overexpressed in the hippocampus of patients with AD41. Similarly, heparan sulfates have been shown in mice to modulate brain amyloid-β clearance and aggregation in AD42. This study pointed to the implication of heparan sulphates to AD.
In a previous large AD study by IGAP15, a proxy to this SNP (r2 = 0.79) had been listed as a suggestive association locus (rs6448799; hg19 chr4:11630049). The gene HS3ST1 is located 197 kb from rs6448799 and 245 kb from rs6448807. This finding adds another risk variant with a low odds ratio to the polygenic risk factors of AD. rs6448807 and rs644879915 are located in the first intron of non-coding RNA RP11-281P23. The Ensembl gene model for RP11-281P23 is supported by expressed sequence tag evidence from testes and kidney, and RNA-seq data provide evidence of expression in the heart, testes43 and brain (dorsolateral prefrontal cortex)44 although expression levels are low in all these tissues. Based on the available data, it is not possible to conclude on which molecule is driving the association. The genetic evidence would tend to suggest that it is the non-coding RNA that underlies the association given that 1) both the genome-wide significant rs6448807 and the suggestive association locus rs6448799 are located near the transcription start site of the long non-coding RNA, 2) HS3ST1 is 245 kb away from these loci, and 3) there is little linkage disequilibrium between the HS3ST1 and the RP11-281P23 locus. On the other hand, FUMA analyses point to the priority of HS3ST1 through not only eQTLs but also chromatin interactions. HS3ST1 has low but consistent expression in the cerebellar and visual cortex38,39 as well as the differential expression in brain between AD cases and controls37 provide supportive evidence for HS3ST1 playing an important role. Further experimental research will be required to elucidate the biological basis of the association of this locus with AD.
Furthermore, we have boosted three genomic loci from the previous IGAP study into genome-wide significance. Two of the three loci are confirmed in a following study in transethnic meta-analysis study and here we observed them in a CEU population. The two loci are an intron on a non-coding RNA BZRAP1-AS1 and an intergenic SNPs with closest gene RP11-138I18.2.
From the discovery sample, we associated IGHV1-68 to AD, previously not associated with the disease. IGHV1-68 is a pseudogene within the immunoglobulin heavy chain (IGH) locus45. Although the exact role of IGHV1-68 is unknown, IGH is essential for biosynthesis of antibodies, a key component of the adaptive immune response46. Interestingly, a recent GWAS-study associated IGHV1-68 with the aberrant immune response associated with rheumatic heart disease35. However, the association with AD is novel, and warrants further studies into immunological pathways in AD47. A potential weakness of the present study entails the inclusion of younger population controls and the inclusion of MCI cases diagnosed with the Winblad criteria which lack AD biomarker profiling. While the population controls boosted the statistical power in our study, it may include controls that may later develop AD at disease prevalence rate. Thus, this study might not have reached its potential full statistical power. On the inclusion of MCI cases, although not all subjects diagnosed with MCI will ultimately develop AD, a large proportion of those with amnestic MCI and mixed type of MCI are expected to develop AD48. The potential inclusion of non-AD MCI cases may have reduced the strength of our findings. However, this only reduces the chance of finding GWAS loci, i.e. likely induce type-II errors but does not affect the rate of type-I errors.
To conclude, our findings further demonstrate the polygenic architecture of AD, and unveil potential mechanisms including the immune system involved in AD pathobiology that warrant further investigation.
Data Availability
Genotype datasets from the Norwegian DemGene network generated and analysed during the current study are not publicly available due to compliance to privacy. Summary statistics are available from the corresponding author on reasonable request. The summary statistics from IGAP are publicly available at http://web.pasteur-lille.fr/en/recherche/u744/igap/igap_download.php. Data from Icelandic study are available from deCODE Genetics on reasonable request.
References
Ferri, C. P. et al. Global prevalence of dementia: a Delphi consensus study. Lancet 366, 2112–2117 (2005).
Sosa-Ortiz, A. L., Acosta-Castillo, I. & Prince, M. J. Epidemiology of dementias and Alzheimer’s disease. Arch Med Res 43, 600–608 (2012).
Hurd, M. D., Martorell, P., Delavande, A., Mullen, K. J. & Langa, K. M. Monetary costs of dementia in the United States. N Engl J Med 368, 1326–1334 (2013).
Langa, K. M. et al. National estimates of the quantity and cost of informal caregiving for the elderly with dementia. J Gen Intern Med 16, 770–778 (2001).
Braak, H. & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259 (1991).
Sperling, R. A. et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 280–292 (2011).
Bateman, R. J. et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367, 795–804 (2012).
Dean, D. C. III et al. Brain differences in infants at differential genetic risk for late-onset Alzheimer disease: a cross-sectional imaging study. JAMA Neurol 71, 11–22 (2014).
Loy, C. T., Schofield, P. R., Turner, A. M. & Kwok, J. B. Genetics of dementia. Lancet 383, 828–840 (2014).
Bertram, L. & Tanzi, R. E. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci 9, 768–778 (2008).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993).
Poirier, J. et al. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 342, 697–699 (1993).
Meyer, M. R. et al. APOE genotype predicts when–not whether–one is predisposed to develop Alzheimer disease. Nat Genet 19, 321–322 (1998).
Glazier, A. M., Nadeau, J. H. & Aitman, T. J. Finding genes that underlie complex traits. Science 298, 2345–2349 (2002).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452–1458 (2013).
Jun, G. et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 21, 108–117 (2016).
Steinberg, S. et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet 47, 445–447 (2015).
Seshadri, S. et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama 303, 1832–1840 (2010).
Hollingworth, P. et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43, 429–435 (2011).
Guerreiro, R. et al. TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127 (2013).
Jonsson, T. et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. New England Journal of Medicine 368, 107–116 (2013).
Cruts, M., Theuns, J. & Van Broeckhoven, C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33, 1340–1344 (2012).
Wagner, G. P. & Zhang, J. The pleiotropic structure of the genotype-phenotype map: the evolvability of complex organisms. Nature reviews. Genetics 12, 204–213 (2011).
Ridge, P. G., Mukherjee, S., Crane, P. K., Kauwe, J. S. & Alzheimer’s Disease Genetics, C. Alzheimer’s disease: analyzing the missing heritability. PLoS One 8 e79771 (2013).
Gatz, M. et al. Heritability for Alzheimer’s disease: the study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci 52, M117–125 (1997).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Athanasiu, L. et al. Gene variants associated with schizophrenia in a Norwegian genome-wide study are replicated in a large European cohort. J Psychiatr Res 44, 748–753 (2010).
Tian, C., Gregersen, P. K. & Seldin, M. F. Accounting for ancestry: population substructure and genome-wide association studies. Hum Mol Genet 17, R143–150 (2008).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics 81, 559–575 (2007).
Li, Y., Willer, C. J., Ding, J., Scheet, P. & Abecasis, G. R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genetic epidemiology 34, 816–834 (2010).
Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8, 1826 (2017).
Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
CARDIoGRAMplusC4D Consortium et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 45, 25–33 (2013).
Parks, T. et al. Association between a common immunoglobulin heavy chain allele and rheumatic heart disease risk in Oceania. Nat Commun 8, 14946 (2017).
Jun, G. R. et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement 13, 727–738 (2017).
Desikan, R. S. et al. Polygenic Overlap Between C-Reactive Protein, Plasma Lipids, and Alzheimer Disease. Circulation 131, 2061–2069 (2015).
Miller, J. A. et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014).
Allen Institute for Brain Science. BrainSpan Atlas of the Developing Human Brain [Internet]. Available from, http://brainspan.org (2016).
Huynh, M. B. et al. Glycosaminoglycans from aged human hippocampus have altered capacities to regulate trophic factors activities but not Abeta42 peptide toxicity. Neurobiol Aging 33(1005), e1011–1022 (2012).
Sepulveda-Diaz, J. E. et al. HS3ST2 expression is critical for the abnormal phosphorylation of tau in Alzheimer’s disease-related tau pathology. Brain 138, 1339–1354 (2015).
Liu, C. C. et al. Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer’s disease. Sci Transl Med 8, 332ra344 (2016).
Ruiz-Orera, J. et al. Origins of De Novo Genes in Human and Chimpanzee. PLoS Genet 11, e1005721 (2015).
Scheckel, C. et al. Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. Elife 5 (2016).
Matsuda, F. et al. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. The Journal of experimental medicine 188, 2151–2162 (1998).
Lefranc, M.-P. & Lefranc, G. The immunoglobulin factsbook, (Academic press, 2001).
Heppner, F. L., Ransohoff, R. M. & Becher, B. Immune attack: the role of inflammation in Alzheimer disease. Nature Reviews Neuroscience 16, 358 (2015).
Fladby, T. et al. Detecting At-Risk Alzheimer’s Disease Cases. J Alzheimers Dis 60, 97–105 (2017).
Acknowledgements
The project has received funding from The Research Council of Norway (RCN) Grant Nos. 213837, 223273, 225989, 248778, and 251134 and EU JPND Program ApGeM RCN Grant No. 237250, the South-East Norway Health Authority Grant No. 2013-123, the Norwegian Health Association, and KG Jebsen Foundation. The RCN FRIPRO Mobility grant scheme (FRICON) is co-funded by the European Union’s Seventh Framework Programme for research, technological development and demonstration under Marie Curie grant agreement No 608695. European Community’s grant PIAPP-GA-2011-286213 PsychDPC. We thank the International Genomics of Alzheimer’s Project (IGAP) for providing summary results data for these analyses. The investigators within IGAP contributed to the design and implementation of IGAP and/or provided data but did not participate in analysis or writing of this report. IGAP was made possible by the generous participation of the control subjects, the patients, and their families. The i–Select chips was funded by the French National Foundation on Alzheimer’s disease and related disorders. EADI was supported by the LABEX (laboratory of excellence program investment for the future) DISTALZ grant, Inserm, Institut Pasteur de Lille, Université de Lille 2 and the Lille University Hospital. GERAD was supported by the Medical Research Council (Grant no. 503480), Alzheimer’s Research UK (Grant no. 503176), the Wellcome Trust (Grant no. 082604/2/07/Z) and German Federal Ministry of Education and Research (BMBF): Competence Network Dementia (CND) grant no. 01GI0102, 01GI0711, 01GI0420. CHARGE was partly supported by the NIH/NIA grant R01 AG033193 and the NIA AG081220 and AGES contract N01–AG–12100, the NHLBI grant R01 HL105756, the Icelandic Heart Association, and the Erasmus Medical Center and Erasmus University. ADGC was supported by the NIH/NIA grants: U01 AG032984, U24 AG021886, U01 AG016976, and the Alzheimer’s Association grant ADGC–10–196728.
Author information
Authors and Affiliations
Contributions
O.A.A. designed the study. A.W. and O.A.A. wrote manuscript. A.W., T.H., F.B., Y.W., L.A., S. Ba., S.L.-H., S.G., A.M.D. and O.A.A. performed analysis on DemGene data. A.R., I.S.A., I.D.U., L.R.W., G.S., E.S., F.A., A.B., I.S., K.E., S. Be., G.B., N.B., S.B.S., D.A., S.D., T.F. and O.A.A. collected DemGene data. S.S., H.S., J.N., K.S. provided data and analysis in Icelandic cohort. A.W., A.R., A.E., T.H., F.B., A.M.D., S.S., H.S., R.S.D., K.S., D.A., S.D., T.F. and O.A.A. interpreted the results. All authors commented on and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Witoelar, A., Rongve, A., Almdahl, I.S. et al. Meta-analysis of Alzheimer’s disease on 9,751 samples from Norway and IGAP study identifies four risk loci. Sci Rep 8, 18088 (2018). https://doi.org/10.1038/s41598-018-36429-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36429-6
This article is cited by
-
The 3-O sulfation of heparan sulfate proteoglycans contributes to the cellular internalization of tau aggregates
BMC Molecular and Cell Biology (2022)
-
Associations of Alzheimer’s disease risk variants with gene expression, amyloidosis, tauopathy, and neurodegeneration
Alzheimer's Research & Therapy (2021)
-
Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes
Nature Genetics (2021)
-
Long runs of homozygosity are associated with Alzheimer’s disease
Translational Psychiatry (2021)
-
Epigenome-wide association study of Alzheimer’s disease replicates 22 differentially methylated positions and 30 differentially methylated regions
Clinical Epigenetics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
