
Abstract
Inflammation is considered as a major contributor to brain injury following cerebral ischemia. The therapeutic potential of both MLC601/MLC901, which are herbal extract preparations derived from Chinese Medicine, has been reported both in advanced stroke clinical trials and also in animal and cellular models. The aim of this study was to investigate the effects of MLC901 on the different steps of post-ischemic inflammation in focal ischemia in mice. In vivo injury was induced by 60 minutes of middle cerebral artery occlusion (MCAO) followed by reperfusion. MLC901 was administered in post-treatment 90 min after the onset of ischemia and once a day during reperfusion. MLC901 treatment resulted in a reduction in infarct volume, a decrease of Blood Brain Barrier leakage and brain swelling, an improvement in neurological scores and a reduction of mortality rate at 24 hours after MCAO. These beneficial effects of MLC901 were accompanied by an inhibition of astrocytes and microglia/macrophage activation, a drastically decreased neutrophil invasion into the ischemic brain as well as by a negative regulation of pro-inflammatory mediator expression (cytokines, chemokines, matrix metalloproteinases). MLC901 significantly inhibited the expression of Prx6 as well as the transcriptional activity of NFκB and the activation of Toll-like receptor 4 (TLR4) signaling, an important pathway in the immune response in the ischemic brain. MLC901 effects on the neuroinflammation cascade induced by cerebral ischemia probably contribute, in a very significant way, in its potential therapeutic value.
Similar content being viewed by others

Introduction
The focal hypoperfusion of the brain induced by ischemic stroke leads to excitotoxic cell death, to oxidative damage and to inflammation1. Inflammatory processes are present at all steps of the ischemic cascade and participate to both stroke-induced brain damage and to brain repair2.
Due to the disruption of the blood-brain-barrier (BBB), the recruitment and the infiltration of circulating leukocytes (neutrophils, lymphocytes and monocytes) in the ischemic brain are central events of post-ischemic inflammation. On the other hand, microglia or resident brain macrophages are the major inflammatory cell types in the CNS. Activation of microglia takes place after ischemic stroke and peaks at 2–3 days. The process lasts for weeks after the initial injury3. The ischemia-induced activation of both microglia and astocytes2 leads to release of pro-inflammatory cytokines such as interleukin 1 β (IL-1β) and tumor necrosis factor α (TNFα)4,5,6 as well as of chemokines7,8. These pro-inflammatory mediators facilitate BBB damage that favors the migration of circulating leukocytes into the ischemic brain3. However, both microglia and astrocytes have a dual function in ischemic stroke. Microglia can also produce TGF-β1, which has a neuroprotective role and astrocytes can induce the production of a number of molecules which activate anti-inflammatory responses9.
Cerebral ischemia triggers activation of Toll-like receptors (TLR)2, via endogenous ligands2 of the danger-associated molecular patterns (DAMPS), that are released from ischemic tissues10. TLR4 seems to be particularly associated with ischemic stroke11. Among the DAMPS released after stroke, there are members of the peroxiredoxin (Prx) family, and particularly Prx5 and Prx6 (increased 12–24 h after stroke onset). Both Prx5 and Prx6 induce activation of infiltrating macrophages and of inflammatory cytokines from invading T lymphocytes12. Ischemia-induced TLR4 signaling leads to activation of the transcription factor NF-κB and subsequent transcriptional induction of pro-inflammatory genes, such as cellular adhesion molecules, growth factors, matrix metalloproteinases (MMPs) and cytokines from immune cells, resulting in potent post-ischemic neuroinflammation and brain injury10.
The inflammatory process linked to brain ischemia is definitively complex but now well explored and provides new potential targets for future treatments for a brain disease that is very much at present in search of novel therapeutic approaches.
The recent failure of so many “classical” therapeutic strategies for stroke patients, despite promising preclinical data13,14,15, has encouraged a search on the potential therapeutic effects of cocktails of extracts of natural plants which are the active constituents of Traditional Chinese Medicine (TCM). This “medicine” has been playing an important role in health protection and disease control for thousands of years in Asia. Its potential therapeutic efficacy is usually attributed to the synergistic property of multiple herbal constituents that provide a combinational therapeutic strategy that improves the efficacy through hitting multiple targets. In the field of stroke, MLC601 (extracts of 14 natural ingredients) and MLC901 (extracts of 9 herbal components) have recently emerged as a promising treatment for improving functional recovery of patients after ischemic stroke16,17,18,19,20,21,22,23,24,25,26,27. The CHInese Medicine neuroaid Efficacy on Stroke recovery (CHIMES) trial in patients with stroke of intermediate severity reported that MLC601 is a safe treatment that reduces the early recurrent vascular events and vascular deaths in post-stroke patients17,28,29,30,31. In the Philippines cohort of the CHIMES trial (378 patients), which included more patients with predictors of poorer prognosis, MLC601 improves their functional recovery22. A recent extension study of the CHIMES trial (CHIMES-E) revealed that a 3-month treatment with MLC601 improved the functional outcome for up to 2 years among patients with stroke of intermediate severity22,32. All these clinical results are encouraging to pursue further exploration of MLC601 and MLC901 as a new efficient therapeutic strategy against stroke. Clinical trials have been successfully carried out with MLC901 for patients with traumatic brain injury33,34 and are ongoing for vascular cognitive impairment (Neurites)35. Consistent with clinical observations of the benefit of this TCM in humans, MLC601 and MLC901 have been extensively studied in vitro and in vivo using rodent models of focal and global ischemia as well as of traumatic brain injury36,37,38,39,40. The neuroprotective and neuroregenerative properties of the two mixtures have been reported to be comparable36. These preclinical studies have pointed out a marked beneficial effect of MLC901 on activation of ATP-sensitive potassium channels41, a process known to be neuroprotective against stroke42 as well on the repair process including neurogenesis, neurite outgrowth and angiogenesis36,43.
Because inflammation and its signaling pathways are considered to play key roles in the ischemic cascade as well as in the repair process, this study investigates the potential anti-inflammatory effects of MLC901 in a model of transient focal cerebral ischemia in mice, and analyzes the effect of the TCM on the different mechanisms that trigger the inflammatory process.
Results
MLC901 protects ischemia-induced brain damage, BBB breakdown, cerebral edema and neurological dysfunction
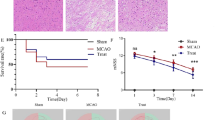
The flowchart illustrating the experimental design is shown in Fig. 1A. To confirm the protective effects of MLC901 with the dose and the protocol used in this work, survival rate, cerebral infarction, BBB disruption, cerebral edema and neurological dysfunction induced by 60 min MCAO were evaluated 30 hours following stroke, according to our previous works on focal ischemia36,43. The mortality rate was reduced in MLC901-treated mice compared to vehicle-treated animals (Fig. 1B, 66.7% survival in MLC901 group as compared to 83.3% survival in the vehicle group,). MLC901 also attenuated markedly the infarction size produced by 60 min of ischemia (Fig. 1C; 43.9 ± 0.2 mm3 in the MLC901 group versus 59.4 ± 0.3 mm3 in the vehicle group, #P < 0.05). MLC901 strongly reduced the early BBB leakage (Fig. 1D and reduced brain edema (Fig. 1E). The FITC-dextran staining appeared inhomogeneous in cortical and subcortical regions, reflecting probably the regular presence of ‘mini cores and penumbras’ in the ischemia-affected area44. These results fully confirm previous observations on the decrease of cerebral infarction induced by a MLC901 treatment36,43,45.

MLC901 reduced mortality rate, cerebral infarct volume, BBB leakage, brain edema and neurologic deficits induced by focal 60-min ischemia. (A) Flowchart illustrating the experimental design. (B) Percentage of survival rate and (C) Infarct volume (mm3) measured 30 hours post-ischemia (n = 10 per experimental group). (D) Representative images of FITC-dextran leakage (in green) in brain sections of mice submitted to focal ischemia and observed 120 min after MCAO (n = 4 per experimental group). (E) Percentage of brain water content in ipsilateral cortical segments measured 30 hours post-ischemia. (n = 6 per experimental group,). (F) Neurological score 30 hours post-ischemia (n = 10 per experimental group). MLC901 was intraperitoneally injected with a single dose of MLC901 (40 μg/kg) diluted in saline (as vehicle) 30 min after the onset of ischemia. Data are reported as mean ± SEM. #P < 0.05 versus vehicle ischemic group (Mann & Whitney test).
The neurological outcome was also improved by MLC901. Ischemia-induced neurological deficits, assessed by neurological Bederson scores, were significantly attenuated in MLC901-treated mice 30 hours after MCAO (Fig. 1F, 1.8 ± 0.5 versus 3.15 ± 0.2 in the vehicle group, #P < 0.05) suggesting a better functional recovery of animals. This result is in line with previous data from our laboratory showing MLC901 efficacy to decrease the impact of stroke on neurological deficits by using a variety of behavioral tests including rotarod, pole test, grip test, Morris water maze test37,38,43 in addition to Bederson score36.
MLC901 decreased the stroke-induced pro-inflammatory infiltration of phagocytes
Brain ischemia induces an early microglial activation and an infiltration of circulating leukocytes to the brain2,3,46. Using specific surface marker labeling coupled to a detailed flow cytometry analysis at 24 hours post-ischemia, we were able to distinguish CNS-inflammatory phagocytes and microglia based on differential antigen expression in ischemic brains47. Microglia and phagocytes were essentially identified based on their CD11b and CD45 expression levels. Microglia express high levels of CD11b and low levels of CD45 (CD11b+/CD45low+) while the heterogeneous population of CNS-associated phagocytes including brain macrophages express high levels of both CD11b and CD45 (CD11b+/CD45high+)47. With the CD11b and CD45 labeling (Fig. 2A), three different populations were identified in the ischemic brains: CD11b+/CD45high+ (CNS-associated phagocytes), CD11b+/CD45low+ (microglia), and CD11b−/CD45 high+ cells (immune cells) and their number changed in function of treatment and reperfusion. Figure 2A,B shows the results obtained at 24 hours following stroke. Compared to the sham group, MCAO induced a strong increase of CNS-associated phagocytes both in saline- and MLC901-treated mice (***P < 0.01 versus the sham group). Reperfusion alone did not change the number of CNS-associated phagocytes in vehicle-treated mice. However, compared to vehicle, MLC901 induced a significant decrease of phagocytes infiltration for animals which have reperfused ($$$P < 0.001 versus the vehicle-treated group) (Fig. 2B). MLC901 also preserved the normal pool of microglial cells that was strongly decreased after MCAO in vehicle-treated mice. The number of microglia cells was close to that of sham animals in the MLC901-treated group. There was no effect of reperfusion but a strong effect of MLC901.

MLC901 reduced the increase in CNS-associated phagocytes induced by focal 60-min ischemia in mouse brains. (A) Representative bivariate dot plots of Percoll isolated brain cells illustrating on the identification of microglia (CD11b+/CD45low+), CNS-associated phagocytes (CD11b+/CD45high+) and immune cells (CD11b−/CD45high+). (B) Histograms of average percentage of CNS- associated phagocytes (left) and microglia (right) in live single immune cells from reperfused (R) or not reperfused (NR) brains of mice submitted to 60-min focal ischemia and treated with vehicle (black bars) or MLC901 (ip injection, 40 μg/kg) (n = 6 to 10 per experimental group) and sacrificed 24 hours after MCAO. Data are reported as mean ± SEM. **P < 0.01, ***P < 0.001 versus sham-operated group and $$P < 0.01, $$$P < 0.001 versus vehicle ischemic group, #P < 0.05 versus non-reperfused MLC901-group (Kruskal-Wallis test, H(4,54) = 35.39).
To further characterize which cell types migrate from periphery to the brain and the effect of MLC901, we performed a detailed analysis of CD11b+/CD45 high+ cells by flow cytometry47. Ly6C and Ly6G surface marker labeling allowed us to discriminate between three subpopulations among immune brain CD11b+/CD45high+ cells (phagocytes). Neutrophils were identified as CD11b+/CD45 high+/Ly6C intermediate+/Ly6G high+ cells and macrophages were identified as CD11b+/CD45 high+/Ly6G neg− cells47. Among them, resident macrophages were identified as Ly6C neg- and inflammatory monocytes as Ly6C high+ cells (Fig. 3A). Neutrophils are the first leukocyte subpopulation to be recruited to the ischemic brain. As compared to the sham group, an extensive and significant infiltration of neutrophils was observed 24 hours after MCAO in the vehicle group (Fig. 3B, ***P < 0.001). Reperfusion alone had no effect. Interestingly, MLC901 strongly decreased the percentage of neutrophils, particularly when there was reperfusion (Fig. 3B, left panel). The percentage of inflammatory monocytes increased in the vehicle-treated mice as compared to values observed in sham animals (**P < 0.01). Reperfusion alone had no influence on the percentage of inflammatory monocytes whatever the group. MLC901 completely prevented the selective migration of inflammatory monocytes to the brain ($$P < 0.01). The percentage of resident macrophage was not affected by MCAO. Neither reperfusion alone nor MLC901 had any effect on the proliferation of resident macrophages (Fig. 3B, right panel). These results, in fact, do not exclude the possibility of a proliferation of resident macrophages induced by MCAO, but it could be that the effect, if it exists, is in fact masked by the dramatic infiltration of neurophils in the brain following stroke or else be delayed in time47.

MLC901 reduced the infiltration of neutrophils and inflammatory monocytes induced by focal 60-min ischemia in mouse brains. (A) Representative bivariate dot plots of CNS-phagocytes stained for Ly6G and Ly6C illustrating a gating strategy to identify Ly6Cintermediate+/Ly6Ghigh+ neutrophils, Ly6C−/Ly6G− resident macrophages and Ly6Chigh+/Ly6G− inflammatory monocytes. (B) Histograms of average percentage of neutrophils (left), inflammatory monocytes (middle) and resident macrophages (right) in CNS-associated phagocyte cell population from reperfused (R) or not reperfused (NR) brains of mice submitted to 60-min focal ischemia and treated with vehicle (black bars) or MLC901 (ip injection, 40 μg/kg) (n = 6 to 10 per experimental group) and sacrificed 24 hours after MCAO. Data are reported as mean ± SEM. **P < 0.01, ***P < 0.001 versus sham-operated group and $$P < 0.01 versus vehicle ischemic group (Kruskal-Wallis test, H(4,56) = 23.16).
The brain infiltration of lymphocytes 24 hours after MCAO has also been analyzed. Among the CD11b−/CD45 high+ population, we compared CD3−/B220+ (B lymphocytes), B220−/CD3+/CD4+ expressing cells (CD4+ lymphocytes) and B220−/CD3+/CD8+ (CD8+ lymphocytes) expressing cells47 in ischemic brains from vehicle- and MLC901-treated mice. While we observed no statistical difference for B-lymphocytes, we detected a significant infiltration of T- lymphocytes. There was no effect of reperfusion or MLC901 treatment at this time point (data not shown).
To further analyze neutrophil infiltration to the ischemic brain we used myeloperoxidase (MPO) as biomarker. MPO is a pro-oxidative and pro-inflammatory enzyme, mainly released by activated neutrophils48. One day after MCAO, immunohistochemical MPO staining showed that MPO-positive cells were predominantly detected in the penumbra as previously reported46. MLC901 significantly reduced MPO staining, an indication of reduced neutrophil infiltration (Fig. 4B, 115 ± 41 in MLC901 group versus 148 ± 38 AU pixel intensity in vehicle group, P < 0.05 as compared to the vehicle-treated group). Glial fibrillary acid protein (GFAP) is a biomarker for astrocyte activation. One day after MCAO, GFAP was highly expressed in the peri-ischemic zone of the vehicle-treated animals (Fig. 4C). Treatment with MLC901 resulted in a significant decrease in the number of activated astrocytes compared with vehicle-treated ischemic mice (380 ± 42 in MLC901 group versus 770 ± 58 AU pixel intensity, P < 0.05, Mann & Whitney test, Fig. 4C).

MLC901 decreased microglial activation and neutrophil recruitment in the reperfused brain 24 hours following MCAO. (A–D) Representative immunohistochemical staining for Iba1 microglia marker (A) myeloperoxidase (MPO) (B) and GFAP astrocytic marker (C) in brains of vehicle- and MLC901-treated mice 24 hours after MCAO. (A) Representative views of Iba1 immunopositive cells in ischemic penumbra, ischemic core and contralateral control side of vehicle- and MLC901-treated mice. Diagram boxes indicate the regions where the images in middle and bottom panels at higher magnification were acquired (B,C) Representative immunostaining of MPO (B) and GFAP (C) in ischemic penumbra of vehicle- and MLC901-treated mice. (D) Quantitation of Iba1 expression (AU positive microglia pixel intensity) in ischemic penumbra and ischemic core of vehicle- and MLC901-treated mice. Scale bar, 20 μm (top panels), 50 μm (middle and bottom panels) (n = 6 per experimental group).
Iba-1 is specifically expressed in microglia/macrophages and is up-regulated during activation of these cells by ischemia i.e. when they switch morphologically from a ramified type with retracted processes extending from the cell perikarya to an activated amoeboid form49. One day after MCAO, Iba1 expression in microglia was strongly increased in both penumbra and ischemic core regions of vehicle-treated mice (Fig. 4A,D) as compared to contralateral control (Fig. 4A,D). In the ischemic core area of the vehicle group, the Iba1-positive cells were a mixing of ramified and amoeboid cells, while most of Iba1 cells were ramified in the penumbra (Fig. 4A). In the penumbra, the MLC901-treatment resulted in a significant decrease in the number of Iba1-positive cells when compared with the ischemic vehicle group (Fig. 4A,D). However, MLC901 induced no significant effect on microglia activation in the ischemic core (Fig. 4A,D).
MLC901 decreased the expression of pro-inflammatory mediators induced by stroke
Then, it became important to analyze whether MLC901 was able to alter the gene expression of different cytokines and chemokines induced by ischemia (Fig. 5). In the vehicle-treated group, stroke induced a many-fold increase of levels of M1-type50 pro-inflammatory mRNA-encoding cytokines, particularly IL11 (in the reperfused group, only), IL1β, IL6, and TNFα (in reperfused and not reperfused groups) as well as the levels of chemokines such as CCL2, CCL3, CCL4, CCL7, CCL11 and CXCL1 (in both groups). Reperfusion, in this case, played a key role on the reduction of M1-type pro-inflammatory cytokines and chemokines, particularly for the efficiency of MLC901. MLC901 decreased the IL-11, IL-1β, IL-6 and TNFα mRNA levels by 93.2, 43.1, 74.9 and 75.5% respectively, in the animals with reperfusion versus 43.9, 34.3, 60.7, and 68.7% in the group without reperfusion (Fig. 5A–C). Interestingly, MLC901 also increased the levels of ILR-4Rα expression, a specific marker of the anti-inflammatory M2 state50. The same observation was made for chemokines. For example, the transcriptional levels of CCL11, CCL2, CCL7 and CXCL1 was decreased by 10.0-, 3.2-, 3.4- and 3.2-folds, respectively in the reperfused group versus 1.2-, 1.4-, 1.1 and 1.6-fold in the non-reperfused mice (Fig. 5B–D). Reperfusion induced a 6.5-fold up-regulation of the anti-inflammatory cytokine, IL10, but in this case, MLC901 had no further effect on this cytokine expression.

Analysis of the effect of MLC901 treatment in inflammatory marker gene expression in mouse brains after MCAO. Arrays of interleukin, cytokine (A,B, left panels) and CC chemokine ligand and receptor (A, B, right panels) gene expression in the injuried cortex of vehicle- and MLC901-treated mice 24 hours after MCAO with (A,C) or without reperfusion (B,D) (n = 6 per experimental group). MCAO-induced fold upregulation versus sham group was calculated using the ∆∆CT method according to the manufacturer protocol. Data are expressed as mean ± SEM. #P < 0.05, ##P < 0.01 versus sham and $P < 0.05 versus vehicle-group (n = 5 per experimental group) (Mann & Whitney test).
The protein expression of M1-type pro-inflammatory cytokines/chemokines in the ischemic brains from vehicle- and MLC901-treated mice 24 hours after MCAO is presented in Fig. 6. MCAO induced a large increase of IL6, CCL2 and TNFα in the vehicle-treated group, as previously observed5. Reperfusion, by itself, decreased significantly the elevated pro-inflammatory cytokine/chemokine protein profile (#P < 0.05 versus Saline-MCAO R and *P < 0.05 versus MLC901-MCAO R). MLC901 significantly reduced the ischemia-induced activation of IL6, CCL2 and TNFα in the reperfused and non-reperfused groups ($P < 0.05 versus the vehicle groups (NR and R)). As previously observed for the real time PCR results, the best effect of MLC901 was obtained with reperfused animals.

MLC901 decreased the increase of pro-inflammatory cytokine/chemokine protein concentration in mouse brains 24 hours after MCAO. The pro-inflammatory cytokines IL6, TNFα and chemokine CCL2 were quantified by cytometric bead array. Data were obtained from reperfused (R) and not reperfused (NR) brains. Bars represent the mean ± SEM. $P < 0.05 versus vehicle-group, #P < 0.0 versus non-reperfused vehicle-group, *P < non-reperfused MLC901-group (n = 6 to 10 per experimental group) (Mann & Whitney test).
Expression and activation of matrix metalloproteases (MMP-9) following cerebral ischemia are closely associated with a disruption of BBB resulting from infiltrated peripheral neutrophils51. Figure 7A shows that 24 h after focal ischemia the increase of MMP-9 expression induced by MCAO in the reperfused vehicle-treated group was significantly reduced by MLC901 treatment (#P < 0.05 versus the reperfused vehicle group).

MLC901 decreased the protein expression of matrix metalloproteinase-9 (MMP-9), peroxiredoxin 6 (Prx6), Toll-like receptor 4 (TLR4) and the phosphorylation of NFκB (pNFκB) in reperfused (R) brains 24 hours after MCAO. (A–D) Representative images from Western blotting analysis of MMP-9 (A) Prx6 (B) TLR4 (C) and phosphorylated and total NFκB (left panels). Optical densitometry quantitation for MMP-9 (92 kDa), Prx6 (25 kDa), TLR4 (85 kDa) normalized to β actin and for pNFκB (65 kDa) normalized to total NFκB and β actin in injured cortical tissue (right panels). β actin was used as internal control for the loading of protein level. Data are representative of 3 separate experiments (n = 4 per group). Values (mean ± SEM) are expressed as percentage of control (#P < 0.05, ##P < 0.01 versus vehicle ischemic group) (Mann & Whitney test).
MLC901 inhibits MCAO-induced expression of peroxiredoxin 6, TLR4 signaling and phosphorylation of NFkB
Peroxiredoxins (Prx’s), and particularly Prx6 are important contributors to immunomodulation and neuroinflammation after ischemic stroke12. As shown in Fig. 7B (left panel), the expression of Prx6 was strongly increased in the ischemic brain of reperfused vehicle-treated group compared with that observed in sham control group. MLC901 markedly decreased Prx6 expression 24 hours after MCAO (#P < 0.05 versus the vehicle-group, Fig. 7B right panel).
TLR4 is also related to inflammation and is associated with the pathological progression of cerebral ischemia2,11. As shown in Fig. 7C (left panel) and as already reported11, cerebral ischemia significantly induced TLR4 expression. This increased TLR4 expression did not take place in MLC901-treated mice (##P < 0.05 versus the vehicle group, Fig. 7C, right panel).
The NFκB signaling pathway is well-known to be associated with immune responses in the ischemic brain52 and NFκB activation is a major signaling system associated with TLR453. Activation of NFκB was observed in response to MCAO (Fig. 7D, left panel), involving phosphorylation of the p65 NFκB subunit53. In the ischemic brain of vehicle-treated animals, the level of phosphorylated NFκB was strongly increased in the ischemic group as compared to the sham group, while the level of total NFκB was not changed (Fig. 7D, left panel). This increase of NFκB phosphorylation was significantly decreased by MLC901 treatment (#P < 0.05 versus the vehicle-treated group, Fig. 7D, right panel).
Discussion
Accumulating evidence from animal studies36,37,38,39,40,54 and clinical trials17,22,23,26,28,29,31,55 indicates that MLC601 and its derivative product MLC901 probably represent a promising therapeutic strategy for brain injuries, and particularly for stroke treatment. The molecular and cellular mechanisms associated with the beneficial effects of this TCM in cellular and animal models of stroke are multifaceted. MLC901 has neuroprotective effects by acting on key players controlling the ischemic cell death. It activates ATP sensitive potassium channels (KATP)41, a process that is neuroprotective56,57 and is also involved in the beneficial effects of preconditioning57,58. MLC901 decreases excitotoxicity36,41 and stimulates the Akt survival pathway. It reduces apoptosis, oxidative stress and free radical generation37. MLC901 is also known to improve neurorepair by stimulating BDNF production, neurogenesis, synaptogenesis and angiogenesis36,43.
The present work confirms that MLC901 administered 90 min after MCAO onset, and then during the entire time of recovery, significantly protects mice against brain injury after focal ischemia. MLC901 increases survival after MCAO, reduces the infarct volume and improves the neurological score. All these results are in line with previous preclinical results36,37,38,45 and also with clinical trials17,22,23,28,29,31,55.
The breakdown of the BBB during post-ischemic reperfusion is associated with interdependent mechanisms including inflammation, oxidative and nitrosative stress, and induction of matrix-metalloproteinases59,60. Preclinical studies examining BBB opening after ischemia have shown the existence of a biphasic permeability response (1–4 h leading to cytotoxic edema and 22–46 hours leading to vasogenic edema)60,61, while others, using MRI have found a continuous BBB leakage during one week62. In humans, the situation is quite comparable with a continuous BBB opening for up to 4 days after stroke63. In previous studies, we have reported the beneficial MLC901 effect on BBB disruption at a later stage (30 hours) both after focal ischemia43 and TBI38. In the present work, we analyzed the early BBB leakage with the FITC-dextran post-ischemia and observed that MLC901 attenuates the severe BBB breakdown as early as 3 hours post-MCAO and the subsequent hemispheric swelling.
While, in the present work, MLC901 was injected 90 min after MCAO onset, previous studies on animal models have reported a window of protection for MLC901 up to 3 h37, a long delay for mice and a delay which seems compatible with the clinical situation (early stroke management of patients in stroke centers and mobile stroke units). The anti-inflammatory effects of MLC901 probably persist for longer times, particularly for patients who continue to take the compound for periods as long as three months.
In the present study we extended the analysis of the potential beneficial effects of MLC901 in stroke treatment by examining more specifically the effects of MLC901 on inflammation in the MCAO model. Indeed, post-ischemic inflammation is now considered as an important potential target for therapeutic intervention1,2. Clinically, systemic inflammation seems to increase the risk of stroke and to be associated with a worse prognosis64,65. Stroke patients with systemic inflammation apparently exhibit poorer recovery66.
Intraluminal MCAO with thread filament used in the present work is one of the most relevant and widely used stroke models because it better mimics the pathophysiology of human stroke compared with permanent occlusion models in rats and mice. Most cases of human ischemic stroke have spontaneous or thrombolytic therapy-induced reperfusion67. Preclinical stroke studies have demonstrated that reperfusion represents an especially vulnerable period for the brain. It, of course, provides the important benefit of restoring blood flow to the ischemic region and, simultaneously, it induces a massive influx of activated leukocytes into the injured zone68,69. Reperfusion hastens inflammation70. This is why we have analyzed the effects of MLC901 against inflammation in different states of brain reperfusion.
Focal ischemia induces a rapid activation of resident cells (mainly microglial cells), production of proinflammatory mediators, and infiltration of circulating inflammatory cells (including neutrophils, T cells and monocytes/macrophages), into the ischemic brain tissue3. It is well known that inhibiting the inflammatory response decreases infarct size and improves neurological deficits in experimental stroke2,71, a result which is also seen in the present work.
Neutrophils are among the first cells in the blood to respond after ischemic stroke, after which they are phagocyted by microglia and macrophages2,3,72,73. Neutrophil infiltration is recognized as an important pathogenic factor following stroke73,74. In the present work, we observed a marked increase in neutrophil infiltration following the ischemic insult in the injured cortex of vehicle-treated mice at 24 hours post-ischemia. MLC901 treatment significantly decreased this neutrophil infiltration, suggesting that this TCM attenuates the neuronal damage caused by stroke, at least in part, via inhibition of harmful neutrophil recruitment. Several drugs targeting neutrophil recruitment such as enlimomab75, Hu23F2G or Leukoarrest76 and UK-27927677 were previously tested in clinical trials but have unfortunately not been successful, in part, for some of them, because of side-effects including leukopenia and immune suppression. MLC901 seems to be in a different category with very satisfactory safety properties observed in large cohorts of patients24 and benefits seen both for patients17,18,22,23,28,29,30,31,32,33,34,35,55,78 and in animal studies36,37,38,39,40. MLC901 provides an interesting therapeutic alternative for preventing neutrophil infiltration in stroke.
In addition to neutrophils, T-lymphocytes are the major leukocyte subpopulation involved in inflammatory brain damage after stroke2. While we observed, at 24 hours following ischemia, a significant infiltration of T - lymphocytes in the vehicle-treated group, MLC901 had no effect. There are many subtypes of T-lymphocytes and the time course of their recruitment remains largely undetermined. Earlier studies suggested that lymphocytes recruitment peaked between 3 and 7 days79 leading to the possibility that MLC901 could have had a positive effect on T lymphocytes accumulation, not in the first 24 hours after ischemia, but in a later phase that has not been analyzed in the present work.
Microglia/macrophages are highly plastic cells that can express two different and apparently opposite phenotypes: a cytotoxic M1 polarization associated to the release of destructive proinflammatory mediators such as pro-inflammatory cytokines, reactive oxygen species (ROS) and proteinases and a protective M2 polarization associated to tissue repair2,50. Therefore, a control of the balance between detrimental and beneficial microglia/macrophage responses has been considered a probable therapeutic strategy against stroke50. Stroke reinforces deleterious M1 polarization and decreases repairing M2 phenotype leading to the inflammatory cascade and to cell injury80,81. IL-4 or IL-10 knock-out mice have a worse outcome after cerebral ischemia due to an enhanced expression of M2 markers82,83. MLC901 significantly reduced the activation of microglia in the ischemic penumbra. We also observed that MLC901 markedly increased mRNA expression of IL-4Rα in microglia and monocytes of reperfused brains, an effect which would favor protective and repair processes. MLC901 seems to favor the shift of microglia from the M1 to the M2 phenotype. This is also indicated by its effects on cytokine and chemokine production induced by stroke. Previous studies in animal models and in stroke patients have shown that continued production of cytokines such as IFNγ, TNFα, IL11, IL-1β, IL-1ra and IL-6 induces and maintains the M1 polarization state3,84. This work also shows that a strong up-regulation of major cytokines (TNFα, IL-1β, IL-6, IL-11) is observed after focal ischemia. MLC901 counteracts this effect.
Ischemic stroke induces the release of different chemokines such as CCL-2, CCL-3, CCL-4, CCL7, CCL-11, CXCL-1, CXCL10, from the ischemic tissue. CCL2, which is the most up-regulated chemokine, in our experimental conditions, is known to be a critical factor regulating post-ischemic inflammation. It is involved in BBB disruption and in monocyte recruitment to the site of lesion after ischemic injury8. CCL2 expression appears in the ischemic cortex as early as 6 hours post-ischemia, peaks within 12 to 48 hours and remains elevated up to 5 days8,85. CCL2 deletion or absence of its receptor (CCR2) limits infarct size, reduces the inflammatory response, BBB permeability and brain edema formation8,86. Taken together these data suggest that inhibiting the CCL2/CCR2 signaling, as MLC901 does very potently (Figs 5 and 6), certainly contributes, in an important way, to brain protection and other beneficial effects of this TCM.
Activation of MMP-9 following cerebral ischemia is closely associated with BBB leakage and microglial activation, and causes severe brain edema or hemorrhagic transformation59,87. Post-ischemic BBB breakdown is reduced after MMP-9 inhibition and MMP-9 gene deletion51. During reperfusion, MMP9 released from neutrophils recruited to the injured brain exhibits pro-inflammatory effects, promotes further recruitment of neutrophils to the lesion site, and finally contributes to post-stroke neuronal damage51. This study demonstrates that MLC901 treatment significantly reduces the expression of MMP-9 in the ischemic brain. Inhibition of neutrophil recruitment by MLC901 could be the origin of this reduction, which certainly contributes to the protective effect of MLC901 against BBB disruption as well as edema formation.
NFκB is an oxidative stress-responsive transcription factor, and its involvement in ischemic injury is well recognized88,89,90. It is a key regulator of the inflammatory response. In the early phase of stroke, infiltrating neutrophils cause excessive production of ROS, resulting in oxidative stress in the injured brain tissue. Oxidative stress promotes the activation of NFκB, which regulates the transcriptional induction of various pro-inflammatory genes52,53. In the current study, we found that MLC901 significantly reduced the level of phosphorylated NFκB, which is a marker of NFκB activation91. Again, because high levels of ROS are well-known to be produced by recruited neutrophils3, the inhibition of neutrophil recruitment by MLC901 would probably lead to prevention of NFκB activation and to a decrease of inflammation via the NFκB pathway.
The TLR4/peroxiredoxin (Prx6) pathway plays a pivotal role in the post-ischemic inflammation12. Cerebral ischemia induces the release of the Prx6 and stimulates macrophage infiltration via TLR412. TLR4-induced activation of NFκB and subsequent production of pro-inflammatory cytokines leads to brain inflammation and ischemic injury12. TLR4-knockout mice show indeed smaller infarct sizes and improved neurological deficits after stroke92,93, and in humans, TLR4 polymorphisms have been associated with ischemic stroke outcome94,95. This work confirms increased Prx6 expression in ischemic brain and the activation of stroke-induced TLR4 signaling, including enhanced TLR4 expression and NFκB activation. All of these events are inhibited by MLC901.
In summary, the major effects of MLC901 are the reduction of neutrophil recruitment, the reduction of microglia activation and the reduction of pro-inflammatory mediators (TNFα, IL6, IL1β, CCL2) production induced by stroke. The modulation of TLR4/Prx6 pathway by MLC901 seems to represent a key step in these anti-inflammatory effects and to contribute to its protective effects against stroke. In the past years a number of anti-inflammatory approaches targeting specific types of immune cells (neutrophils, microglia ….) have proven to be successful in animal models against stroke. However, the transfer to a clinical setting has, so far, been unsuccessful3. There are at least three reasons to be more optimistic with MLC901. The first one is that MLC901 has multiple anti-inflammatory effects on neutrophils, microglia, vascular endothelium and neuronal cells. The second one is that, instead of going, as usual, from the bench to the patient bed, here we started from the bed side (several clinical assays performed) and went successfully from there to the bench to discover the mechanisms that are responsible for the reported beneficial effects of MLC901 against stroke. The third one is that MLC901 is most probably a cocktail of molecules which explains the multifaceted positive effect of this TCM against the inflammatory process along with other types of positive effects such as neurorepair36,45.
Methods
Animals
Seven weeks old C57BL/6 male mice (Janvier) were housed five per cage on inversed 12 H light/dark cycle (light on at 08:00 PM) in animal facility maintained at ambient temperature of 21 ± 1 °C. They were provided with food and beverage ad libitum. Experiments were performed in accordance with the policies on the care and use of laboratory animals of European Community laws (directive 2010/63/EU) and approved by the French ministry of higher education and scientific research (approval number 01314.04). A particular effort was made to minimize the number of animals used per group when not needed. The researchers who carried out the ischemic surgery and all post-stroke experiments were blinded to the treatment code.
Drug treatment
MLC901 combines 9 herbal extracts equivalent to the following composition of raw herbs per capsule: 0.80 g Radix astragali, 0.16 g Radix salvia miltiorrhizae, 0.16 g Radix paeoniae rubra, 0.16 g Rhizoma chuanxiong,0.16 g Radix angelicae sinensis, 0.16 g Carthamus tinctorius, 0.16 g Prunus persica, 0.16 g Radix polygalae, and 0.16 g Rhizoma acori tatarinowii. In these different herbs that are extracted to manufacture MLC901, there are a number of identified molecules such as tetramethylpyrazine (Rhizoma chuanxiong)96,97, ferulic acid (Radix angelicae sinensis, Rhizoma chuanxiong)98, ligustilide and butylidenephtalide (Radix angelicae sinensis, Rhizoma chuanxiong)99,100, astralagoside IV (Radix astragali)101, salvianolic acid B and tanshinone IIB (Radix salvia miltiorrhizae)102,103 that are known to be neurobeneficial and for some of them, anti-inflammatory. MLC901 (batch BRAINS provided by Moleac, Singapore) was intraperitoneally injected with a single dose of 40 μg/kg MLC901 diluted in saline (as vehicle) 90 min after the onset of ischemia and once a day during reperfusion until the sacrifice of animals as previously described33. The dose for MLC901 used in this work is the most efficient dose determined in previous studies for both cerebral ischemia and traumatic brain injury in rodents36,37,38. The animals were equally and randomly divided into five groups as follows: Control (sham-operated), Sal-MCAO-R (with reperfusion), Sal-MCAO-NR (no reperfusion), MLC901-MCAO-R and MLC901-NR.
Ischemic stroke model (MCAO)
The ischemic stroke model (focal ischemia) was performed on seven weeks old male mice by transient (60 min) right middle cerebral artery occlusion (MCAO) and reperfusion, using a 6-0 coated filament (Doccol, Redlands, CA, USA) as previously described36. The regional cerebral blood flow was monitored by laser-Doppler flowmetry (Perimed, Craponne, France) to control MCAO severity and reperfusion. Animals presenting with sustained CBF reduction >70% during ischemia or a severe brain hemorrhage after MCAO were excluded from the study (<1%). Mice received MLC901 in post-treatment until the sacrifice of animals. Sham-operation was performed by inserting the thread into the common carotid artery without advancing it to occlude MCA. The animals were allowed to regain full consciousness on a nursing cage during 24 hours before returning to the home cage. The researchers who did the MCAO surgery were blinded to the treatment code.
Neurological deficit scoring
Neurological deficits of mice were assessed 30 hours post-ischemia in a blinded fashion according to a scoring scale in a postural reflex test104: Grade 0 = no visible deficits; Grade 1 = forelimb flexion; Grade 2 = unidirectional circling when the animal is pulled by the tail; Grade 3 = circling and rolling movement; Grade 4 = decreased level of consciousness; Grade 5 = death.
Measurements of infarct volumes, blood brain barrier (BBB) leakage and brain edema
To assess the infarct volume, as previously described36, mice were euthanized by decapitation 30 hours after reperfusion. Coronal frozen brain sections (20 μm thick) were stained using a solution of 1% cresyl violet in 0.25% acetic acid and mounted with Entellan. The striatal and cortical areas of infarction, outlined in light, were measured on each section using a computer image analysis system and corrected for brain edema according to Golanov and Reis105. Infarct volume, expressed in mm3 was calculated by a linear integration of the corrected lesions areas as previously described36.
The early phase of BBB breakdown BBB degeneration following MCAO was assessed 3 hours after MCAO by the extravasation of fluorescein isocyanate-conjugated dextran (FITC-dextran, 2000 Da, Sigma)106,107. 0.3 ml of 5% FITC-dextran (wt/vol in sterile PBS) was injected through the left femoral vein. All mice were reperfused for 10 min to allow sufficient circulation of FITC-dextran to the ischemic brain. The FITC-dextran leakage was visualized under confocal microscope at 488 nm. Mice were transcardially perfused with normal saline to wash away the remaining dye in the blood vessels. Brains were removed and coronal sections from bregma-1 to 1 mm were performed to visualize the blue staining at 30 hours after MCAO.
To measure the vasogenic edema, animals were sacrificed 30 hours after ischemia. As previously described36, the brains were divided into the ipsilateral hemisphere (ischemic side) and contralateral hemisphere. The ipsilateral hemisphere was weighed to obtain the wet weight and then dried at 110 °C for 24 hours. The brain water content in the ipsilateral hemisphere was calculated as follows: water content = (wet weight – dry weight)/wet weight.
RNA isolation and Polymerase chain reaction array for cytokines, interleukins and chemokines
Total mRNA from ischemic brains was isolated according to the current Chomczynski method using Fast Prep apparatus (Q-Biogene, France). Two micrograms of total mRNAs were denatured at 65 °C for 5 min in the presence of 0.5 mM dNTP and oligodT primers (25 ng/μl, Promega, France).
RT2 Profiler Mouse Inflammatory Cytokines and Receptors Real time PCR arrays (PAMM-011F) (n = 6) were used to analyze the expression of a focused panel of genes. Data analysis was performed using the ∆∆CT method according to the manufacturer’s protocol (SABio-Sciences/Qiagen, France). In each assay, PCR were performed in duplicate. Relative quantities of target genes were determined by comparison with results for the housekeeping gene GAPDH.
Isolation of immune cells from mouse brains
Immune brain cells were isolated from ipsi- and contralateral brain hemispheres homogenates as previously described47. Mice were transcardially perfused with ice-cold PBS (pH 7.4, 1 mg/ml/EDTA). Collected brains were roughly homogenized in PBS, resuspended in PBS containing collagenase D (3 mg/ml) and incubated 30 minutes at 37 °C. Then, brain homogenates were filtered, centrifuged (10 min, 2000 rpm), washed and resuspended in 6 ml of 38% isotonic Percoll before centrifugation (20 min, 2000 rpm). Myelin and debris were discarded. Cell pellets containing brain immune cells were collected, washed and labeled for subsequent cell sorting and flow cytometry analysis.
Brain immune cell staining, flow cytometry and cell sorting
Staining of brain immune cell surface antigens was performed as previously described47. Fc receptors were blocked with 2.4G2 antibody. Cells were incubated with the appropriate combination of conjugated antibodies: CD11b-PercP-Cy5.5, CD45-APC-Cy7, Ly6C-PE-Cy7, Ly6G-pacific blue, CD3-FITC, CD8-APC, major histocompatibility complex class II-Alexa700, CD80-V450, CD86-eFluor605, CD14-APC, TLR4-Alexa488, CD124/IL-R4α-Biotin and streptavidin-PE-Cy7 (BD Biosciences), CD4-Viogreen (Miltenyi Biotec), CCR2-PE (R&D Systems) or isotype control antibodies for 30 minutes. Cells were washed and resuspended in PBS containing 0.5% bovine serum albumin (BSA) for analysis and cell sorting with FACS Aria III (BD Biosciences).
Cytokine measurement by CBA
Ischemic brains from vehicle- or MLC901-treated mice were homogenized in NP-40 containing buffer (10 mM Tris-HCL, ph 8, 150 mM NaCl, 1% NP-40, 10% glycerol, 5 mM EDTA and protease inhibitor cocktail (Roche Diagnostics)) as previously described47. Supernatants from brain homogenates or ex vivo cultured cells were harvested and the concentration of secreted cytokines (TNFα, IL1β, IL6 and CCL2) was detected using a CBA according to the manufacturer’s instructions (BD Biosciences). For comparison, data were normalized relative to the protein concentration of relative brain homogenate.
Western blotting
As previously described34, brain tissues (vehicle- and MLC901-treated, sham-operated controls, n = 3 per group) were collected after 24 hours of reperfusion, and the fresh brains were cut into 2-mm-thick coronal sections 6 to 8 mm from the frontal pole, and carefully separated into ipsilateral and contralateral hemispheres, with respect to the infarct location. Samples were homogenized in four volumes of cold lysis buffer (20 nmol/l Tris pH: 7.5, 137 mmol/l NaCl, 2 mmol/l EDTA, 1% Triton X-100, 10% glycerol, and protease inhibitor cocktail) on ice. The homogenates were centrifuged at 12,000 g for 30 min at 4 °C. The supernatant was stored at −70 °C until further use. Protein concentrations were measured using conventional Bradford’s method. Fifty micrograms of proteins from each experimental group were applied to 10% SDS PAGE gels and electrophoresed for 1 h at 100 mA. Proteins were transferred onto a PVDF membrane in blotting buffer (156 mmol/l Tris, 1 mol/l glycine, PBS) for 90 min at 80 mA and blocked with 5% skim milk (Regilait) in PBS for 2 h at room temperature. The blotted membrane was then incubated overnight at 4 °C with the different primary antibodies: rabbit monoclonal phospho-NFκB p65 (Ser536) antibody (mAb#3033, diluted 1:1000; Cell Signal Technology) or rabbit monoclonal total NFκB p65 (D14E12) antibody (mAb#8242, diluted 1:1000; Cell Signal Technology) or rabbit polyclonal antibody against MMP9 (Millipore, AB 19026, 1/200). Western blots were incubated with horseradish-peroxidase conjugated anti-rabbit IgG (Jackson ImmunoResearch, diluted 1/15,000) for 1 h at room temperature. Specific bands were detected using the ECL Western blotting substrate and exposed to LAS-4000 image detection system. To control for sample loading, stripped membranes were rehybridized with a β-actin antibody (1:2000, Proteintech Group, USA) as internal control. Films with specific bands were scanned and quantified using an imaging densitometer. The optical densities of specific bands were analyzed with QUANTITY ONE software (Bio-Rad). Western blots were duplicated with 3 independent sets.
Immunohistochemistry
Floating brain sections (25 μm thick) were immersed in 0.3% H2O2/PBS for 10 min, permeabilized in 0.1% Triton/PBS for 10 min and blocked with 3% goat serum/PBS for 2 h at room temperature as previously described33. After a PBS rinse, sections were incubated overnight with the following primary antibodies: rabbit polyclonal antibody against IL6 (Abcam, A6672, 1/500), GFAP (Dako, Z0334, 1/300) and MMP9 (Millipore, AB 19026, 1/200), mouse monoclonal antibody against Iba1 (Abcam 15690, 1/400), MPO (R&D, MAB3174) and rabbit monoclonal against phospho-NFκB p65 (Ser536) antibody (mAb#3033, diluted 1:1000; Cell Signal Technology), goat polyclonal antibody against TNFα (R&D, AF410—NA, 10μl/ml) and IL1β (R&D, AF401, 10μl/ml) and rabbit monoclonal antibody against Iba1 (Wako, 1/1000). After the primary incubation and three rinses in PBS, sections were then incubated in biotinylated horse anti-rabbit IgG (Jackson ImmunoResearch, diluted 1/15,000) for 2 h at room temperature. Protein expression was visualized by 3, 3-diaminobenzidine (DAB) staining using VectaStain ABC kit (Biovalley). All sections were washed and mounted with Entellan. Sections were observed using conventional microscopy. Signal specificity was assessed in negative control coverslips by omitting primary antibody. Images were acquired as single trans-cellular optical sections and averaged over at least four scans per frame. Analysis of the positive color intensity was performed by using the NIH Image J software (http://rsbwebnih.gov/ij), which allowed to measure the intensity levels of cells of each image saved as a 16-bit TIFF file. Five random and non-overlapping microscopic fields per section were examined at the cortical border of the positively stained area under 200X magnification in a blind fashion Results are given as mean of positive pixel ± SEM of three experiments.
Statistical analysis
Statistical analysis was performed using Statistica softwareTM and SigmaStat 2.3. Statistical values are presented as mean ± SEM. Significant differences between two groups of data (Vehicle group versus MLC901 group) were determined using a Mann & Whitney test for non-parametric data. For FACS experiment analyses, we chose to use a non-parametric permutation statistical test to overcome the variability of absolute values obtained between the replicates (3 independent experiments with 4 brains per experimental group). We used the non parametric Kruskal-Wallis one-way ANOVA test followed by Tukey’s multiple comparison to compare the five groups (Sham, Saline-MCAO NR, Saline-MCAO R, MLC901-MCAO NR and MLC901-MCAO R). In all analyses, the level of significance was set at P < 0.05.
Data Availability Statement
Materials, data and associated protocols are available to readers.
References
Chamorro, A., Dirnagl, U., Urra, X. & Planas, A. M. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol 15, 869–881, https://doi.org/10.1016/S1474-4422(16)00114-9 (2016).
Iadecola, C. & Anrather, J. The immunology of stroke: from mechanisms to translation. Nat Med 17, 796–808, https://doi.org/10.1038/nm.2399 (2011).
Jin, R., Yang, G. & Li, G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87, 779–789, https://doi.org/10.1189/jlb.1109766 (2010).
Banati, R. B., Gehrmann, J., Schubert, P. & Kreutzberg, G. W. Cytotoxicity of microglia. Glia 7, 111–118, https://doi.org/10.1002/glia.440070117 (1993).
Rothwell, N. J., Loddick, S. A. & Stroemer, P. Interleukins and cerebral ischaemia. Int Rev Neurobiol 40, 281–298 (1997).
Schnell, L., Fearn, S., Schwab, M. E., Perry, V. H. & Anthony, D. C. Cytokine-induced acute inflammation in the brain and spinal cord. J Neuropathol Exp Neurol 58, 245–254 (1999).
Anthony, D. C., Walker, K. & Perry, V. H. The therapeutic potential of CXC chemokine blockade in acute inflammation in the brain. Expert Opin Investig Drugs 8, 363–371, https://doi.org/10.1517/13543784.8.4.363 (1999).
Hughes, P. M. et al. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab 22, 308–317, https://doi.org/10.1097/00004647-200203000-00008 (2002).
Cekanaviciute, E. & Buckwalter, M. S. Astrocytes: Integrative Regulators of Neuroinflammation in Stroke and Other Neurological Diseases. Neurotherapeutics 13, 685–701, https://doi.org/10.1007/s13311-016-0477-8 (2016).
Zuany-Amorim, C., Hastewell, J. & Walker, C. Toll-like receptors as potential therapeutic targets for multiple diseases. Nat Rev Drug Discov 1, 797–807, https://doi.org/10.1038/nrd914 (2002).
Macrez, R. et al. Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol 10, 471–480, https://doi.org/10.1016/S1474-4422(11)70066-7 (2011).
Shichita, T. et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med 18, 911–917, https://doi.org/10.1038/nm.2749 (2012).
Dirnagl, U. & Macleod, M. R. Stroke research at a road block: the streets from adversity should be paved with meta-analysis and good laboratory practice. Br J Pharmacol 157, 1154–1156, https://doi.org/10.1111/j.1476-5381.2009.00211.x (2009).
Gribkoff, V. K. & Kaczmarek, L. K. The need for new approaches in CNS drug discovery: Why drugs have failed, and what can be done to improve outcomes. Neuropharmacology 120, 11–19, https://doi.org/10.1016/j.neuropharm.2016.03.021 (2017).
Corbett, D., Jeffers, M., Nguemeni, C., Gomez-Smith, M. & Livingston-Thomas, J. Lost in translation: rethinking approaches to stroke recovery. Prog Brain Res 218, 413–434, https://doi.org/10.1016/bs.pbr.2014.12.002 (2015).
Bavarsad Shahripour, R. et al. The effect of NeuroAiD (MLC601) on cerebral blood flow velocity in subjects’ post brain infarct in the middle cerebral artery territory. Eur J Intern Med 22, 509–513, https://doi.org/10.1016/j.ejim.2011.01.002 (2011).
Chen, C. et al. Danqi Piantang Jiaonang (DJ), a traditional Chinese medicine, in poststroke recovery. Stroke 40, 859–863, STROKEAHA.108.531616 (2009).
Chen, C. L. et al. Chinese Medicine Neuroaid Efficacy on Stroke Recovery: A Double-Blind, Placebo-Controlled, Randomized Study. Stroke 44, 2093–2100, STROKEAHA.113.002055 (2013).
Gan, R. et al. Danqi Piantan Jiaonang does not modify hemostasis, hematology, and biochemistry in normal subjects and stroke patients. Cerebrovasc Dis 25, 450–456, https://doi.org/10.1159/000126919 (2008).
Harandi, A. A. et al. Safety and Efficacy of MLC601 in Iranian Patients after Stroke: A Double-Blind, Placebo-Controlled Clinical Trial. Stroke Res Treat 2011,, 721613, https://doi.org/10.4061/2011/721613 (2011).
Kong, K. H. et al. A double-blind, placebo-controlled, randomized phase II pilot study to investigate the potential efficacy of the traditional chinese medicine Neuroaid (MLC 601) in enhancing recovery after stroke (TIERS). Cerebrovasc Dis 28, 514–521, https://doi.org/10.1159/000247001 (2009).
Navarro, J. C. et al. CHIMES-I: sub-group analyzes of the effects of NeuroAiD according to baseline brain imaging characteristics among patients randomized in the CHIMES study. Int J Stroke 8, 491–494, https://doi.org/10.1111/ijs.12044 (2013).
Navarro, J. C., Molina, M. C., Baroque Ii, A. C. & Lokin, J. K. The Use of NeuroAiD (MLC601) in Postischemic Stroke Patients. Rehabil Res Pract 2012, 506387, https://doi.org/10.1155/2012/506387 (2013).
Siddiqui, F. J., Venketasubramanian, N., Chan, E. S. & Chen, C. Efficacy and safety of MLC601 (NeuroAiD(R)), a traditional Chinese medicine, in poststroke recovery: a systematic review. Cerebrovasc Dis 35(Suppl 1), 8–17, https://doi.org/10.1159/000346231 (2013).
Siow, C. H. Neuroaid in stroke recovery. Eur Neurol 60, 264–266, https://doi.org/10.1159/000155220 (2008).
Suwanwela, N. C. et al. Effect of Combined Treatment with MLC601 (NeuroAiDTM) and Rehabilitation on Post-Stroke Recovery: The CHIMES and CHIMES-E Studies. Cerebrovasc Dis 46, 82–88, https://doi.org/10.1159/000492625 (2018).
Young, S. H. et al. Safety profile of MLC601 (Neuroaid) in acute ischemic stroke patients: A Singaporean substudy of the Chinese medicine neuroaid efficacy on stroke recovery study. Cerebrovasc Dis 30, 1–6, https://doi.org/10.1159/000313398 (2010).
Chen, C. L., Venketasubramanian, N., Lee, C. F., Wong, K. S. & Bousser, M. G. Effects of MLC601 on Early Vascular Events in Patients After Stroke: The CHIMES Study. Stroke 44, 3580–3583, https://doi.org/10.1161/STROKEAHA.113.003226 (2013).
Venketasubramanian, N. et al. A double-blind, placebo-controlled, randomized, multicenter study to investigate CHInese Medicine Neuroaid Efficacy on Stroke recovery (CHIMES Study). Int J Stroke 4, 54–60, https://doi.org/10.1111/j.1747-4949.2009.00237.x (2009).
Venketasubramanian, N. et al. Prognostic Factors and Pattern of Long-Term Recovery with MLC601 (NeuroAiD) in the Chinese Medicine NeuroAiD Efficacy on Stroke Recovery - Extension Study. Cerebrovasc Dis 43, 36–42, https://doi.org/10.1159/000452285 (2017).
Venketasubramanian, N. et al. Chinese medicine NeuroAiD efficacy stroke recovery-extension study (CHIMES-E study): an observational multicenter study to investigate the longer-term efficacy of NeuroAiD in stroke recovery. Cerebrovasc Dis 35(Suppl 1), 18–22, https://doi.org/10.1159/000346233 (2013).
Navarro, J. C. et al. Durability of the beneficial effect of MLC601 (NeuroAiD) on functional recovery among stroke patients from the Philippines in the CHIMES and CHIMES-E studies. Int J Stroke 12, 285–291, https://doi.org/10.1177/1747493016676615 (2017).
Theadom, A. et al. MLC901 (NeuroAiD II) for cognition after traumatic brain injury: a pilot randomized clinical trial. Eur J Neurol. https://doi.org/10.1111/ene.13653 (2018).
Theadom, A., Bhattacharjee, R., Parmar, P. & Feigin, V. A randomized, placebo-controlled pilot trial to investigate the safety and efficacy of MLC901 (NEUROAID IITM) in adults following mild or moderate traumatic brain injury. Journal of the Neurological Sciences/Abstracts 381, 54–180 (2017).
Chen, C. L. et al. TheNeuroAiD II (MLC901) in vascular cognitive impairment study (NEURITES). Cerebrovasc Dis 35(Suppl 1), 23–29, https://doi.org/10.1159/000346234 (2013).
Heurteaux, C. et al. Neuroprotective and neuroproliferative activities of NeuroAid (MLC601, MLC901), a Chinese medicine, in vitro and in vivo. Neuropharmacology 58, 987–1001, https://doi.org/10.1016/j.neuropharm.2010.01.001 (2010).
Quintard, H. et al. MLC901, a traditional Chinese medicine protects the brain against global ischemia. Neuropharmacology 61, 622–631, https://doi.org/10.1016/j.neuropharm.2011.05.003 (2011).
Quintard, H., Lorivel, T., Gandin, C., Lazdunski, M. & Heurteaux, C. MLC901, a Traditional Chinese Medicine induces neuroprotective and neuroregenerative benefits after traumatic brain injury in rats. Neuroscience 277, 72–86, https://doi.org/10.1016/j.neuroscience.2014.06.047 (2014).
Chio, C. C., Lin, M. T., Chang, C. P. & Lin, H. J. A positive correlation exists between neurotrauma and TGF-beta1-containing microglia in rats. Eur J Clin Invest 46, 1063–1069, https://doi.org/10.1111/eci.12693 (2016).
Tsai, M. C. et al. Therapeutic efficacy of Neuro AiD (MLC 601), a traditional Chinese medicine, in experimental traumatic brain injury. J Neuroimmune Pharmacol 10, 45–54, https://doi.org/10.1007/s11481-014-9570-0 (2015).
Moha Ou Maati, H. et al. Activation of ATP-sensitive potassium channels as an element of the neuroprotective effects of the Traditional Chinese Medicine MLC901 against oxygen glucose deprivation. Neuropharmacology 63, 692–700, https://doi.org/10.1016/j.neuropharm.2012.05.035 (2012).
Zhang, Y., Pan, S., Zheng, X. & Wan, Q. Cytomembrane ATP-sensitive K(+) channels in neurovascular unit targets of ischemic stroke in the recovery period. Exp Ther Med 12, 1055–1059, https://doi.org/10.3892/etm.2016.3373 (2016).
Gandin, C., Widmann, C., Lazdunski, M. & Heurteaux, C. MLC901 Favors Angiogenesis and Associated Recovery after Ischemic Stroke in Mice. Cerebrovasc Dis 42, 139–154, https://doi.org/10.1159/000444810 (2016).
del Zoppo, G. J., Sharp, F. R., Heiss, W. D. & Albers, G. W. Heterogeneity in the penumbra. J Cereb Blood Flow Metab 31, 1836–1851, https://doi.org/10.1038/jcbfm.2011.93 (2011).
Heurteaux, C. et al. NeuroAiD: properties for neuroprotection and neurorepair. Cerebrovasc Dis 35(Suppl 1), 1–7, https://doi.org/10.1159/000346228 (2013).
Zhou, W. et al. Postischemic brain infiltration of leukocyte subpopulations differs among murine permanent and transient focal cerebral ischemia models. Brain Pathol 23, 34–44, https://doi.org/10.1111/j.1750-3639.2012.00614.x (2013).
Cazareth, J., Guyon, A., Heurteaux, C., Chabry, J. & Petit-Paitel, A. Molecular and cellular neuroinflammatory status of mouse brain after systemic lipopolysaccharide challenge: importance of CCR2/CCL2 signaling. J Neuroinflammation 11, 132, https://doi.org/10.1186/1742-2094-11-132 (2014).
Barone, F. C. et al. Time-related changes in myeloperoxidase activity and leukotriene B4 receptor binding reflect leukocyte influx in cerebral focal stroke. Mol Chem Neuropathol 24, 13–30 (1995).
Kloss, C. U., Bohatschek, M., Kreutzberg, G. W. & Raivich, G. Effect of lipopolysaccharide on the morphology and integrin immunoreactivity of ramified microglia in the mouse brain and in cell culture. Exp Neurol 168, 32–46, https://doi.org/10.1006/exnr.2000.7575 (2001).
Hu, X. et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43, 3063–3070, https://doi.org/10.1161/STROKEAHA.112.659656 (2012).
Gidday, J. M. et al. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol 289, H558–568, https://doi.org/10.1152/ajpheart.01275.2004 (2005).
Harari, O. A. & Liao, J. K. NF-kappaB and innate immunity in ischemic stroke. Ann N Y Acad Sci 1207, 32–40, https://doi.org/10.1111/j.1749-6632.2010.05735.x (2010).
Yi, J. H., Park, S. W., Kapadia, R. & Vemuganti, R. Role of transcription factors in mediating post-ischemic cerebral inflammation and brain damage. Neurochem Int 50, 1014–1027, https://doi.org/10.1016/j.neuint.2007.04.019 (2007).
Vincent, A. et al. Acute and long-term cardioprotective effectsof the Traditional Chinese Medicine MLC901 against myocardialischemia-reperfusion injury in mice. Scientific Reports (2017).
Chankrachang, S. et al. Prognostic factors and treatment effect in the CHIMES study. J Stroke Cerebrovasc Dis 24, 823–827, https://doi.org/10.1016/j.jstrokecerebrovasdis.2014.11.017 (2015).
Heurteaux, C., Bertaina, V., Widmann, C. & Lazdunski, M. K+ channel openers prevent global ischemia-induced expression of c-fos, c-jun, heat shock protein, and amyloid beta-protein precursor genes and neuronal death in rat hippocampus. Proc Natl Acad Sci USA 90, 9431–9435 (1993).
Obrenovitch, T. P. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev 88, 211–247, https://doi.org/10.1152/physrev.00039.2006 (2008).
Heurteaux, C., Lauritzen, I., Widmann, C. & Lazdunski, M. Essential role of adenosine, adenosine A1 receptors and KATP channels in cerebral ischemic preconditioning. Proc. Natl. Acad. Sci. USA 92, 4666–4670 (1995).
del Zoppo, G. J. et al. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke 38, 646–651, https://doi.org/10.1161/01.STR.0000254477.34231.cb (2007).
Sandoval, K. E. & Witt, K. A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 32, 200–219, https://doi.org/10.1016/j.nbd.2008.08.005 (2008).
Huang, Z. G., Xue, D., Preston, E., Karbalai, H. & Buchan, A. M. Biphasic opening of the blood-brain barrier following transient focal ischemia: effects of hypothermia. Can J Neurol Sci 26, 298–304 (1999).
Durukan, A. et al. Post-ischemic blood-brain barrier leakage in rats: one-week follow-up by MRI. Brain Res 1280, 158–165, https://doi.org/10.1016/j.brainres.2009.05.025 (2009).
Merali, Z., Huang, K., Mikulis, D., Silver, F. & Kassner, A. Evolution of blood-brain-barrier permeability after acute ischemic stroke. PLoS One 12, e0171558, https://doi.org/10.1371/journal.pone. (2017).
Emsley, H. C. & Hopkins, S. J. Acute ischaemic stroke and infection: recent and emerging concepts. Lancet Neurol 7, 341–353, https://doi.org/10.1016/S1474-4422(08)70061-9 (2008).
McColl, B. W., Allan, S. M. & Rothwell, N. J. Systemic infection, inflammation and acute ischemic stroke. Neuroscience 158, 1049–1061, https://doi.org/10.1016/j.neuroscience.2008.08.019 (2009).
Elkind, M. S., Cheng, J., Rundek, T., Boden-Albala, B. & Sacco, R. L. Leukocyte count predicts outcome after ischemic stroke: the Northern Manhattan Stroke Study. J Stroke Cerebrovasc Dis 13, 220–227, https://doi.org/10.1016/j.jstrokecerebrovasdis.2004.07.004 (2004).
Lo, E. H. Experimental models, neurovascular mechanisms and translational issues in stroke research. Br J Pharmacol 153 Suppl 1, https://doi.org/10.1038/sj.bjp.0707626 (2008).
Prestigiacomo, C. J. et al. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke 30, 1110–1117 (1999).
Zhang, R. L. et al. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke 26, 1438–1442; discussion 1443 (1995).
Clark, R. K. et al. Reperfusion following focal stroke hastens inflammation and resolution of ischemic injured tissue. Brain Res Bull 35, 387–392 (1994).
Wang, X. Investigational anti-inflammatory agents for the treatment of ischaemic brain injury. Expert Opin Investig Drugs 14, 393–409, https://doi.org/10.1517/13543784.14.4.393 (2005).
Jickling, G. C. et al. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab 35, 888–901, https://doi.org/10.1038/jcbfm.2015.45 (2015).
Kolaczkowska, E. & Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13, 159–175, https://doi.org/10.1038/nri3399 (2013).
McColl, B. W., Rothwell, N. J. & Allan, S. M. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci 27, 4403–4412, https://doi.org/10.1523/JNEUROSCI.5376-06.2007 (2007).
Investigators, E. A. S. T. Use of anti-ICAM-1 therapy in ischemic strokes: results of the EnlimomabAcute Stroke Trial. Neurology 57, 1428–1434 (2001).
Becker, K. J. Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr Med Res Opin 18(Suppl 2), s18–22 (2002).
Krams, M. et al. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke 34, 2543–2548, https://doi.org/10.1161/01.STR.0000092527.33910.89 (2003).
Nielsen, M. S., Jacobsen, C., Olivecrona, G., Gliemann, J. & Petersen, C. M. Sortilin/neurotensin receptor-3 binds and mediates degradation of lipoprotein lipase. J Biol Chem 274, 8832–8836 (1999).
Jander, S., Kraemer, M., Schroeter, M., Witte, O. W. & Stoll, G. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab 15, 42–51, https://doi.org/10.1038/jcbfm.1995.5 (1995).
Pan, J. et al. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARgamma-dependent manner. J Neuroinflammation 12, 51, https://doi.org/10.1186/s12974-015-0270-3 (2015).
Won, S., Lee, J. K. & Stein, D. G. Recombinant tissue plasminogen activator promotes, and progesterone attenuates, microglia/macrophage M1 polarization and recruitment of microglia after MCAO stroke in rats. Brain Behav Immun 49, 267–279, https://doi.org/10.1016/j.bbi.2015.06.007 (2015).
Perez-de Puig, I. et al. IL-10 deficiency exacerbates the brain inflammatory response to permanent ischemia without preventing resolution of the lesion. J Cereb Blood Flow Metab 33, 1955–1966, https://doi.org/10.1038/jcbfm.2013.155 (2013).
Xiong, X. et al. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke 42, 2026–2032, https://doi.org/10.1161/STROKEAHA.110.593772 (2011).
Lakhan, S. E., Kirchgessner, A. & Hofer, M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 7, 97, https://doi.org/10.1186/1479-5876-7-97 (2009).
Wang, X., Yue, T. L., Barone, F. C. & Feuerstein, G. Z. Monocyte chemoattractant protein-1 messenger RNA expression in rat ischemic cortex. Stroke 26, 661–665, discussion 665–666 (1995).
Dimitrijevic, O. B., Stamatovic, S. M., Keep, R. F. & Andjelkovic, A. V. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38, 1345–1353, https://doi.org/10.1161/01.STR.0000259709.16654.8f (2007).
Sumii, T. & Lo, E. H. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke 33, 831–836 (2002).
Clemens, J. A. et al. Drug-induced neuroprotection from global ischemia is associated with prevention of persistent but not transient activation of nuclear factor-kappaB in rats. Stroke 29, 677–682 (1998).
Schneider, A. et al. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med 5, 554–559, https://doi.org/10.1038/8432 (1999).
Blondeau, N., Widmann, C., Lazdunski, M. & Heurteaux, C. Activation of the nuclear factor-kappaB is a key event in brain tolerance. J Neurosci 21, 4668–4677, 21/13/4668 (2001).
Baeuerle, P. A. & Baltimore, D. NF-kappa B: ten years after. Cell 87, 13–20, S0092-8674(00)81318-5 (1996).
Caso, J. R. et al. Toll-like receptor 4 is involved in subacute stress-induced neuroinflammation and in the worsening of experimental stroke. Stroke 39, 1314–1320, STROKEAHA.107.498212 (2008).
Caso, J. R. et al. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 115, 1599–1608, https://doi.org/10.1161/CIRCULATIONAHA.106.603431 (2007).
Brea, D. et al. Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J Cereb Blood Flow Metab 31, 1424–1431, https://doi.org/10.1038/jcbfm.2010.231 (2011).
Lin, Y. C. et al. Toll-like receptor 4 gene C119A but not Asp299Gly polymorphism is associated with ischemic stroke among ethnic Chinese in Taiwan. Atherosclerosis 180, 305–309, https://doi.org/10.1016/j.atherosclerosis.2004.12.022 (2005).
Kao, T. K. et al. Neuroprotection by tetramethylpyrazine against ischemic brain injury in rats. Neurochem Int 48, 166–176, https://doi.org/10.1016/j.neuint.2005.10.008 (2006).
Liao, S. L. et al. Tetramethylpyrazine reduces ischemic brain injury in rats. Neurosci Lett 372, 40–45, https://doi.org/10.1016/j.neulet.2004.09.013 (2004).
Cheng, C. Y. et al. Ferulic acid reduces cerebral infarct through its antioxidative and anti-inflammatory effects following transient focal cerebral ischemia in rats. Am J Chin Med 36, 1105–1119, https://doi.org/10.1142/S0192415X08006570 (2008).
Kuang, X., Du, J. R., Liu, Y. X., Zhang, G. Y. & Peng, H. Y. Postischemic administration of Z-Ligustilide ameliorates cognitive dysfunction and brain damage induced by permanent forebrain ischemia in rats. Pharmacol Biochem Behav 88, 213–221, https://doi.org/10.1016/j.pbb.2007.08.006 (2008).
Liao, S. J. et al. Enhanced angiogenesis with dl-3n-butylphthalide treatment after focal cerebral ischemia in RHRSP. Brain Res 1289, 69–78, https://doi.org/10.1016/j.brainres.2009.06.018 (2009).
Luo, Y. et al. Astragaloside IV protects against ischemic brain injury in a murine model of transient focal ischemia. Neurosci Lett 363, 218–223, https://doi.org/10.1016/j.neulet.2004.03.036 (2004).
Wang, S. X., Hu, L. M., Gao, X. M., Guo, H. & Fan, G. W. Anti-inflammatory activity of salvianolic acid B in microglia contributes to its neuroprotective effect. Neurochem Res 35, 1029–1037, https://doi.org/10.1007/s11064-010-0151-1 (2010).
Yu, X. Y. et al. Tanshinone IIB, a primary active constituent from Salvia miltiorrhza, exhibits neuro-protective activity in experimentally stroked rats. Neurosci Lett 417, 261–265, https://doi.org/10.1016/j.neulet.2007.02.079 (2007).
Bederson, J. B. et al. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 17, 1304–1308 (1986).
Golanov, E. V. & Reis, D. J. Contribution of cerebral edema to the neuronal salvage elicited by stimulation of cerebellar fastigial nucleus after occlusion of the middle cerebral artery in rat. J Cereb Blood Flow Metab 15, 172–174 (1995).
Jin, X., Liu, J., Yang, Y., Liu, K. J. & Liu, W. Spatiotemporal evolution of blood brain barrier damage and tissue infarction within the first 3h after ischemia onset. Neurobiol Dis 48, 309–316, https://doi.org/10.1016/j.nbd.2012.07.007 (2012).
Chen, B. et al. Severe blood-brain barrier disruption and surrounding tissue injury. Stroke 40, e666–674, https://doi.org/10.1161/STROKEAHA.109.551341 (2009).
Acknowledgements
The authors are very grateful to D. Picard (Moleac Singapore) for helpful discussions concerning the program, and particularly the follow-up of the clinical effects of MLC601/MLC901 and for providing MLC901 capsules and funding. We wish to thank J. Cazareth for providing helpful technical support for the FACS studies. This work is supported by the Centre National de la Recherche Scientifique (CNRS) and CNRS/MOLEAC contract.
Author information
Authors and Affiliations
Contributions
C.H. conceived the design of the study and supervised the study. C.W., A.P.P. and C.G. performed research: C.G. performed surgery, RT-PCR, W.B. and I.H.C. experiments. C.W. and A.P.P. performed FACS experiments. C.H., A.P.P. and C.W. analyzed the data and their presentation. M.L. and C.H. chose the topic and wrote the manuscript. A.P.P. revised the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
This work was supported by a CNRS/MOLEAC contract (SPV120602). A.P.P., C.G., C.W. and C.H. report no disclosure. M.L. was vice-president for research of Moleac.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Widmann, C., Gandin, C., Petit-Paitel, A. et al. The Traditional Chinese Medicine MLC901 inhibits inflammation processes after focal cerebral ischemia. Sci Rep 8, 18062 (2018). https://doi.org/10.1038/s41598-018-36138-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36138-0
This article is cited by
-
Profiling the neurovascular unit unveils detrimental effects of osteopontin on the blood–brain barrier in acute ischemic stroke
Acta Neuropathologica (2022)
-
NeuroAid II (MLC901) and polypharmacy in stroke and the risk of hepatotoxicity: a case report
The Egyptian Journal of Neurology, Psychiatry and Neurosurgery (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
