
Abstract
The Antarctic silverfish (Pleuragramma antarctica) is a critically important forage species with a circumpolar distribution and is unique among other notothenioid species for its wholly pelagic life cycle. Previous studies have provided mixed evidence of population structure over regional and circumpolar scales. The aim of the present study was to test the recent population hypothesis for Antarctic silverfish, which emphasizes the interplay between life history and hydrography in shaping connectivity. A total of 1067 individuals were collected over 25 years from different locations on a circumpolar scale. Samples were genotyped at fifteen microsatellites to assess population differentiation and genetic structuring using clustering methods, F-statistics, and hierarchical analysis of variance. A lack of differentiation was found between locations connected by the Antarctic Slope Front Current (ASF), indicative of high levels of gene flow. However, gene flow was significantly reduced at the South Orkney Islands and the western Antarctic Peninsula where the ASF is absent. This pattern of gene flow emphasized the relevance of large-scale circulation as a mechanism for circumpolar connectivity. Chaotic genetic patchiness characterized population structure over time, with varying patterns of differentiation observed between years, accompanied by heterogeneous standard length distributions. The present study supports a more nuanced version of the genetic panmixia hypothesis that reflects physical-biological interactions over the life history.
Similar content being viewed by others

Introduction
Silverfish life history and connectivity
The Antarctic silverfish (Pleuragramma antarctica) is a critically important forage species in the Southern Ocean that connects higher and lower trophic levels in the continental shelf ecosystem1,2. Having a circumpolar distribution, silverfish dominate the high-Antarctic neritic assemblage in terms of both biomass and abundance3. Atypical for a notothenioid, silverfish have a wholly pelagic life cycle, including a cryopelagic egg and larval phase4. Silverfish have adopted a relatively inactive, energy-conserving life strategy similar to related benthic species, in which their neutral buoyancy allows them to remain suspended in the water column without active swimming3,5. Remaining in the water column throughout their life history exposes silverfish to current and front systems over the slope and shelf that have been hypothesized by Ashford et al.6 to play an integral role in shaping their circumpolar distribution6.
The silverfish life history hypothesis predicts along-shelf connectivity facilitated principally by three major features of the large-scale circulation. The Antarctic Coastal Current (AACC) is typical of coastal buoyant plumes7 and transports water westward between glacial trough systems along the inner shelf. Similarly, a horizontal pressure gradient across the Antarctic Slope Front (ASF) drives westward transport in the Antarctic Slope Front Current. In this paper, ASF refers to this system, including the Antarctic Slope Front Current. It is located over the continental slope and thought to be continuous from the Amundsen Sea along the Ross Sea continental shelf and East Antarctica, forming the southern limb of the Weddell Gyre and reaching waters north of the Antarctic Peninsula8,9 (Fig. 1, overview). In contrast to the AACC and ASF, the Antarctic Circumpolar Current (ACC) transports water eastward. It approaches the continental shelf in the Amundsen Sea where it bifurcates, and its southern boundary is located over the slope off the western Antarctic Peninsula before moving seaward west of the South Shetland Islands10,11 (Fig. 1, overview).

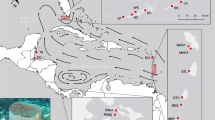
Sampling locations. Overview Map of the Antarctic, showing sampling areas. Sampling locations are color-coded by geographic region. Areas of interest in subsequent maps are indicated in color-coded squares and ordered by movement along the ASF. Inferred position of the ACC and ASF approximated based on Fig. 7 in Orsi et al.11 and Whitworth et al.8, respectively. Ross Sea (RS) Position of the ASF is approximated based on Ashford et al.6 Fig. 10.11. Weddell Sea (WS) Position of the AACC and ASF is approximated based on Fig. 2b in Nicholls et al.56. Larsen Bay (LB) Putative position of the ASF according to Whitworth et al.8. Northern Antarctic Peninsula (NAP) Position of the ACC and AACC are based on Fig. 7 in Orsi et al.11 and Fig. 3 in Thompson et al.47, respectively. Entry of Circumpolar Deep Water from the ACC into the Bransfield Strait through the Boyd Strait is shown according to Savidge and Amft10. Yellow and pink arrows within the Bransfield Strait and Trough area represent components of the Bransfield Gyre with Bellingshausen influence and Weddell influence respectively, based on Fig. 15 in Sangrà et al.87. South Orkney Islands (SOI) Schematic illustration of the circulation drawn from: ACC (Fig. 7 in Orsi et al.11), ASF (Whitworth et al.8, Fig. 6 in Heywood et al.9, Fig. 14 in Azaneu et al.51), AACC (Fig. 3 in Thompson et al.47, Fig. 15 in Sangrà et al.87), and WF (Fig. 6 in Heywood et al.9, Fig. 3 in Thompson et al.47, Fig. 14 in Azaneu et al.51). Putative position of the ASF according to Azaneu et al.51. Western Antarctic Peninsula (WAP) Position of the ACC and AACC is approximated based on both, Fig. 7 in Orsi et al.11 and Fig. 14 in Moffat et al.7 respectively. Empty arrows indicate the putative position of the AACC according to Moffat et al.7. ACC, Antarctic Circumpolar Current; ASF, Antarctic Slope Front; WF, Weddell Front; AACC, Antarctic Coastal Current. Maps created using the Norwegian Polar Institute’s Quantarctica 2.0 package88 in the software QGIS version 2.18.9 http://qgis.osgeo.org 89.
Entrainment of silverfish in transport pathways is facilitated by a life history characterized by vertical migrations associated with different life stages12. Eggs and larvae are found from 0–100 m among the platelet ice layer beneath coastal fast ice13. Descent to deeper layers begins at the post-larval phase, and post-larvae and juveniles up to 2 + years are found at depths up to 400 m3. Adult silverfish, ranging in length between approximately 8–25 cm, are typically found at depths from 400–700 m, exhibiting a diel migration in which a greater abundance of fish are present in the shallower portion of their distribution by night, and in deeper waters by day3,14. These changes in habitat occupancy correlate with ontogenetic shifts in diet composition, such that silverfish occupy several trophic levels throughout their 12–14 year lifespan3,15. While feeding almost exclusively on zooplankton, silverfish have been shown to exhibit dietary plasticity, including cannibalism16,17,18. Thus, early-life dependence on coastal sea-ice zones and later movement into deeper waters in pursuit of prey and avoidance of predators expose fish to hydrographic features along the continental shelf. This results in a complex life history which generates the conditions for a dynamic population structure and underlying patterns of connectivity6.
The silverfish life history hypothesis incorporates these physical-biological interactions, predicting that cross-shelf circulation mediates retention within populations and exposure to along-shelf transport pathways6,19,20. Schools of adult silverfish have been observed moving inshore along the Antarctic Peninsula21. Assemblages of silverfish eggs and larvae have been found in the summer polynya of the eastern Weddell Sea22, in waters along the Antarctic Peninsula23, under sea ice in the western Ross Sea4,12,24, and in the Dumont d’Urville Sea25 (see Fig. 1 for place names). These observations suggest a life cycle in which adults return to coastal areas each winter to spawn. This migration is facilitated by the circulation within glacial trough systems across the continental shelf. While trough outflows aid in the dispersal of developing fish away from hatching sites, inflows function to retain adults by transporting them back to spawning areas. The discovery of newly hatched larvae in trough systems along the Weddell Sea, Ross Sea, and Antarctic Peninsula6,13,22,26 implies this cycle of dispersion and retention occurs in multiple areas around the Antarctic. However, such a cycle would be vulnerable to advective losses along the shelf in the AACC, and along the slope in the ASF or ACC, resulting in transport to locations downstream6.
Genetic structuring and gene flow around the Antarctic
Estimates of genetic differentiation are a principal tool for defining population structure, by assessing gene flow between populations, as well as the extent to which inbreeding and genetic drift contribute to population differentiation27. The first hypotheses concerning population structure in silverfish arose from research carried out in the 1980s in the eastern Weddell Sea and Antarctic Peninsula22,23. Recent advances in our understanding of hydrography and physical-biological interactions provide a contemporary life history framework in which the population genetic structure of silverfish needs to be considered (see Ashford et al.6).
Highly connected populations resulting in genetic panmixia have previously been the null hypothesis to describe genetic structuring in notothenioids. This hypothesis is based on the expected influence of circumpolar currents on the transport of pelagic larvae, which would sustain high levels of gene flow28. This was supported by genetic homogeneity found among multiple species of the families Nototheniidae and Channichthyidae inhabiting the Scotia Arc29,30. At the same time however, this panmictic hypothesis was challenged by evidence of population structuring based on genetic heterogeneity in multiple other species, including Dissostichus mawsoni31,32, Chionodraco hamatus33, Chaenocephalus aceratus34,35, and Champsocephalus gunnari35.
Evidence for gene flow and genetic panmixia has been similarly variable for silverfish. Using mitochondrial DNA markers, Zane et al.36 found little indication of population structuring in silverfish on a circumpolar scale, although differences were found between sampling years in the eastern Weddell Sea, and between one sample in the eastern Weddell Sea and one of two taken in the western Ross Sea36. A more recent study on silverfish connectivity around the Antarctic Peninsula employing microsatellite markers revealed that fish taken from Marguerite Bay and Charcot Island along the western Antarctic Peninsula (WAP) were significantly different from those along the northern Antarctic Peninsula (NAP) at Joinville Island and on the eastern side of the peninsula at Larsen Bay (LB)19 (see Fig. 1, WAP, NAP, and LB for place names). Samples were collected over multiple years, and as in Zane et al.36, evidence for restrictions to gene flow varied between years19.
Variation in recruitment and dispersal, which could explain the observed changes in silverfish distribution, is predicted by the hypothesis of chaotic genetic patchiness37, developed to explain the fluctuations in gene flow between sampling years19. Current theory emphasizes the role of physical-biological interactions in structuring populations and the potential for extensive along-shelf connectivity. Yet to date, there has been no comprehensive investigation of silverfish gene flow and population structure at different spatial scales and over time6, nor re-examination of earlier genetic evidence in the context of recent theoretical advances in understanding.
The present study builds upon the Matschiner et al.28 hypothesis of genetic panmixia in the light of the physical-biological framework provided by Ashford et al.6. It aims to test for population structuring and along-shelf connectivity mediated by large-scale circulation. Employing microsatellite markers, we assessed gene flow between silverfish sampled over 25 years from six different geographic regions around Antarctica (Table 1). The following regions were investigated based on observed and predicted silverfish larval assemblages: the western Ross Sea4,12,24, eastern Weddell Sea22, Larsen Bay26, northern Antarctic Peninsula23, South Orkney Islands38, and western Antarctic Peninsula39. Inclusion of samples from previous studies allowed for a greater number of spatial and temporal comparisons. The expansion of the Agostini et al.19 analysis of Antarctic Peninsular connectivity enabled comparisons to be made on a circumpolar scale. Finally, the use of microsatellite markers increased the analytical power of the Zane et al.36 mitochondrial DNA-based study40. With the present approach, we were able to compare shelf areas located along the ASF from the Ross Sea to the tip of the Antarctic Peninsula, as well as areas located along the ACC from Charcot Island to the South Shetland Islands. In addition, we tested for potential gene flow downstream to the South Orkney Islands along the southern ACC and Weddell Gyre. Finally, standard length (SL) data from over half of the samples collected indicated differences in SL distributions between sampling locations, and we used these to test for chaotic genetic patchiness. Where more than one SL mode existed, sampling locations were further divided into groups of smaller and larger fish to examine for genetic differentiation between and among different cohorts. Although temporal replicates differed between sampling locations, and more locations were sampled in some geographic regions than others, the dataset allowed us to test for genetic panmixia on a circumpolar scale for the first time in silverfish.
Results
Genotyping
Genotypes from 16 microsatellite loci were successfully obtained for all 505 newly sequenced individuals, further supporting that microsatellites developed for C. hamatus19 can be used in P. antarctica. While amplifying successfully in all individuals, one locus (Ch11230) was problematic in terms of accurate genotyping, due to stuttering and interference from the other fluorophores used to detect fragments in sequencing. Subsequent analyses were carried out on both datasets, including and excluding this locus, and no major differences were obtained in the results. Thus, results are presented based on 15 microsatellite loci, excluding locus Ch11230, for verisimilitude. No significant linkage disequilibrium was observed between loci. Significant departures from Hardy Weinberg Equilibrium (HWE) were found at 22 of 304 tests after SGoF + correction for multiple tests (threshold for significance with 304 comparisons P = 0.008) with no locus exhibiting Hardy Weinberg Disequilibrium (HWD) in more than 3 sampling locations and no sampling location containing more than 5 loci in HWD (Table S1). Of the 20 occasions in which the software Micro-Checker found evidence for null alleles at a locus in a given sampling location, only 3 of those coincided with an instance of HWD. Thus, it is likely that the 22 instances of HWD were not representative of an artifactual excess of homozygotes among sampling locations for the loci tested. Evidence for overall heterozygosity excess (HE > HO) was not found by population or by locus, though greater numbers of loci within populations displayed extreme excesses of HE than such excesses of HO (27 comparisons with HE exceeding HO by > 0.1 versus 13 comparisons with HO exceeding HE by > 0.1, out of 304 comparisons). However, only 2 of the 27 instances of extreme HE excess coincided with a significant instance of HWD. Thus, given that the effect size is quite small for the majority (20 of 22) of the instances of HWD, it is unlikely that these departures from HWE are of biological importance41. Neither ARLEQUIN nor LOSITAN identified any locus as a putative outlier, with all simulated FST values falling within neutral expectations. High levels of genetic variation were observed in comparable values of NA, AR, HO, and HE (Table 2, Table S1), showing no significant differences (one-way ANOVA, P > 0.05) across all sampling locations.
Population structure
A power analysis showed that the 15 microsatellites employed in the analysis of genetic differentiation had a 100% chance to detect FST values above 0.0025. The analysis was performed considering various combinations of observed allelic frequencies and sample sizes at multiple iterations of effective population size (Ne) (1000–10,000) and generations (2–201) (Table S4). For FST values greater than 0.001, there was a 94.1–97.4% chance of detecting significant FST values for t = 2, 10, 20 and Ne = 1000, 5000, 10000 using both Fisher’s and Chi-square tests. Thus, the present dataset has the power to detect differences associated with FST higher than 0.001. In the current study, all significant comparisons obtained for FST estimates ranged between 0.00388 and 0.0125, and all near-significant values (for which 0.1 > P > 0.03326, the threshold for significance after SGoF + correction) ranged from 0.00168 to 0.00572. This confirms that all non-significant comparisons can be presumed to be such for the present dataset, and are not non-significant due to the inability of the markers used to detect differences among samples.
Hierarchical AMOVA (Table S3) indicated that partitioning the 19 sampling locations into k = 6 groups based on geographic region (Ross Sea (RS), Weddell Sea (WS), Larsen Bay (LB), northern Antarctic Peninsula (NAP), South Orkney Islands (SOI), and western Antarctic Peninsula (WAP), Table 1) maximized between group heterogeneity (global FCT = 0.00159; P = 0.00218 ± 0.00050) while minimizing within group heterogeneity (global Fsc = 0.00002; P = 0.63050 ± 0.00429). Genetic differentiation was present (global FST = 0.00161; P = 0.00644 ± 0.00087) but very low, with <1% of the total variation due to sampling location. Only 0.16% of variation was attributed to variation among groups. Locus-by-locus AMOVA confirmed that no particular locus had a significantly outsized impact on the calculation of global F-statistics.
Clustering methods failed to indicate a clear structure among the sampling locations analyzed. Clustering methods employed by DAPC and FLOCK (without pre-defined groups) randomly assigned individuals to groups, with no correlation to sampling location or geographic region (Fig. 2, S1). DAPC run without a priori parameters suggested the existence of nine clusters (Fig. 2a), with k = 9 selected based on cluster optimization plotted against Bayesian Information Criteria (Fig. S2a). The same DAPC based on the six geographic regions (RS, WS, LB, NAP, SOI, and WAP), run in an attempt to maximize variation between localities, showed no evidence of cluster formation (Fig. 2b). PCoA on all 19 sampling locations provided limited support for geographic clustering: the first principal coordinate accounted for 39.21% of the variance and clearly distinguished the SOI and WAP samples from all others (Fig. 3a). PCoA on the six geographic regions (RS, WS, LB, NAP, SOI, and WAP) emphasized this delineation further: the first principal coordinate accounted for 81.16% of the variance, and separated the SOI and WAP samples from the rest (Fig. 3b). However, in both cases PCoA failed to distinguish NAP, WS, and LB from one another, and also from the other geographic regions.

Discriminant analysis of principal components (DAPC) clustering results for Pleuragramma antarctica populations. (a) DAPC run without a priori parameters. (b) DAPC run based on geographic region, acronyms are as in Table 1.

Principal coordinate analysis (PCoA) for Pleuragramma antarctica samples based on FST genetic distance. (a) PCoA of 19 sampling locations. (b) PCoA of six geographic groups. Sampling location and geographic group acronyms are as in Table 1.
In contrast to clustering methods, pairwise FST provided support for geographic population clusters. Within sampling locations, there were no significant differences between samples collected over multiple years (FST ranged from −0.0025 to 0.0024, P > 0.05 for all comparisons, Table 3), suggestive of genetic stability over time. In addition, within geographic regions (RS, WS, LB, NAP, SOI, and WAP), there was no significant difference between sampling locations (FST ranged from −0.0051 to 0.0024, P > 0.05 for all comparisons, Table 3), indicative of genetic homogeneity within these regions.
Pairwise comparisons between years and different sampling locations among the geographic groupings displayed limited structure. Patterns of significant differentiation previously found along the Antarctic Peninsula were maintained to some extent (5 significant comparisons out of 36 in the current study and 9 out of 36 in Agostini et al.19 after SGoF + correction for multiple tests, threshold for significance with 171 comparisons P = 0.0332). Such discrepancies are due to the use of 15 loci instead of 16 loci as in Agostini et al.19 as well as to changes in significance thresholds resulting from the increased number of comparisons in the present study (nineteen sampling locations versus nine). Indeed, when the dataset of nine sampling locations from Agostini et al.19 was reanalyzed with the removal of the problematic locus Ch11230, three differences were found with respect to the original analysis (Table S5). Two of these disparities from the original analysis (the comparisons between CI10 and LB07, and between MB01 and JI12) were also seen in the larger analysis between nineteen sampling locations (Table 3). Variation in differentiation between years in comparisons from the same sampling location, as previously documented in the Ross Sea36 and the Antarctic Peninsula19, was also obtained in the current study. For every location for which samples existed from multiple years, patterns of differentiation varied over sampling years (Table 3). This was especially evident at Marguerite Bay, Joinville Island, the South Orkney Islands, Larsen Bay, and the Ross Sea.
Grouping sampling locations by geographic region provided evidence of weak genetic structuring (FST ranged from 0.0001 to 0.0153, Table 4). Significant pairwise FST values were found between all groups and the western Antarctic Peninsula group (also seen in Agostini et al.19), as well as between the South Orkney Islands and all groups except for the Weddell Sea, where the comparison was not significant after SGoF + correction for multiple tests (threshold for significance with 15 comparisons P = 0.0142, Table 4). In addition, the comparison between Larsen Bay and the Ross Sea was not significant after correction (P = 0.0312, Table 4).
Comparison by length modes
Of the 1067 samples analyzed in this study, standard length (SL) data were available for 671 (63%) samples. SL ranged from 3.7–21.5 cm. Post-larvae and juveniles (SL < 9 cm)3 composed 15% of all fish for which SL data were available. To control for the possibility of bias presented by the wide range of SL represented in the present study, both DAPC and pairwise FST analyses were performed on SL-derived groups independent of sampling location. Six groupings were tested, separating fish by SL into groups divided by 1, 2, 3, 4, 5, and 6 cm increments (Table S6a). No evidence of clustering was seen in the DAPC analysis of the length groupings (Fig. S3), nor was any pattern of genetic differentiation associated with life stage observed (Table S6b–g).
Standard length (SL) distributions varied considerably between sampling locations (Fig. 4). Agostini et al.19 observed a pronounced bimodal distribution in the sample taken from Joinville Island 2010 (JI10), with modes at SL = 5 cm and SL = 10 cm. After dividing the JI10 group into small and large fish representative of more and less recent cohorts, pairwise FST analysis revealed that the small fish (n = 68) and large fish (n = 72) were significantly different. Seven of eight comparisons were significant between all groups and JI10 small, whereas three of eight comparisons were significant between all groups and JI10 large after SGoF + correction for multiple tests19 (threshold for significance with 190 comparisons P = 0.0453). When this analysis was extended to the sampling locations considered in the current study, this result was largely replicated: six of eight comparisons were significant between the original AP groups and JI10 small, and two of eight comparisons were significant between the original AP groups and JI10 large after SGoF + correction for multiple tests, (Table S7). However, SOI was significantly different from JI10 small (FST = 0.0045, P = 0.0265) and JI10 large (FST = 0.0042, P = 0.0312), in contrast to the original analysis. Consistent with results seen in the initial and replicated AP analysis between JI10 small and Larsen Bay 2007 (n = 46), JI10 small significantly differentiated after SGoF + correction for multiple tests from Larsen Bay 2011 (n = 52, FST = 0.0037, P = 0.0361), a comparison that was not significant when analyzing the undivided JI10 group. Similarly, the comparison between JI10 small and Ross Sea 1997 (n = 84) was significant after SGoF + correction for multiple tests (FST = 0.0044, P = 0.0061), despite no indication of differentiation with the undivided JI10 group. Finally, seven additional pairwise comparisons were significant after SGoF + correction for multiple tests in the JI10 cluster analysis, despite not involving the newly introduced JI10 small or large clusters (see Table S7 for details).

Standard length (SL) distributions of Pleuragramma antarctica around the Antarctic Peninsula and Weddell Sea. Sampling locations and collection years are specified above the graphs, as well as group sizes (n). When several collections were performed within the same year and area, sampling stations are listed after the collection year. Frequency indicated on the y-axis, SL in cm indicated on the x-axis, and the size interval is 2 cm.
Spatially recurring length modes among sampling locations collected in the eastern Weddell Sea in 2014 provided evidence for episodic connectivity (Fig. 4). Thus, a separate pairwise FST analysis was performed including only sampling locations collected in 2014 within the eastern Weddell Sea. No significant differences were seen between any of the sampling areas, which included west Filchner Trough (n = 23), east Filchner Trough (n = 27), Coats Land (n = 29), Halley Bay (n = 52), and Atka Bay (n = 25) (FST ranged from −0.0062 to 0.0027, P > 0.05 for all comparisons, Table S8). With such low levels of differentiation between areas despite the diverse SL distributions, a final pairwise FST analysis was performed on groupings of fish from the eastern Weddell Sea that were based solely on SL range, independent of sampling location. SL among specimens from the eastern Weddell Sea ranged from 7.0–21.5 cm. First, fish were divided into six length-based groups (<10 cm, 10–12 cm, 12–14 cm, 14–16 cm, 16–18 cm, >18 cm), with group n ranging from 14–43 individuals. When no significant differences were found between length-based groups after SGoF + correction for multiple tests (FST ranged from −0.0081 to 0.01019), an exploratory analysis of various pools of the six original length-based groups was conducted. The smallest length-based pool was defined as SL ≤ 12 cm and then subsequently as SL ≤ 14 cm. The largest length-based pool was defined as SL < 16 cm and various combinations of the intermediate groups were created. Testing these length-based pools allowed for the control of the influence of group size, and none of the pairwise comparisons were significant after SGoF + correction for multiple tests (FST ranged from −0.0081 to 0.0103).
Discussion
The present study tests the hypothesis of genetic panmixia28 in the light of the physical-biological framework provided by Ashford et al.6 for Antarctic silverfish. Evidence of high gene flow between regions connected by the Antarctic Slope Front (ASF) and associated Antarctic Slope Front Current on a circumpolar scale was accompanied by indications of reduced gene flow in regions not reached by the ASF. The Discriminant Analysis of Principle Components (DAPC), revealed evidence of nine clusters, but these were independent of geographic region. Instead, they may have arisen due to historical extinction and recolonization events predicted by Ashford et al.6, related to glacial periods that recurrently structured past populations of silverfish. Overall levels of genetic differentiation were low (maximum FST = 0.0153), and no significant differences were found from the Ross Sea westward to the northern Antarctic Peninsula. However, limited evidence of genetic structuring was found. We corroborated the findings from Agostini et al.19 that Charcot Island and Marguerite Bay represent a homogeneous population with significantly reduced gene flow from populations around the AP and beyond, while also discovering significant reductions in gene flow to the population at the South Orkney Islands. These results were broadly consistent with the Matschiner et al.28 prediction of gene flow facilitated by the large-scale circulation. However, the genetic structuring we found corresponded to discontinuities between major current systems along the shelf. Accordingly, we argue for a more nuanced version of the genetic panmixia hypothesis that incorporates physical-biological interactions over silverfish life history.
Along-shelf gene flow and connectivity
Similarity between samples from the Ross Sea, eastern Weddell Sea, Larsen Bay and Joinville Island indicated gene flow along the shelf corresponding to the ASF. Pairwise FST analysis provided evidence for six possible silverfish populations among the geographic regions analyzed. Moreover, the Ross Sea did not differ from the Weddell Sea, Larsen Bay or Joinville Island. Joinville Island and the South Shetland Islands formed a cluster, which did not differ from the others, suggesting gene flow along the ASF into the Bransfield Strait. Notably, comparisons made by Agostini et al.19 showed evidence of differentiation between Larsen Bay and Joinville Island, whereas in the present analysis, not a single comparison among sampling locations from Joinville (JI07, JI10, and JI12) and Larsen Bay (LB07 and LB11) revealed significant differentiation. This is consistent with the predictions made in La Mesa et al.26 of connectivity between Larsen Bay and the Bransfield Strait. Thus, the inclusion of the additional samples from Larsen Bay (LB11) clarified connectivity along the ASF between the western Weddell Sea and Bransfield Strait.
In contrast, regions not connected by the ASF showed evidence of reduced gene flow. Along the western Antarctic Peninsula, where westward flow inshore along the AACC contrasts with eastward flow along the slope in the southern ACC, Charcot Island (CI) and Marguerite Bay (MB) represented one cluster, which differentiated genetically from all other geographic regions. Discontinuity in silverfish distribution between the northern and southern regions of the western Antarctic Peninsula was first discussed in Agostini et al.19 based in part on the decrease of silverfish in Adélie penguin diets at Palmer Station42. The loss of intermediate populations that may have previously facilitated connectivity between the northern and southern extents of the western Antarctic Peninsula was further supported by the total absence of silverfish in sampling surveys at Anvers and Renaud Islands and the Croker Passage midway up the western Antarctic Peninsula in 2010 (see Fig. 1, WAP for place names) despite having previously been the dominant fish species in these areas since the 1980s14. In addition, the South Orkney Islands differentiated from all other regions except for the eastern Weddell Sea. Differentiation between years within certain sampling locations echoed evidence for chaotic genetic patchiness seen in the Ross Sea by Zane et al.36 and in the Antarctic Peninsula by Agostini et al.19. This was further indicated by the variability in SL distributions between years in particular sampling locations, providing evidence for variation in recruitment and dispersal of different cohorts37.
The clearest example of interannual variability was seen in the western Antarctic Peninsula (WAP). Specimens from MB01 and MB02 exhibited a bimodal distribution of SL representative of more and less recent cohorts. In contrast, specimens from MB10 and CI10 exhibited a much tighter unimodal distribution around SL = 15 cm, as well as MB11, which had a similarly tight unimodal distribution around SL = 10 cm (Fig. 4). This disparity between cohorts was reflected in patterns of genetic differentiation within the WAP area between years, with MB01 and MB02 differentiating from two of the nineteen other sampling locations, while MB11 differed from six of the nineteen other sampling locations (Table 3). However, position with respect to local hydrographic features must also be considered. Despite being collected only a year before the strongly differentiated MB11 sample, MB10 shares the weak differentiation pattern of nearby MB01 and MB02, which were all collected in the Labeuf Fjord closer to the coast (Fig. 1, WAP, Table 3). This greater connectivity with populations outside of the WAP is consistent with gene flow down the Antarctic Peninsula promoted by the AACC19. Furthermore, the position of MB11 along the trough inflow in Marguerite Trough (Fig. 1, WAP) raises the possibility that the sample consisted of fish resulting from connectivity along the slope, where entrainment in the southern ACC potentially transports fish from the Bellingshausen and Amundsen Seas23. The influx of migrants from populations further upstream may contribute to the stronger pattern of differentiation found between MB11 and sampling locations beyond the Antarctic Peninsula.
Differences between the previously published Antarctic Peninsula study19 and the current results are likely attributable to changes in significance thresholds resulting from the increased number of comparisons in the present study, as well as the use of 15 instead of 16 loci due to the removal of one problematic locus. In the original Antarctic Peninsula analysis, Agostini et al.19 found 9 out of 36 significant comparisons around the Antarctic Peninsula, while in the current analysis, only four out of the thirty-six Antarctic Peninsula comparisons were significant. This was mainly due to weaker differentiation between WAP locations and JI10 and JI12. Discrepancies in patterns of differentiation between the current study and the original investigation of population connectivity in Zane et al.36 can be attributed to their use of mtDNA as opposed to microsatellite markers to resolve patterns of genetic heterogeneity40, in addition to differences in sample size (Table 3). Given the small differences geographically, variation associated with reproduction, differential survival, and recruitment can lead to changes in resolution in subsequent investigations6. These results highlight the importance of continued, targeted sampling that incorporates hydrography in order to monitor changes in population structure over time. The integration of multidisciplinary techniques, notably circulation and movement modelling43, otolith chemistry44, and genomics45, will also help in resolving population structure and connectivity over the life history.
Population structure and life history connectivity
The silverfish physical-biological population hypothesis6 provides a mechanistic explanation consistent with the patterns of gene flow and genetic structuring we observed. When fish in outflows from local trough systems, for instance in the Ross Sea20 and eastern Weddell Sea22, reach the shelf break, entrainment in the ASF and westward transport has the potential to introduce fish into trough systems downstream. Samples from Terra Nova Bay in the Ross Sea collected in 1996 and 1997 were genetically very similar to those collected in the Weddell Sea and Antarctic Peninsula, only differentiating from samples taken along the western Antarctic Peninsula and the South Orkney Islands. This high level of connectivity between regions on opposite sides of the continent may be attributed to two main factors. The first is related to the extent and importance of the Terra Nova Bay silverfish nursery, where silverfish comprise 98% of the ichthyoplankton4,13. The second is related to potential silverfish populations along East Antarctica. Despite the extensive geographic purview of this study, significant gaps remain for this circumpolar species, especially along East Antarctica. Substantial distributions of silverfish have been found across the continental shelf along East Antarctica3, as well as in association with the Ninnis and Adélie Troughs off Wilkes Land46 (Fig. S4, East Antarctica). Other trough systems along East Antarctica in Lützow-Holm Bay, Iceberg Alley, Nielsen Basin, and Prydz Bay may also provide suitable habitats for coherent silverfish populations6 (Fig. S4, East Antarctica). The existence of a network of local populations of silverfish associated with trough systems along East Antarctica provides a mechanism for the exchange of individuals via the westward-flowing ASF. Such a mechanism would explain the high levels of gene flow observed between sampling locations as geographically separated as Terra Nova Bay and Joinville Island.
Moreover, our results lend further support for connectivity beyond Joinville Island into the region around the Bransfield Strait. Fish at Joinville could be transported there by either: (1) the AACC, which could transport fish from Larsen Bay26,47; or (2) the ASF, which could transport enough individuals from the eastern Weddell Sea and beyond to render the Joinville population genetically indistinguishable from upstream populations6,9,48. Although the path of the AACC south of Joinville Island is undefined, buoyancy forcing is likely to generate connectivity that is shallow and close to the coast7. This could result in larval transport north from Larsen Bay which would explain the differentiation between size modes in the Antarctic Sound (Fig. 1., NAP).
The results of the current study raise the possibility of a coherent silverfish population at the South Orkney Islands. Surveys carried out between 1967–1981 revealed the presence of silverfish larvae at the South Orkney Islands38, consistent with a local breeding population. Furthermore, a recent bathymetric analysis of the South Orkney Islands microcontinent found seven trough systems on the northern and southern sides of the islands49. Features supportive of a locally breeding silverfish population were found in Signy Trough between Coronation and Signy Island49, including active glaciers at the trough head, thought to be necessary for early-life stages6 (Fig. 1, SOI). Evidence from otolith chemistry analyses has revealed that fish from the northern Antarctic Peninsula significantly differed from those at the South Orkney Islands, indicating that fish in these areas were sourced from different spawning grounds50. This is supported by our findings of genetic differentiation between the northern Antarctic Peninsula and South Orkney Islands populations.
However, the lack of differentiation between specimens from the South Orkney Islands and specimens from the eastern Weddell Sea means connectivity between these two regions cannot be discounted based on these data. The existence of migrants or non-breeding vagrants at the South Orkney Islands would be predicated by a clear and consistent transport system from the eastern Weddell Sea to the South Orkney Islands microcontinent. Although fish carried to the shelf slope along trough outflows in the eastern Weddell Sea can come into contact with the ASF6, subsequent transport to the South Orkney Islands microcontinent remains unclear. In addition to the distance along the ASF, fish are potentially exposed to inflows at the mouths of the Hughes, Ronne (Fig. 1, WS), Jason, and Robertson (Fig. 1, LB) Troughs that can draw them away from the slope towards the inner shelf. Continued entrainment in the ASF north of Larsen Bay carries fish around Joinville Island, where the AACC merges with the ASF. Subsequently, the currents separate again, the AACC forming the southern branch of the Bransfield Gyre10 (Fig. 1, NAP) and the ASF moving north along the South Scotia Ridge9,47,51. However, upon entering the Hesperides Trough, increased influence by Weddell-Scotia Confluence waters combined with local bathymetric complexity impede continued identification of the ASF51. Thus, the ASF does not provide a direct pathway to the South Orkney Islands. Alternatively, the Weddell Front, which forms east of the Joinville Ridge and rounds the Powell Basin to reach the South Orkney Islands microcontinent, may represent a path from the Weddell Sea to the South Orkney Islands. Future studies will require an interdisciplinary approach to assess the impact of episodic connectivity on genetic similarity between the South Orkney Islands and the eastern Weddell Sea. The hydrography between these areas needs to be elaborated in order to determine the eventual fate of the ASF. Otolith chemistry analyses52 and dispersal modelling53 of fish from the South Orkney Islands and the eastern Weddell Sea will be able to clarify how much the genetic similarity between these regions is due to episodic connectivity into the local population.
Hydrography may also explain the chaotic genetic patchiness observed in the differentiation patterns between the two Larsen Bay samples and the eastern Weddell Sea and South Orkney Islands (Table 3). It is important to note that the LB07 and LB11 samples were collected from different parts of Larsen Bay (Fig. 1, LB). LB07 was collected in the Larsen B embayment off Scar Inlet, near one of the heads of the Larsen Inlet Trough. The LB11 samples were collected in the Larsen A embayment, just north of the Seal Nunataks Ice Shelf, at the head of the Robertson Trough. Since the disintegration of the Larsen A and B shelves, runoff into the Larsen embayments has been derived from smaller glaciers along the peninsula that contribute to disparate patterns of freshwater influx between the Larsen A and B areas54,55. Differences in seasonal melting patterns between the two areas are likely to directly impact coastal buoyancy and connectivity between trough systems in Larsen Bay. Considering the evidence of gene flow between the eastern Weddell Sea and both the Larsen Bay and South Orkney Islands, the lack of differentiation between LB11 and SOI11 may be due to changes in the influx of eastern Weddell Sea migrants into these areas. This is in contrast to the hypothesis that the observed gene flow results from the direct exchange of individuals between Larsen Bay and the South Orkney Islands. Changes in recruitment may occur in tandem with the influx of eastern Weddell Sea migrants into the respective Larsen A and Larsen B embayments. This would result in the variations of gene flow observed between the eastern Weddell Sea, South Orkney Islands, and the LB07 and LB11 sampling locations.
The pattern of homogeneity observed among fish across all sampling locations within the eastern Weddell Sea fails to explain the variation in SL distributions. This suggests population mixing between local and migrant fish, seen from Atka Bay in the northeast, to Halley Bay and Coats Land, and finally to the eastern and western sides of the Filchner Trough in the southeast. Aspects of local hydrography help to explain these observations. In the southeast Weddell Sea, the AACC and the ASF are functionally equivalent given the narrowness of the continental shelf in this area9. Seasonal and interannual variability in the AACC in the southeast Weddell Sea56 may promote and restrict connectivity along the shelf from Atka Bay to Filchner Trough. Fish that become entrained in the ASF upstream are introduced to the Weddell Sea east of Atka Bay, as it becomes the southern limb of the Weddell Gyre8,57. Having formed in the Amundsen Sea, the ASF has the potential to connect local trough systems in the Ross Sea and East Antarctica6,8. While it remains a more consistent feature than the AACC, the position and strength of the ASF is nevertheless sensitive to wind stress forcing on seasonal and interannual time scales in the northwest Weddell Sea51,58,59. Such variations in the intensity of the ASF could explain fluctuations in the influx of migrants into eastern Weddell Sea populations and provide a general mechanism for the chaotic genetic patchiness observed among silverfish populations.
While the present dataset is unique in both its temporal and spatial extents, as well as in its inclusion of life history data, it is important to keep in mind its limitations and what types of analyses and sampling could fill the present gaps. Variation in collection gear by research vessel between sampling locations (Table 1) may have introduced bias into the allele frequencies measured between location. However, the high levels of homogeneity found between locations sampled by different research vessels, coincident with differences between locations sampled by the same research vessels, imply that any gear biases did not have a consequential impact on the present results. Subsequent studies analyzing otolith nucleus chemistry in silverfish along the eastern Weddell Sea will allow us to distinguish between areas more heavily composed of locally spawned fish and migrants. Furthermore, enhanced oceanographic data would improve dispersal modelling, allowing for a more targeted approach in defining connectivity hypotheses. Contiguous sampling from regions that could contribute migrating fish to the ACC and the ASF are needed in order to better define in which ways the various current systems influence eastern Weddell Sea population mixing.
Nevertheless our results show no basis for rejecting along-shelf connectivity predicted by Ashford et al.6, and are consistent with the cross-shelf component of their hypothesis, examined earlier by Brooks et al.20 in the Drygalski Trough. Moreover, the implications of observed structure and gene flow in Antarctic silverfish is seen in other Antarctic species. Krill (Euphasia superba), a keystone species in waters beyond the continental shelf60, exhibit a similar lack of population structure, as shown in studies using mtDNA and microsatellites61, as well as restriction-site-associated DNA sequencing62. Such panmixia observed in Antarctic krill was explained by very large population sizes and ongoing gene flow61,62. However, it was stressed that genetically indistinguishable populations do not imply ecologically independent populations61,63. Evidence for a recent population expansion observed in analysis of mtDNA in Bortolotto et al.61 emphasized the differential impact of glaciations on the population structure of pelagic versus benthic Antarctic species. Thus, impacts of climate change may result in range restrictions that will have the opposite effect of glacial expansions on pelagic species, cutting off intermediate populations which can result in cascading effects on the connectivity of ecologically interdependent populations64,65.
Antarctic silverfish are a pillar of the Southern Ocean ecosystem2. Understanding their population structure and connectivity in the context of their life history and the impact of hydrography is critical to assessments of Southern Ocean ecosystem health.
Materials and Methods
Sample collection and DNA extraction
A total of 1067 individuals from 19 sampling locations collected by multiple institutions between 1989 and 2014 were included in the analysis (Fig. 1, Table 1). Of these, 249 individuals collected between 1989 and 1997 from the Ross Sea (RS96, RS97), Halley Bay (HB89, HB91), and South Shetland Islands (SSI96) were included from a previous study on population structure using mitochondrial DNA36. A further 562 individuals collected between 2001 and 2012 from Larsen Bay (LB07), Charcot Island (CI10), Marguerite Bay (MB01, MB02, MB10, MB11), and Joinville Island (JI07, JI10, JI12) were included from a previous connectivity study focused on the Antarctic Peninsula19. The remaining 256 individuals collected in 2011 and 2014 from the South Orkney Islands (SOI11), Larsen Bay (LB11), Filchner Trough (FT14), Atka Bay (AB14), and Halley Bay (HB14) have not previously been examined. Table 1 outlines sampling details, including research vessel information and collection gear. All samples, including those obtained in 2011 and 2014 that had not been previously examined, were collected during authorized scientific cruises carried out by nations legally committed to the Convention for the Conservation of Antarctic Marine Living Resources66. No manipulations were carried out on live samples.
Standard length (SL) data were available from all individuals except from those collected from RS96, RS97, and HB91. SL data from HB89 and SSI96 represent the length distribution of fish collected at the sampling stations, but lack identification numbers to correspond them to the tissue samples collected for genetic analysis. The distribution of SL was used to identify modes for consideration in the eventual genetics analysis.
Muscle tissue or fin clips were preserved in 95–100% ethanol at the time of sampling. Genomic DNA was extracted from specimens using a standard salting-out procedure67. Genomic DNA was extracted from all previously unexamined samples, as well as from tissue samples remaining from individuals on which the mitochondrial DNA analysis was performed in Zane et al.36. Concentration and quality of the extracted DNA (260/280 nm and 260/230 nm) was checked using a NanoDrop UV–Vis spectrophotometer (Thermo Scientific) prior to PCR amplification. All extracted DNA was of high enough quality to use in subsequent PCR reactions.
DNA amplification and microsatellite genotyping
Individuals were genotyped using 16 published EST-linked microsatellites developed in Chionodroco hamatus68,69, that had been previously shown to cross-amplify successfully in P. antarctica19. The 16 loci were amplified in two multiplex PCR reactions (see Table S2 for primer sequences and final conditions for all loci). Multiplex PCR reaction volume was 10 µL, containing 1x QIAGEN Multiplex PCR Master mix (HotStartTaq DNA Polymerase, Multiplex PCR Buffer, dNTP Mix; QIAGEN, Hilden, Germany), 0.2 µM primer mix and 100 ng of genomic DNA. The PCR amplification profile for all loci consisted of: (1) an initial activation step of 15 min at 95 °C; (2) 30 cycles of denaturation at 94 °C for 30 s, annealing at 57 °C for 90 s, and extension at 72 °C for 60 s; and (3) a final extension of 30 min at 60 °C.
PCR products were prepared for microsatellite genotyping and sent to an external service (BMR Genomics, http://www.bmr-genomics.com/), where they were sequenced using an ABI 3730xl automated sequencer (LIZ 500 as internal size ladder, Applied Biosystems, Waltham, MA, USA). Microsatellites were analyzed using GeneMarker ver. 2.6 (SoftGenetics). Genotypes of individuals included in the microsatellite analysis published by Agostini et al.19 were integrated into this analysis. To ensure that the datasets were comparable, two individual samples from Agostini et al.19 were processed alongside samples from the present study as positive controls during each amplification and sequencing run. This allowed the comparison of the raw microsatellite data between the previous and current study and confirmed that there was no change in microsatellite sizing. Binning was automated with FlexiBin 270 and refined by eye to assure accuracy with the corresponding binnings established in Agostini et al.19.
Data analysis
A suite of programs was used to assess population differentiation and genetic structuring based on the microsatellite genotypes. Input files were created in the appropriate software-specific formats using CREATE ver 1.3771. Linkage Disequilibrium (LD) probability was tested with GENEPOP online72. Deviations from Hardy-Weinberg equilibrium were calculated with the R package diveRsity ver. 1.9.8973. The software Micro-Checker 2.2.3 was used to detect the presence of null alleles and scoring errors due to large allele drop-out and stuttering74. Significance levels for multiple comparisons were adjusted using the program SGoF+ 75.
Detection of loci under selection was carried out using the methods implemented in LOSITAN ver. 1.0.076 and ARLEQUIN 3.5.1.377. Both software generate coalescent simulations based on observed data and a chosen demographic model of migration to create the expected distribution of FST versus HE with neutral markers. Outlier loci are detected based on the presence of significantly higher or lower FST values compared to neutral expectations. In LOSITAN, 1,000,000 simulations were run under neutral mean FST, simulating 19 groups (for each sampling location), confidence interval of 0.95% and a false discovery rate of 0.01 assuming a Stepwise Mutation Model (SMM)19. In ARLEQUIN, genetic structure was assessed by running 50,000 simulations, simulating 19 groups (for each sampling location) and 6 groups (corresponding to the number of geographic regions), and assuming 100 demes per group under the hierarchical island model78.
Within-sampling location genetic variability was assessed by calculating observed (HO) and expected (He) heterozygosity, number of alleles (NA), and allelic richness (AR) using the R package diveRsity ver. 1.9.8973. Genetic variability was compared between sampling locations using a one-way ANOVA performed in R79.
The extent to which sampling locations could be clustered into the same group was assessed using various methods. Discriminant analysis of principal components (DAPC), a multivariate method in which fish are assigned to known groups, and Principle Coordinate Analysis (PCoA), a multidimensional scaling method in which the matrix of dissimilarities was built from FST comparisons, were carried out using the R package ADEGENET 2.0.180 and GenAlEx 6.50181,82, respectively, to provide a visual assessment of differentiation between sampling locations. Estimation of the number of groups into which genotypes from the various sampling locations cluster, ignoring a priori information about provenance, was tested using FLOCK 3.183. FLOCK employs a non Markov Chain Monte Carlo (MCMC) algorithm that uses an iterative method to divide genotypes into k clusters. This method has been shown to allocate clusters more accurately than common Bayesian methods when population structure is weak84.
The power of microsatellite loci to detect differences in the sample sizes analyzed was assessed applying Fisher’s and Chi-square tests in POWSIM ver 4.185. Observed sample sizes and allele frequencies taken from the 19 sampling locations were used to gauge the statistical power of the 15 microsatellites to detect FST values ≤ 0.01 using 1000 replicates. In accordance with the POWSIM manual, several combinations of Ne (1000–10,000) and generations (t = 2–201) were tested to account for variability in the estimation of Ne.
Population differentiation was determined by calculating pairwise genetic distances represented by FST86 between sampling locations as well as between SL-defined groups. Hierarchical analysis of molecular variance (AMOVA) was also used to assess population differentiation. Pairwise genetic differences and AMOVA were calculated in ARLEQUIN 3.5.1.377. Significant differentiation was determined with 10,000 permutation tests. The significance threshold was adjusted according to SGoF + correction for multiple tests75.
Data Availability
The datasets generated and analyzed during the current study are available from the corresponding author on request.
References
Duhamel, G. et al. In Biogeographic Atlas of the Southern Ocean. (ed. De Broyer, C. et al.) Ch. 7, 328–362 (Scientific Committee on AntarcticResearch, 2014).
Koubbi, P. et al. In The Antarctic Silverfish: a Keystone Species in a Changing Ecosystem (eds Marino Vacchi, Eva Pisano & Laura Ghigliotti) 287–305 (Springer International Publishing, 2017).
La Mesa, M. & Eastman, J. T. Antarctic silverfish: Life strategies of a key species in the high-Antarctic ecosystem. Fish and Fisheries 13, 241–266 (2012).
Vacchi, M. et al. A nursery area for the Antarctic silverfish Pleuragramma antarcticum at Terra Nova Bay (Ross Sea): First estimate of distribution and abundance of eggs and larvae under the seasonal sea-ice. Polar Biology 35, 1573–1585, https://doi.org/10.1007/s00300-012-1199-y (2012).
Wöhrmann, A. P. A., Hagen, W. & Kunzmann, A. Adaptations of the Antarctic silverfish Pleuragramma antarcticum (Pisces: Nototheniidae) to pelagic life in high-Antarctic waters. Marine Ecology Progress Series 151, 205–218 (1997).
Ashford, J., Zane, L., Torres, J. J., La Mesa, M. & Simms, A. R. In The Antarctic Silverfish: a Keystone Species in a Changing Ecosystem (eds Marino Vacchi, Eva Pisano & Laura Ghigliotti) 193–234 (Springer International Publishing, 2017).
Moffat, C., Beardsley, R. C., Owens, B. & van Lipzig, N. A first description of the Antarctic Peninsula CoastalCurrent. Deep Sea Research Part II: Topical Studies in Oceanography 55, 277–293, https://doi.org/10.1016/j.dsr2.2007.10.003 (2008).
Whitworth, T., Orsi, A. H., Kim, S. J., Nowlin, W. D. & Locarnini, R. A. In Ocean, Ice, and Atmosphere: Interactions at the Antarctic Continental Margin 1–27 American Geophysical Union, 1998).
Heywood, K. J., Naveira Garabato, A. C., Stevens, D. P. & Muench, R. D. On the fate of the Antarctic Slope Front and the origin of the Weddell Front. Journal of Geophysical Research: Oceans 109, C06021, https://doi.org/10.1029/2003JC002053 (2004).
Savidge, D. K. & Amft, J. A. Circulation on the West Antarctic Peninsula derived from 6 years of shipboard ADCP transects. Deep-Sea Research Part I: Oceanographic Research Papers 56, 1633–1655, https://doi.org/10.1016/j.dsr.2009.05.011 (2009).
Orsi, A. H., Whitworth, T., III & Nowlin, W. D., Jr. On the meridional extent and fronts of the Antarctic Circumpolar Current. Deep-Sea Research Part I 42, 641–673, https://doi.org/10.1016/0967-0637(95)00021-W (1995).
La Mesa, M. et al. Influence of environmental conditions on spatial distribution and abundance of early life stages of antarctic silverfish, Pleuragramma antarcticum (Nototheniidae), in the Ross Sea. Antarctic Science 22, 243–254, https://doi.org/10.1017/S0954102009990721 (2010).
Guidetti, P., Ghigliotti, L. & Vacchi, M. Insights into spatial distribution patterns of early stages of the Antarctic silverfish, Pleuragramma antarctica, in the platelet ice of Terra Nova Bay, Antarctica. Polar Biology 38, 333–342, https://doi.org/10.1007/s00300-014-1589-4 (2014).
Lancraft, T. M., Reisenbichler, K. R., Robison, B. H., Hopkins, T. L. & Torres, J. J. A krill-dominated micronekton and macrozooplankton community in Croker Passage, Antarctica with an estimate of fish predation. Deep-Sea Research Part II: Topical Studies in Oceanography 51, 2247–2260, https://doi.org/10.1016/j.dsr2.2004.07.004 (2004).
Giraldo, C. et al. Lipid dynamics and trophic patterns in Pleuragramma antarctica life stages. Antarctic Science 27, 429–438, https://doi.org/10.1017/S0954102015000036 (2015).
Eastman, J. T. Pleuragramma antarcticum (Pisces, Nototheniidae) as food for other fishes in McMurdo Sound, Antarctica. Polar Biology 4, 155–160, https://doi.org/10.1007/BF00263878 (1985).
La Mesa, M., Eastman, J. T. & Vacchi, M. The role of notothenioid fish in the food web of the Ross Sea shelf waters: A review. Polar Biology 27, 321–338, https://doi.org/10.1007/s00300-004-0599-z (2004).
Kellermann, A. Food and feeding ecology of postlarval and juvenile Pleuragramma antarcticum (Pisces; Notothenioidei) in the seasonal pack ice zone off the Antarctic peninsula. Polar Biology 7, 307–315, https://doi.org/10.1007/BF00443949 (1987).
Agostini, C. et al. Genetic differentiation in the ice-dependent fish Pleuragramma antarctica along the Antarctic Peninsula. Journal of Biogeography 42, 1103–1113, https://doi.org/10.1111/jbi.12497 (2015).
Brooks, C. M. et al. Early life history connectivity of Antarctic silverfish (Pleuragramma antarctica) in the Ross Sea. Fisheries Oceanography 27, 1–14, https://doi.org/10.1111/fog.12251 (2018).
Daniels, R. A. & Lipps, J. H. Distribution and Ecology of Fishes of the Antarctic Peninsula. Journal of Biogeography 9, 1–9, https://doi.org/10.2307/2844726 (1982).
Hubold, G. Spatial distribution of Pleuragramma antarcticum (Pisces: Nototheniidae) near the Filchner- and Larsen ice shelves (Weddell Sea/Antarctica). Polar Biology 3, 231–236, https://doi.org/10.1007/BF00292628 (1984).
Kellermann, A. Geographical distribution and abundance of postlarval and juvenile Pleuragramma antarcticum (Pisces, Notothenioidei) off the Antarctic Peninsula. Polar Biology 6, 111–119, https://doi.org/10.1007/BF00258262 (1986).
Vacchi, M., La Mesa, M., Dalu, M. & Macdonald, J. Early life stages in the life cycle of Antarctic silverfish, Pleuragramma antarcticum in Terra Nova Bay, Ross Sea. Antarctic Science 16, 299–305, https://doi.org/10.1017/S0954102004002135 (2004).
Koubbi, P. et al. Spatial distribution and inter-annual variations in the size frequency distribution and abundances of Pleuragramma antarcticum larvae in the Dumont d’Urville Sea from 2004 to 2010. Polar Science 5, 225–238, https://doi.org/10.1016/j.polar.2011.02.003 (2011).
La Mesa, M., Piñones, A., Catalano, B. & Ashford, J. Predicting early life connectivity of Antarctic silverfish, an important forage species along the Antarctic Peninsula. Fisheries Oceanography 24, 150–161, https://doi.org/10.1111/fog.12096 (2015).
Lowe, W. H. & Allendorf, F. W. What can genetics tell us about population connectivity? Molecular ecology 19, 3038–3051, https://doi.org/10.1111/j.1365-294X.2010.04688.x (2010).
Matschiner, M., Hanel, R. & Salzburger, W. Gene flow by larval dispersal in the Antarctic notothenioid fish Gobionotothen gibberifrons. Molecular ecology 18, 2574–2587, https://doi.org/10.1111/j.1365-294X.2009.04220.x (2009).
Papetti, C. et al. Population genetic structure and gene flow patterns between populations of the Antarctic icefish Chionodraco rastrospinosus. Journal of Biogeography 39, 1361–1372, https://doi.org/10.1111/j.1365-2699.2011.02682.x (2012).
Damerau, M., Matschiner, M., Salzburger, W. & Hanel, R. Comparative population genetics of seven notothenioid fish species reveals high levels of gene flow along ocean currents in the southern Scotia Arc, Antarctica. Polar Biology 35, 1073–1086, https://doi.org/10.1007/s00300-012-1155-x (2012).
Parker, R. W., Paige, K. N. & DeVries, A. L. Genetic variation among populations of the Antarctic toothfish: Evolutionary insights and implications for conservation. Polar Biology 25, 256–261, https://doi.org/10.1007/s00300-001-0333-z (2002).
Kuhn, K. L. & Gaffney, P. M. Population subdivision in the Antarctic toothfish (Dissostichus mawsoni) revealed by mitochondrial and nuclear single nucleotide polymorphisms (SNPs). Antarctic Science 20, 327–338, https://doi.org/10.1017/S0954102008000965 (2008).
Patarnello, T., Marcato, S., Zane, L., Varotto, V. & Bargelloni, L. Phylogeography of the Chionodraco genus (Perciformes, Channichthydae) in the Southern Ocean. Molecular Phylogenetics and Evolution 28, 420–429, https://doi.org/10.1016/S1055-7903(03)00124-6 (2003).
Papetti, C., Susana, E., Patarnello, T. & Zane, L. Spatial and temporal boundaries to gene flow between Chaenocephalus aceratus populations at South Orkney and South Shetlands. Marine Ecology Progress Series 376, 269–281, https://doi.org/10.3354/meps07831 (2009).
Damerau, M., Matschiner, M., Salzburger, W. & Hanel, R. Population divergences despite long pelagic larval stages: lessons from crocodile icefishes (Channichthyidae). Molecular ecology 23, 284–299, https://doi.org/10.1111/mec.12612 (2014).
Zane, L. et al. Demographic history and population structure of the Antarctic silverfish Pleuragramma antarcticum. Molecular ecology 15, 4499–4511, https://doi.org/10.1111/j.1365-294X.2006.03105.x (2006).
Johnson, M. S. & Black, R. Chaotic genetic patchiness in an intertidal limpet, Siphonaria sp. Marine Biology 70, 157–164, https://doi.org/10.1007/BF00397680 (1982).
Efremenko, V. N. Atlas of fish larvae of the Southern Ocean. Cybium 7, 1–74 (1983).
La Mesa, M., Riginella, E., Mazzoldi, C. & Ashford, J. Reproductive resilience of ice-dependent Antarctic silverfish in a rapidly changing system along the Western Antarctic Peninsula. Marine Ecology 36, 235–245, https://doi.org/10.1111/maec.12140 (2015).
Cuéllar-Pinzón, J., Presa, P., Hawkins, S. J. & Pita, A. Genetic markers in marine fisheries: Types, tasks and trends. Fisheries Research 173(Part 3), 194–205, https://doi.org/10.1016/j.fishres.2015.10.019 (2016).
Waples, R. S. Testing for Hardy–Weinberg Proportions: Have We Lost the Plot? Journal of Heredity 106, 1–19, https://doi.org/10.1093/jhered/esu062 (2015).
Emslie, S. D., Fraser, W., Smith, R. C. & Walker, W. Abandoned penguin colonies and environmental change in the Palmer Station area, Anvers Island, Antarctic Peninsula. Antarctic Science 10, 257–268 (1998).
Goethel, D., Quinn, T. & Cadrin, S. Incorporating Spatial Structure in Stock Assessment: Movement Modeling in Marine Fish Population Dynamics. Reviews in Fisheries Science 19, 119–136, https://doi.org/10.1016/j.fishres.2012.07.025 (2011).
Hawkins, S. J. et al. Fisheries stocks from an ecological perspective: Disentangling ecological connectivity from genetic interchange. Fisheries Research 179, 333–341, https://doi.org/10.1016/j.fishres.2016.01.015 (2016).
Fuentes-Pardo, A. P. & Ruzzante, D. E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Molecular ecology. https://doi.org/10.1111/mec.14264 (2017).
Moteki, M., Koubbi, P., Pruvost, P., Tavernier, E. & Hulley, P.-A. Spatial distribution of pelagic fish off Adélie and George V Land, East Antarctica in the austral summer 2008. Polar Science 5, 211–224, https://doi.org/10.1016/j.polar.2011.04.001 (2011).
Thompson, A. F., Heywood, K. J., Thorpe, S. E., Renner, A. H. H. & Trasviña, A. Surface Circulation at the Tip of the Antarctic Peninsula from Drifters. Journal of Physical Oceanography 39, 3–26, https://doi.org/10.1175/2008JPO3995.1 (2009).
Orsi, A. H., Nowlin, W. D. & Whitworth, T. On the circulation and stratification of the Weddell Gyre. Deep Sea Research Part I: Oceanographic Research Papers 40, 169–203, https://doi.org/10.1016/0967-0637(93)90060-G (1993).
Dickens, W. A. et al. A new bathymetric compilation for the South Orkney Islands region, Antarctic Peninsula (49°-39°W to 64°-59°S): Insights into the glacial development of the continental shelf. Geochemistry, Geophysics, Geosystems 15, 2494–2514, https://doi.org/10.1002/2014GC005323 (2014).
Ferguson, J. W. Population structure and connectivity of an important pelagic forage fish in the Antarctic ecosystem, Pleuragramma antarcticum, in relation to large scale circulation. MS Thesis, Old Dominion University, Norfolk, VA (2012).
Azaneu, M., Heywood, K. J., Queste, B. Y. & Thompson, A. F. Variability of the Antarctic Slope Current System in the Northwestern Weddell Sea. Journal of Physical Oceanography 47, 2977–2997, https://doi.org/10.1175/JPO-D-17-0030.1 (2017).
Ashford, J. et al. Does large-scale ocean circulation structure life history connectivity in Antarctic toothfish (Dissostichus mawsoni)? Canadian Journal of Fisheries and Aquatic Sciences 69, 1903–1919, https://doi.org/10.1139/f2012-111 (2012).
Young, E. F. et al. Oceanography and life history predict contrasting genetic population structure in two Antarctic fish species. Evolutionary Applications 8, 486–509, https://doi.org/10.1111/eva.12259 (2015).
Seehaus, T., Marinsek, S., Helm, V., Skvarca, P. & Braun, M. Changes in ice dynamics, elevation and mass discharge of Dinsmoor-Bombardier-Edgeworth glacier system, Antarctic Peninsula. Earth and Planetary Science Letters 427, 125–135, https://doi.org/10.1016/j.epsl.2015.06.047 (2015).
Wuite, J. et al. Evolution of surface velocities and ice discharge of Larsen B outlet glaciers from 1995 to 2013. The Cryosphere 9, 957–969, https://doi.org/10.5194/tc-9-957-2015 (2015).
Nicholls, K. W., Østerhus, S., Makinson, K., Gammelsrød, T. & Fahrbach, E. Ice-ocean processes over the continental shelf of the southern Weddell Sea, Antarctica: A review. Reviews of Geophysics 47, RG3003, https://doi.org/10.1029/2007RG000250 (2009).
Schröder, M. & Fahrbach, E. On the structure and the transport of the eastern Weddell Gyre. Deep-Sea Research Part II: Topical Studies in Oceanography 46, 501–527, https://doi.org/10.1016/S0967-0645(98)00112-X (1999).
Meijers, A. J. S. et al. Wind-driven export of Weddell Sea slope water. Journal of Geophysical Research: Oceans 121, 7530–7546, https://doi.org/10.1002/2016JC011757 (2016).
Youngs, M. K., Thompson, A. F., Flexas, M. M. & Heywood, K. J. Weddell Sea Export Pathways from Surface Drifters. Journal of Physical Oceanography 45, 1068–1085, https://doi.org/10.1175/JPO-D-14-0103.1 (2015).
Vacchi, M., Koubbi, P., Ghigliotti, L. & Pisano, E. In Adaptation and Evolution in Marine Environments, Volume 1: The Impacts of Global Change on Biodiversity (eds Guido di Prisco & Cinzia Verde) 51–73 (Springer Berlin Heidelberg, 2012).
Bortolotto, E., Bucklin, A., Mezzavilla, M., Zane, L. & Patarnello, T. Gone with the currents: Lack of genetic differentiation at the circum-continental scale in the Antarctic krill Euphausia superba. BMC Genetics 12, https://doi.org/10.1186/1471-2156-12-32 (2011).
Deagle, B. E., Faux, C., Kawaguchi, S., Meyer, B. & Jarman, S. N. Antarctic krill population genomics: apparent panmixia, but genome complexity and large population size muddy the water. Molecular ecology 24, 4943–4959, https://doi.org/10.1111/mec.13370 (2015).
Waples, R. S. & Gaggiotti, O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Molecular ecology 15, 1419–1439, https://doi.org/10.1111/j.1365-294X.2006.02890.x (2006).
Young, E. F. et al. Stepping stones to isolation: Impacts of a changing climate on the connectivity of fragmented fish populations. Evolutionary Applications. https://doi.org/10.1111/eva.12613 (2018).
Mintenbeck, K. & Torres, J. J. In The Antarctic Silverfish: a Keystone Species in a Changing Ecosystem (eds Marino Vacchi, Eva Pisano, & Laura Ghigliotti) 253-286 (Springer International Publishing, 2017).
CCAMLR. Convention on the Conservation of Antarctic Marine Living Resources. https://www.ccamlr.org/en/organisation/camlr-convention (1980).
Patwary, M. U., Kenchington, E. L., Bird, C. J. & Zouros, E. The use of random amplified polymorphic DNA markers in genetic studies of the sea scallop Placopecten magellanicus (Gmelin, 1791). Journal of Shellfish Research 13, 547–553 (1994).
Molecular Ecology Resources Primer Development Consortium et al. Permanent genetic resources added to Molecular Ecology Resources Database 1 October 2010-30 November 2010. Molecular ecology resources 11, 418–421, https://doi.org/10.1111/j.1755-0998.2010.02970.x (2011).
Agostini, C. et al. Permanent genetic resources added to molecular ecology resources database 1 April 2013-31 May 2013. Molecular ecology resources 13, 966–968, https://doi.org/10.1111/1755-0998.12140 (2013).
Amos, W. et al. Automated binning of microsatellite alleles: Problems and solutions. Molecular Ecology Notes 7, 10–14, https://doi.org/10.1111/j.1471-8286.2006.01560.x (2007).
Coombs, J. A., Letcher, B. H. & Nislow, K. H. create: a software to create input files from diploid genotypic data for 52 genetic software programs. Molecular ecology resources 8, 578–580, https://doi.org/10.1111/j.1471-8286.2007.02036.x (2008).
Rousset, F. GENEPOP'007: A complete re-implementation of the GENEPOP software for Windows and Linux. Molecular ecology resources 8, 103–106, https://doi.org/10.1111/j.1471-8286.2007.01931.x (2008).
Keenan, K., McGinnity, P., Cross, T. F., Crozier, W. W. & Prodöhl, P. A. DiveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods in Ecology and Evolution 4, 782–788, https://doi.org/10.1111/2041-210X.12067 (2013).
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M. & Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes 4, 535–538, https://doi.org/10.1111/j.1471-8286.2004.00684.x (2004).
Carvajal-Rodriguez, A. & de Uña-Alvarez, J. Assessing Significance in High-Throughput Experiments by Sequential Goodness of Fit and q-Value Estimation. PloS one 6, e24700, https://doi.org/10.1371/journal.pone.0024700 (2011).
Antao, T., Lopes, A., Lopes, R. J., Beja-Pereira, A. & Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9, 323, https://doi.org/10.1186/1471-2105-9-323 (2008).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular ecology resources 10, 564–567, https://doi.org/10.1111/j.1755-0998.2010.02847.x (2010).
Excoffier, L., Hofer, T. & Foll, M. Detecting loci under selection in a hierarchically structured population. Heredity 103, 285–298, https://doi.org/10.1038/hdy.2009.74 (2009).
R: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria, 2016).
Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405, https://doi.org/10.1093/bioinformatics/btn129 (2008).
Peakall, R. O. D. & Smouse, P. E. GenALEx 6: Genetic Analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6, 288–295, https://doi.org/10.1111/j.1471-8286.2005.01155.x (2006).
Peakall, R. O. D. & Smouse, P. E. GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539, https://doi.org/10.1093/bioinformatics/bts460 (2012).
Duchesne, P. & Turgeon, J. FLOCK: a method for quick mapping of admixture without source samples. Molecular ecology resources 9, 1333–1344, https://doi.org/10.1111/j.1755-0998.2009.02571.x (2009).
Duchesne, P. & Turgeon, J. FLOCK provides reliable solutions to the “number of populations” problem. Journal of Heredity 103, 734–743, https://doi.org/10.1093/jhered/ess038 (2012).
Ryman, N. & Palm, S. POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Molecular Ecology Notes 6, 600–602, https://doi.org/10.1111/j.1471-8286.2006.01378.x (2006).
Weir, B. S. & Cockerham, C. C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 38, 1358–1370, https://doi.org/10.2307/2408641 (1984).
Sangrà, P. et al. The Bransfield current system. Deep-Sea Research Part I: Oceanographic Research Papers 58, 390–402, https://doi.org/10.1016/j.dsr.2011.01.011 (2011).
Matsuoka, K., Skoglund, A. & Roth, G. Quantarctica [Data set] (Norwegian Polar Institute, 2018).
QGIS Development Team. QGIS Geographic Information System. Open Source Geospatial Foundation Project, https://qgis.org/en/site/index.html (2018).
Donnelly, J. & Torres, J. J. Pelagic fishes in the Marguerite Bay region of the West Antarctic Peninsula continental shelf. Deep-Sea Research Part II: Topical Studies in Oceanography 55, 523–539, https://doi.org/10.1016/j.dsr2.2007.11.015 (2008).
Ferguson, J. et al. Connectivity and population structure in Pleuragramma antarcticum. CCAMLR Scientific Papers Working Group on Fish Stock Assessment WG-FSA-11/19 (2011).
Ruck, K. E., Steinberg, D. K. & Canuel, E. A. Regional differences in quality of krill and fish as prey along the Western Antarctic Peninsula. Marine Ecology Progress Series 509, 39–55, https://doi.org/10.3354/meps10868 (2014).
Acknowledgements
We would like to thank Nils Koschnick (AWI) and Emilio Riginella (University of Padua) for collecting the 2012 AP and 2014 WS samples during ‘Polarstern’ ANT-XXVIII/4 and ANT-XXIX/9 cruises. We thank Luca Schiavon (University of Padua) for helping to run the POWSIM analysis, and Mathieu Casado (AWI, Potsdam) for assistance in the creation of the map figure. We are indebted to two anonymous reviewers who greatly improved the quality of the final version of the manuscript. This work was supported by the National Program for Antarctic Research (PNRA16_307) to L.Z.; J.A.C. is a PhD student in Evolution, Ecology, and Conservation at the University of Padua, with funding from a Cariparo Fellowship for foreign students and additional support from an Antarctic Science International (ASI) Bursary, a Scientific Committee for Antarctic Research (SCAR) Fellowship, and an Erasmus + Student Traineeship. C.P. acknowledges financial support from the University of Padua (BIRD164793/16) and from the European Marie Curie project “Polarexpress” Grant No. 622320. Funding for J.R.A. was provided by the National Science Foundation (Grant No. 0741348).
Author information
Authors and Affiliations
Contributions
C.P., M.W. and R.K. collected the samples; L.Z., J.R.A. and J.A.C. conceived the ideas; J.A.C. collected the data, J.A.C, C.P., and L.Z. analyzed the data; C.P., J.R.A. and L.Z. contributed to interpretation of results; J.A.C., J.R.A., C.P. and L.Z. wrote the manuscript. All authors contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Caccavo, J.A., Papetti, C., Wetjen, M. et al. Along-shelf connectivity and circumpolar gene flow in Antarctic silverfish (Pleuragramma antarctica). Sci Rep 8, 17856 (2018). https://doi.org/10.1038/s41598-018-36030-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36030-x
This article is cited by
-
Spatial structuring in early life stage fish diversity in the Scotia Sea region of the Southern Ocean
Polar Biology (2024)
-
Climate drives long-term change in Antarctic Silverfish along the western Antarctic Peninsula
Communications Biology (2022)
-
Genetic pattern and demographic history of cutlassfish (Trichiurus nanhaiensis) in South China Sea by the influence of Pleistocene climatic oscillations
Scientific Reports (2022)
-
Growth and early life stage of Antarctic silverfish (Pleuragramma antarctica) in the Amundsen Sea of the Southern Ocean: evidence for a potential new spawning/nursery ground
Polar Biology (2022)
-
Pellet analysis evidences flexibility in the diet of Brown Skua (Stercorarius antarcticus) during breeding at Esperanza/Hope Bay (Antarctic Peninsula)
Polar Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
