
Abstract
Severe Malarial Anemia (SMA), a life-threatening childhood Plasmodium falciparum malaria syndrome requiring urgent blood transfusion, exhibits inflammatory and hemolytic pathology. Differentiating between hypo-haptoglobinemia due to hemolysis or that of genetic origin is key to understand SMA pathogenesis. We hypothesized that while malaria-induced hypo-haptoglobinemia should reverse at recovery, that of genetic etiology should not. We carried-out a case-control study of children living under hyper-endemic holoendemic malaria burden in the sub-Saharan metropolis of Ibadan, Nigeria. We show that hypo-haptoglobinemia is a risk factor for childhood SMA and not solely due to intravascular hemolysis from underlying schizogony. In children presenting with SMA, hypo-haptoglobinemia remains through convalescence to recovery suggesting a genetic cause. We identified a haptoglobin gene variant, rs12162087 (g.-1203G > A, frequency = 0.67), to be associated with plasma haptoglobin levels (p = 8.5 × 10−6). The Homo-Var:(AA) is associated with high plasma haptoglobin while the reference Homo-Ref:(GG) is associated with hypo-haptoglobinemia (p = 2.3 × 10−6). The variant is associated with SMA, with the most support for a risk effect for Homo-Ref genotype. Our insights on regulatory haptoglobin genotypes and hypo-haptoglobinemia suggest that haptoglobin screening could be part of risk-assessment algorithms to prevent rapid disease progression towards SMA in regions with no-access to urgent blood transfusion where SMA accounts for high childhood mortality rates.
Similar content being viewed by others

Introduction
The annual global incidence of clinical malaria is estimated at about 216 million cases leading to approximately 445,000 deaths in 20161, a slight increase in morbidity over what was previously reported1,2. The mortality rate of malaria was estimated to be 90% in Africa, and 80% of these deaths occur in sub-Saharan Africa and mostly in the under five years–of-age group due to severe, and often-overlapping syndromes such as Severe Malarial Anemia (SMA) and Cerebral Malaria (CM)1,2. A quarter of the global cases and a third of malaria-attributable childhood deaths occur in Nigeria, the most populous country of Africa3,4,5.
Malaria is characterized by increased inflammation and intravascular hemolysis due to schizogony leading to the release of free Hemoglobin (fHb) into the plasma6,7,8. Heme, the molecule bearing the iron moiety of fHb, can be oxidized by nitric oxide (NO) leading to oxidative stress-mediated damage through mediators such as lipid and hydrogen peroxides9,10. To avoid this, fHb is scavenged by plasma Haptoglobin (HP)10,11 forming a complex, thereby preventing fHb to be oxidized and potentially preventing further heme-mediated nitric oxide damage, renal excretion of iron and further oxidative damage10,11,12.
The HP gene is comprised of two co-dominant alleles, HP1 and HP2, located on the long arm of chromosome 1612,13,14,15. The HP1 allele has five exons and the HP2 allele has seven exons (Supplementary Fig. 1). The HP2 allele contains a 1.7 Kb intragenic duplication. HP is synthesized mainly by hepatocytes in response to increased levels of interleukins such as IL-6 and IL-1 and Tumor Necrosis Factor (TNF-α) during an acute inflammatory response11,16. The HP-fHb complex, formed by the binding of HP to fHb, is removed from circulation by CD163 receptors expressed on the surface of macrophages followed by internalization, where fHb is degraded and heme catabolized by heme oxygenase-117.
A high level of fHb during severe hemolysis has been reported to be responsible for low HP plasma levels18,19 and therefore the circulatory level of HP has been used as a surrogate marker of intravascular hemolysis18,20,21,22,23. Similarly, Lactate Dehydrogenase (LDH) is known to be abundant in erythrocytes and its plasma level is elevated in the event of erythrocyte lysis. An elevated plasma level of LDH is also a surrogate marker of hemolysis in vascular pathophysiology24,25,26,27.
In some individuals, a complete lack of HP (ahaptoglobinemia) or low circulating HP (hypo-haptoglobinemia) can be observed28. Circulatory HP level shows ethnic and geographic variation with a high prevalence of hypo-haptoglobinemia in Africans compared with Europeans. Recent studies report that the average frequency of hypo-haptoglobinemia in Africa might be as high as 40%18,29 although with different and unknown etiologies18,20,30,31,32.
Studies on hypo-haptoglobinemia have provided conflicting views regarding its etiology in childhood malaria. On one hand, the low level of plasma HP found in malaria infections has been proposed to have a genetic basis18,31,33,34,35, while on the other hand the low level has been attributed to the malaria infection causing intravascular hemolysis18,28,36 and other immune pathological processes37. HP levels have been reported to recover in 75% in a study population following anti-malarial treatment38. While malaria infection induced hypo-haptoglobinemia should be reversed with anti-malarial treatment18,20,30,31,32, that of genetic etiology should not. Differentiating between these two etiological scenarios should provide insights into the role of HP in the establishment of mild or severe malaria syndromes.
We hypothesized that if low HP levels are exclusively a consequence of malaria infection they would return to normal on patients’ recovery. To test this hypothesis, we carried out a prospective case-control study of children (with well-defined follow-up through convalescence to recovery) living in the sub-Saharan 3-million inhabitants densely-populated urban metropolis of Ibadan, Nigeria, that is under hyper-endemic and holoendemic malaria burden. We assayed for levels of plasma HP and the markers of intravascular hemolysis fHb and LDH from admission through convalescence to recovery. Furthermore, we performed high throughput amplicon-based sequencing of the HP genes to identify genotypes associated with both circulatory HP levels and the malarial disease syndromes studied.
Here we show, by longitudinal follow-up of malaria-positive pediatric cohorts, that hypo-haptoglobinemia is a risk factor for acute onset childhood SMA, rather than the unique consequence of this syndrome, and it is likely of genetic etiology. We report for the first time a variant in the distal upstream region of the HP gene rs12162087 to be associated with circulatory HP level. We provide evidence that the variant is also associated with SMA.
Materials and Methods
Ethical Statement
The internationally recognized ethical committee at the Institute for Advanced Medical Research and Training (IMRAT) of the College of Medicine, University of Ibadan (COMUI) approved this research on the platform of the Childhood Malaria Research Group (CMRG) within the academic Department of Paediatrics, University College Hospital (UCH), as well as at school and Primary Care centers throughout the city of Ibadan with permit number: UI/EC/10/0130.
Ethics, Consent and Permissions
Parents and/or guardians of study participants gave informed written consent in accordance with the World Medical Association ethical principles for research involving human subjects.
Study Site
All study participants were recruited under the auspices of the CMRG at the 800-bed tertiary hospital, UCH in the city of Ibadan, Nigeria, in west sub-Saharan Africa. UCH Ibadan academic prestige, quality of care, costs and excellent accessibility from any point in the large metropolis makes it the preferred choice for all socio-economic sectors of the population. UCH Ibadan provides tertiary, secondary and it is associated with primary care centers across the city.
Ibadan is the second most densely populated urban metropolis in Nigeria with nearly 3 Million inhabitants. The city has a lengthy 8-month rainy season, with the average of 10 rainy days per month between May and October. Malaria transmission and severe disease occur throughout the year. Although severe malaria syndromes are predominant in children under 5 years-of-age, there is a significant large burden of severe disease in children up to 16 years-of-age39.
Study Design and Case Definitions
As part of a larger case-control study of severe malaria4,5,39,40, we recruited children with median age of 26–48 months-of-age between 2010 and 2013. The defined clinical cases for the malaria-positive groups include Uncomplicated Malaria (UM), Severe Malaria Anemia (SMA) and Cerebral Malaria (CM) according to WHO criteria for diagnosis of P. falciparum complications41,42. UM cases were defined as patients with fever and P. falciparum parasitemia who did not require hospital admission. The SMA group was defined as conscious children with packed cell volume (PCV) <16%, or hemoglobin concentration <5 g/dL, with asexual forms of P. falciparum in circulation and no other overt etiology. All SMA patients were transfused within two hours of admission and confirmation of malaria diagnosis. However, samples were collected prior to the blood transfusion. CM cases were defined as children with unarousable coma persisting for more than an hour with generalized convulsion in the presence of asexual forms of P. falciparum. A Blantyre coma score <2 was used to define coma status. Children presenting with both CM and SMA, belonging to the CM-SMA clinical group, did not form part of this study.
The UM group was recruited as part of daily routine malaria parasite screening at our children outpatient clinics at UCH Ibadan. All severe CM and SMA cases were recruited on admission from our emergency department at UCH Ibadan, the Otunba Tunwase Children’s Emergency Ward (OTCHEW). The Community Control (CC) group was comprised of malaria-negative healthy children.
All children diagnosed with malaria were treated and followed through convalescence to recovery following Nigerian antimalarial guidelines which are based on the WHO guidelines43. Uncomplicated malaria is treated with a 3-day oral course of Artemisinin-based Combination Therapy (ACT). On the other hand, severe malaria is treated with intravenous artesunate for at least 24 hours and until they can tolerate oral therapy after which treatment is then completed with three days of oral ACT. Overall, successful antimalarial treatment is completed in less than one week. At our clinics at UCH Ibadan, an urban academic tertiary-care center, guidelines are strictly followed and monitored weekly.
All cohorts excluded children presenting with: moderate to severe malnutrition and/or iron deficiency; hypochromic microcytic anemia; spherocytosis; sickle cell anemia (SS); haemoglobin (SC) disease; known beta-thalassemia status. Only the alpha-thalassemia –α3.7 deletion, which is of little or no clinical significance, is observed in Nigerians44. Beta-thalassemia trait is known to have a very low gene frequency of 0.008 in this population45 and recent authors report inconclusive higher prevalence within healthy individuals46.
The study was prospectively performed on subjects recruited from 2010 to 2012 (Discovery Cohort) and 2013 (Validation Cohort). The entire cohort is predominantly of the same Yoruba ethnicity and within the same population geopolitical zone.
All clinical malaria-positive groups received anti-malaria treatment. A child was considered recovered if, and only if, the PCV and reticulocyte counts returned to normal ranges; there were no clinical nor laboratory signs of hemolysis with fHb and LDH within normal range and; with a negative malaria Giemsa blood-film at day 28–35, day 28 (PD28) on the average. All these are well-defined clinical and physiological signs that a child has recovered from the acute malaria episode and are consistent with previous studies showing that Hb, reticulocyte counts and haptoglobin levels return to normal 2–3 weeks after commencement of antimalarial treatment31,47,48,49,50. As shown by these studies and our data, the PD28 recovery time-point is sufficient for the HP level and other clinical parameters to normalize as it happens in the CM and UM clinical groups.
Clinical Data and Sample Collection
Clinical data were collected using a malaria-tailored questionnaire designed by the CMRG4,5,39,40 at University College Hospital Ibadan. The experiments were conducted using samples from 3 well-defined cohorts namely the discovery, the validation and the High-throughput Sequencing (HTS) cohorts.
For the discovery cohort, plasma samples were collected at acute onset of the disease termed as PD0 (Plasma at Day zero) as well as from parasite-negative healthy community controls; CC.
For the validation cohort plasma samples were collected at acute onset of the disease at day zero (PD0) during convalescence at day 7 (PD7) and day 14 (PD14); and at recovery at day 28–32 (PD28).
The HTS cohort is drawn from both the discovery and validation cohorts, including malaria-negative healthy controls, whose HP genes were sequenced.
All plasma samples from malaria-positive clinical groups within all these cohorts were collected prior to onset of anti-malarial drug treatment or blood transfusion in the case of SMA.
A 2.5 ml blood sample was obtained from each participant. A very short needle was used to collect the blood into a tube, which was firmly screwed to the syringe. This was to avoid dispensing blood into EDTA tubes through the syringe, which might have caused hemolysis of some red blood cells. Blood samples in an EDTA collection tube were kept on ice and plasma for each subject was harvested following centrifugation. Plasma for this study was harvested by centrifugation (1000 g, 10 minutes), aliquoted and stored at −80 °C within 2 hours of collection.
PCV was measured using micro-hematocrit capillary tubes centrifuged at 12,000 g for 5 min. The cell volume was calculated as a percentage of the whole tube volume.
Malaria Diagnosis
Malaria parasites (MPs) were detected and counted by microscopy following Giemsa staining of thick and thin blood films. The criterion for declaring a participant to be malaria parasite-free was no detectable parasites in 100 high-power (1000x) fields in both thick and thin films. We validated the diagnosis outcome by randomly selecting one in ten thick blood films for independent review by local experienced senior microscopists who were not part of the study research team4,5,39,40.
Quantitative Determination of Plasma Haptoglobin
HP levels in plasma were quantified using an HP sandwich enzyme-linked immunosorbent assay (sELISA) kit (Genway). Plasma samples diluted 1 in 20 in phosphate buffered saline (PBS) as stock were further diluted to 1/50, 1/100, 1/250, 1/5,000, 1/10,000, 1/20,000 and 1/40,000. The low to high dilutions were to ensure detection of HP that could be present at low to high concentration, respectively. Standards were prepared according to the manufacturer’s instructions but diluted in PBS. Anti-human HP antibody (Genway) was diluted in coating buffer (0.05M sodium carbonate-bicarbonate, pH 9.6) to 5 µg/mL. Plates were incubated with this antibody for 60 minutes at room temperature. Thereafter, a blocking step was performed with 400 μL of blocking buffer (50 mM Tris-HCl, 140 mM NaCl, 1% BSA, pH 8.0) followed by a washing step in washing buffer (0.05% Tween 20 in PBS, pH 7.4). Diluted plasma samples were added to the plates and incubated for 60 minutes, followed by another washing step. Bound HP was detected with a secondary antibody coupled with horseradish peroxidase, which was diluted 1 in 2,000 in 50 mM Tris-HCl containing 140 mM NaCl, 1% BSA (wt/vol), and 0.05% Tween 20 (vol/vol), pH 8.0. The substrate, OPD was prepared at 1 mg/mL by dissolving one OPD tablet (Acros Organics) in 20 mL citrate-phosphate buffer (50 mM) to which 20 µL of 30% H2O2 was added. A 2M tetraoxosulfate-VI-acid solution was used as the stop solution. Plates were read within 5 minutes at 490 nm using a FLUorStar-Omega (BMG-Labtech) ELISA plate reader. HP concentrations were computed using the five to four parameters logistic (5/4-PL) curve-fit (MasterPlex ReaderFit v2.0, MiraiBio Group, Hitachi Solutions America, Ltd.).
Determination of HP genotypes and protein phenotypes
We performed genotyping and phenotyping of HP in individual subjects and ascertained whether the circulatory level of HP varies with the HP1-1, HP2-1 and HP2-2 phenotypes.
For HP gene isoform genotyping, a forward primer, Sa5 (5′ GAGGGGAGCTTGCCTTTCCATTG 3′) and a reverse primer, Sa6 (5′ GAGATTTTTGAGCCCTGGCTGGTG 3′) were designed to amplify both HP1 and HP2 alleles (Supplementary Fig. 1) to detect the 1.7 kb and 3.4 kb fragments diagnostic of HP1-1 and HP2-2 genotypes, respectively (Supplementary Fig. 2a). The forward, LS1 (5′ TGAGCACTTAAGAGAGCAGGC 3′) and reverse, LS2 (5′ CTTCACATTCAGGAAGTTTATCTCC 3′) primers were designed for specific genotyping of the HP2 allele, taking advantage of the additional sequence in the HP2 allele not found in HP1. These specific primers, LS1/LS2, for the HP2 allele were necessary because it was difficult to obtain a good amplification of the 3.4 kb fragment for the HP2 allele in HP2-1 subjects in which the 1.7 kb fragment was also amplified. Hence, LS1/LS2 primers were designed to amplify a 1.7 kb fragment specific for the HP2 allele and not the HP1 allele (Supplementary Fig. 1 and Fig. 2b). Therefore, while the Sa5/Sa6 primer pair could not amplify efficiently the HP2 allele from HP2-1 DNA for unknown reasons, it could detect the HP1 allele, and the HP2 allele in the HP2-1 genotype was detected by LS1/LS2 primers. The PCR mix consisted of Kapa HiFi (Kapa Biosystem) DNA polymerase (0.5 μL); 10 mM Kappa dNTP mix (0.75 μL); Kapa HiFi fidelity buffer (0.5 μL); forward and reverse primers (10 μM each); template DNA (50 ng) and distilled water to a total reaction volume of 25 μL. The PCR for LS1 and LS2 was performed at 95 °C for 3 minutes followed by 35 cycles of 98 °C for 20 seconds, 61.5 °C for 15 seconds and 72 °C for 2 minutes with a final extension step at 72 °C for 2 minutes. The PCR conditions for the Sa5 and Sa6 primers were similar to that for LS1 and LS2 but with an additional 2 mM of MgCl2 and at an annealing temperature of 69.2 °C or 71.5 °C. Amplified products were resolved by agarose gel electrophoresis and detected by fluorescence following staining with ethidium bromide. Subjects with only the 1.7 kb band produced with primers Sa5/Sa6 are designated as having the HP1-1 genotype, and subjects with a 1.7 kb band from both the Sa5/Sa6 and LS1/LS2 primers sets are designated as having the HP2-1 genotype while those with a 3.4 kb band from the Sa5/Sa6 and a 1.7 kb band from the LS1/LS2 primer pairs are designated as having the HP2-2 genotype (Supplementary Fig. 2a,b).
Protein HP phenotyping was carried out by western blotting and immunoprobing. Protein blotting to PVDF or nitrocellulose membrane following SDS-PAGE was performed in a Bio-Rad trans-blot cell using electro-blotting buffer (39 mM glycine, 48 mM Tris-base, 0.0375% SDS, 20% methanol and distilled water) and at a constant voltage of 30 V for approximately 15–16 hours. Polyclonal rabbit anti-human HP (Dako-Cytomation) diluted 1 in 10,000 in blocking solution (PBS containing 0.1% Tween 20 and 2–3% dried milk) was used as the primary antibody at room temperature for 2 hours or at 4 °C overnight. Alexa Fluor 488-labelled goat anti-rabbit diluted 1 in 5000 in blocking solution or HRP-conjugated goat anti-rabbit IgG diluted 1 in 10,000 in blocking solution were used as secondary antibody. The blots were then placed in blocking solution for one hour and washed in PBS-Tween. The protein bands were visualized using the Pharos FX Plus (Bio-Rad) molecular imager or chemi-luminescent detection (Supplementary Fig. 3). The western blot assay displayed the different HP phenotypes. The ~40 kDa β-chain was present in all the samples (Supplementary Fig. 3). The presence of both the α1 and α2 chains is diagnostic of the HP2-1 phenotype while the presence of only α2 or α1 indicates the HP2-2 and HP1-1 phenotypes, respectively (Supplementary Fig. 3).
Quantitative Determination of plasma Lactate Dehydrogenase (LDH)
The level of LDH was measured by sELISA using a specific antibody (Cloud-clone Corp.) as follows. Plasma samples used for the assay were diluted 1:500 in PBS and the absorbance measured at 450 nm. LDH concentrations were calculated from the standard curve using the best-fit parameter in the MasterPlex ReaderFit v2.0 software (MiraiBio Group, Hitachi Solutions America, Ltd.).
Quantitative Determination of plasma free-Hemoglobin (fHb)
The level of fHb was quantified by sELISA using a human fHb-specific antibody (Mybiosource) assay MBS700075 and the manufacturer’s instructions. Plasma samples were diluted 1:800 in PBS, the absorbance measured at 450 nm and fHb concentrations calculated from the standard curve using the 5-PL function in the MasterPlex ReaderFit v2.0 software (MiraiBio Group, Hitachi Solutions America, Ltd.).
High Throughput HP gene sequencing
The DNA used for the HP gene high throughput sequencing was prepared from packed blood cells with a QIamp blood kit as described previously51. Primers specific to different locations on the HP gene were designed to produce overlapping amplicons for the entire HP gene (Supplementary Fig. 1) in a series of monoplex PCR reactions. The primer pairs were designed to cover extensively the promoter region of the HP gene; extending from about 3.4 kb upstream of the translation start site to beyond exon 7 of the HP2 allele and exon 5 of the HP1 allele (Supplementary Fig. 1). This covered the region 16:72083574-72095400 in the human genome database version GRCh37 or hg19. The HP2 allele was used as the reference gene for primer design. However, the primers also recognize the corresponding region of the HP1 allele, producing shorter PCR products (Supplementary Fig. 1). The KAPA HiFi HotStart ReadyMix PCR kit (KapaBiosystems) and 50–100 ng of template DNA were used for the PCR mix.
The lyophilized primers purchased from Sigma-Aldrich were reconstituted in distilled water (dH2O) to a concentration of 100 μM, and then further diluted 1:10 to a working concentration of 10 μM. The final concentration of each primer in the PCR mix was about 0.3 μM in a 25 μL reaction volume.
The quality and concentration of each amplicon were analyzed by Glomax and Bioanalyzer prior to library preparation. The library was prepared and sequenced following the Illumina Nextera XT and the Illumina MiSeq machine user guides. A Phred quality score; >Q30 and paired-end sequencing was used. The FastQ output file obtained from the machine was used for further data analysis.
Statistical Analyses
We carried-out Principal Components Analysis (PCA) using MatLab to visualize a projection that best represents the structure of the multivariate data from all of the clinical groups of the Discovery Cohort (PD0). The multivariate data has five dimensions including the following parameters at admission: age; PCV; reticulocyte count; plasma haptoglobin and; plasma fHb). Clinical group labels and parasite-density values were excluded from the PCA analysis as it would strongly bias the results towards a malaria-positive vs. malaria-negative projection. The two orthogonal components that captured most of the variance were then plotted on two-dimensional space and clinical groups where imposed thereafter by labelling as follows: UM (blue); SMA (red); CM (grey) and; CC (yellow) (Fig. 1).

Principal Component Analysis (PCA) projection of multivariate clinical and laboratory plasma parameters of Discovery Cohort study groups at PD0. The first two principal components (PCA1 and PCA 2) were plotted in a two-dimensional scatter-plot after which the clinical group labels CC (yellow), UM (blue), CM (grey) and SMA (red) were color imposed thereafter. The projection shows the internal structure of the multivariate data while preserving its orthogonal variance. (UM) = Uncomplicated Malaria; (SMA) = Severe Malarial Anemia; (CM) = Cerebral Malaria; (CC) = healthy malaria-negative Community Controls; PD0 = at admission prior to drug treatment and/or blood transfusion.
Unpaired non-parametric Mann-Whitney tests were used to compare clinical demographic data between two disease groups (Table 1). Non-parametric Kruskal-Wallis tests were performed to determine differences between disease groups using Dunn’s multiple comparison correction (Figs 2–4). Paired non-parametric Wilcoxon tests were used to compare paired data within a disease group (Fig. 3). Non-parametric Spearman tests were used to correlate the levels of fHb and HP with parasite density in malaria-positive clinical groups (Supplementary Table 1). Linear regression was used to determine independency of HP levels with age in the malaria-negative CC group (Supplementary Fig. 4).

Analysis of plasma haptoglobin and plasma free-hemoglobin of discovery cohort study groups at PD0. (a) Box plot of Log [plasma haptoglobin (ng/ml)] at admission (PD0) in the different study groups of the discovery cohort at PD0. SMA < CC (p < 0.0001); SMA < CM (p < 0.001); SMA < UM (p < 0.0001). (b) Box plot of Log [plasma free-hemoglobin (ng/ml)] at admission (PD0) in the different study groups of the discovery cohort at PD0. (p > 0.05 for all comparisons). The box plots show the IQR (box); the median (box line); and the 10–90 percentile (whiskers). Non-parametric Kruskal-Wallis with Dunn’s multiple comparison correction. (UM) = Uncomplicated Malaria; (SMA) = Severe Malarial Anemia; (CM) = Cerebral Malaria; (CC) = healthy malaria-negative Community Controls; PD0 = at admission prior to drug treatment and/or blood transfusion. IQR = Interquartile Q3-Q1 Range. For discovery cohort clinical groups details see Table 1.

Analysis of plasma HP, fHb and LDH of validation cohort study groups from admission (PD0) through convalescence (PD7-PD14) to recovery (PD28). (a) Log [plasma haptoglobin (ng/ml)] from admission (PD0) through convalescence (PD7-PD14) to recovery (PD28). Asterisks show: HP SMA@PD0 < HP CC (p < 0.001); HP SMA@PD7 < HP CC (p < 0.05); HP SMA@PD14 < Plasma HP CC (p < 0.05); HP SMA@PD28 < HP CC (p < 0.001). Ampersand (&) shows: HP SMA@PD0 < HP SMA@PD28 (p < 0.01) (b) Log [plasma free-hemoglobin (ng/ml)] from admission (PD0) through convalescence (PD7-PD14) to recovery (PD28). c) Log [plasma LDH (ng/ml)] from admission (PD0) through convalescence (PD7-PD14) to recovery (PD28). Plasma LDH in all malaria-positive groups at PD0 > CC (p < 0.001). Plots show mean (circles) and standard error of the mean (whiskers). Asterisk: *(p < 0.05); **(p < 0.01); ***(p < 0.001). Non-parametric Kruskal-Wallis with Dunn’s multiple comparison correction (between group comparisons). Paired non-parametric Wilcoxon tests were used to compare paired data within a disease group. For validation cohort clinical group details see Table 1. (UM) = Uncomplicated Malaria (blue line); (SMA) = Severe Malarial Anemia (red line); (CM) = Cerebral Malaria (black line); (CC) = healthy malaria-negative Community Controls (yellow dotted-line); HP = haptoglobin; fHB = free hemoglobin; LDH = lactate dehydrogenase; PD0 = at admission prior to drug treatment and/or blood transfusion; PD7 = Plasma sample collected at the 7th day after PD0; PD14 = Plasma sample collected at 14th day after PD0; PD28 = Plasma level collected on 28th day after PD0 (recovery).

Haptoglobin isoform frequencies and plasma HP of HTS Cohort study groups. (a) Frequency of haptoglobin isoforms HP1-1 (dark-grey square); HP2-1 (grey square) and HP2-2 (white square) for the clinical groups. Sizes of the square are drawn to scale and depicts the frequency of the isoform written with the square. HP1-1, HP2-1 and HP2-2 are HP phenotypes. *The HP1-1 isoform is associated with CM (p = 0.038) with an estimated OR of 2.1 (1.04–4.4). (b) Bar plot of Log [plasma HP (ng/ml)] per isoforms HP1-1 (black bar); HP2-1 (grey bar) and HP2-2 (white bar) for the clinical groups. Bar plot shows mean and standard error of the mean (whiskers). Non-parametric Kruskal-Wallis with Dunn’s multiple comparison correction; ns = not significant (p > 0.05). (UM) = Uncomplicated Malaria; (SMA) = Severe Malarial Anemia; (CM) = Cerebral Malaria; (CC) = healthy malaria-negative Community Controls. For HTS Cohort clinical group details see Table 1.
For the genomic data and the identification of variants, FASTQ output files were aligned to the reference genome using Burrows-Wheeler Aligner (BWA). Samtools was used on the BAM file to produce the output VCF file, containing the variants. This was used for downstream computation such as the measure of association (Odds Ratio; OR), 95% CI and genotype frequency. Minimum Allele Frequency greater or equals 10% (MAF ≥ 10%) were used to select variants (Fig. 5). We used Mendelian randomization with the assumption that the inheritance of SNP is essentially random, to determine the effect of the SNP on HP level and disease. The CC subjects were genotyped for rs12162087 polymorphism and the frequency of each genotype (AA, GA, GG) was determined together with the HP levels (Fig. 5). Each participant genotype for the observed SNP were measured and the frequency determined relative to the CC. chi-Square tests were used to determine whether the genotype distributions in the control group were conformed to the Hardy-Weinberg equilibrium (Fig. 5) GenePop software (web version 4.2 option 1). The genotypes effect on HP levels were estimated by ANOVA and linear regression analysis while univariate analyses of HP variants (binary logistic regression) were carried out with SPSS version 10.1 to examine association between the genotype and SMA (Fig. 5).

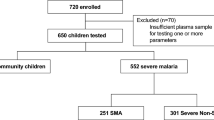
Flow chart showing the variant filtering strategies and association studies used in the Ibadan HTS-Cohort. High throughput sequencing of Haptoglobin gene has been performed on 401 samples. We used an allele frequency filtering to select the variants with a MAF ≥ 10%. ANOVA and linear regression analysis were then used to evaluate their association with the haptoglobin level in CC group. One variant rs12162087 was selected upon this association. GenePop software was used to determine the Hardy Weinberg equilibrium in control group. We performed a binary logistic regression to test the association of this variant and SMA. The allele and the genotype frequencies for rs12162087 are indicated for each disease group. (UM) = Uncomplicated Malaria; (SMA) = Severe Malarial Anemia; (CM) = Cerebral Malaria; (CC) = healthy malaria-negative Community Controls; (N) = number of subjects; MP(+ve) = malaria parasite positive; MP(−ve) = malaria parasite negative, MAF = Minor allele frequency. For HTS Cohort clinical group details see Table 1 and details for variants see Table 2.
Results
Study Participants
The discovery cohort (Table 1) consists of 545 subjects in total with 371 malaria-positive patients with either UM, CM or SMA whose plasma samples were collected at acute onset of the disease (PD0) and 174 malaria-negative healthy community controls (CC).
The validation cohort (Table 1) consists of 147 malaria-positive patients with either UM, CM or SMA and 19 healthy malaria-negative community controls. This cohort was independently recruited to validate the data obtained at PD0 in the discovery cohort and more importantly to observe trends within the measured parameters through convalescence to recovery.
The HTS cohort (Table 1) consists of a total 401 subjects with 300 malaria-positive patients and 101 healthy malaria-negative community controls drawn from both the discovery and validation cohorts and whose HP genes were sequenced to determine whether an association between genetic variation, clinical syndromes and observed plasma HP levels exist.
As expected, PCV values were lowest in the SMA clinical group. In SMA cases, low PCV at recruitment (PD0) was attributed to the anemia as the consequence of the malaria infection. In all clinical malaria-positive groups receiving anti-malaria treatment the PCV returned to normal at recovery (PD28) Table 1. The median age of children with SMA were slightly lower but not significantly different (p > 0.05) to other disease groups and the healthy control (CC). It is well-known within the sub-Saharan West African holoendemic high-transmission malaria region that SMA is observed in younger children5,39,40,48,49. Although G6PD deficiency has been associated with protection against SMA (G- males only) in a previous cohort from Ibadan50, we did not observe this in our present cohort (data not shown).
The mean parasite densities were not significantly different among the malaria-positive groups; the CM, SMA and UM. Females and males were reasonably represented in this study. The clinical demographics of the subjects within the Discovery Cohort (Table 1), Validation Cohort (Table 1) and HTS Cohort (Table 1) were not different (p > 0.05).
Children presenting with SMA have significantly lower plasma HP than any other study group at hospital admission in the discovery cohort
To explore the structure of the multivariate data and to obtain the most visually useful projection of its variance in two-dimensional space, we created a scatter-plot with the two most informative principal components coefficients of all study groups in the discovery cohort (Fig. 1) followed by imposing their respective group label UM (blue), SMA (red), CM (grey) and CC (yellow). To facilitate visualization of the structure of the data the two-dimensional PCA space is divided clock-wise in four quadrants (Fig. 1: Q1, Q2, Q3 and Q4). Children with SMA mostly aggregated inside the Q3 quadrant and in a manner that is more homogeneous than that of the other groups (Fig. 1 – Q3 red). There is little overlap of children with SMA (red) in Q3 and those within the UM (blue), CM (grey) and CC (yellow) groups (Fig. 1). Children with UM (blue) spread more heterogeneously across all quadrants with the majority in Q2, Q1, and Q4 respectively (Fig. 1). Children with CM (grey) aggregated predominantly within Q2 and Q3 and overlap greatly with the UM (blue) group in this projection of the data. Children in the CC (yellow) group spread across Q1, Q2, Q4 (Fig. 1) and clearly overlap with the UM (blue) group in this projection of the data (Fig. 1). Overall, the PCA projection shows the SMA group as more homogeneous and clearly amenable of linear separation from the rest of the groups (Fig. 1). Observed overlap of variance of UM, CM and CC groups in the PCA projection suggests that non-linear methods are required.
The plasma HP levels at admission in the SMA group are significantly lower when compared to the levels in the other malaria groups SMA < CC (p < 0.0001); SMA < CM (p < 0.001); SMA < UM (p < 0.0001) (Fig. 2a). All children with SMA presented with hypo-haptoglobinemia with plasma HP levels below 2 × 104 ng/ml, median of 4 × 103 ng/ml and mean of 6 × 103 ng/ml (Fig. 2a).
The low plasma HP level in the SMA group does not appear to be dependent on the fHb level since the levels of fHb did not differ among the malaria groups (Fig. 2b). HP plasma levels do not correlate with age in the healthy malaria-negative CC group (Supplementary Fig. 4).
Plasma HP level in children with SMA remained low through convalescence to recovery in the validation cohort
The validation cohort was used to test whether the low HP levels at PD0 observed in the discovery cohort would remain low through convalescence to recovery. As shown in the discovery cohort (Fig. 2a), HP levels were significantly lower at PD0 in SMA (mean of 7 × 103 ng/ml) compared to the other clinical groups CM, UM and CC (p < 0.001) (Fig. 3a). Overall, the HP levels in the SMA group remain significantly lower at all time points compared to levels in both other malaria groups and the parasite-negative CC group (Fig. 3a). Although SMA HP levels at PD28 are significantly higher than that at PD0 (p < 0.01), these levels at recovery still remain significantly lower (mean of 5 × 104 ng/ml) than those of the UM, CM and CC group (mean 105 ng/ml) (p < 0.001) (Fig. 3a). The validation cohort also shows that SMA HP plasma level remains low through convalescence to recovery (Fig. 3a) despite stabilized levels of plasma fHb (Fig. 3b) and decreasing levels, to normal range, of LDH at recovery (Fig. 3c). Unlike the SMA group, plasma HP levels through convalescence to recovery in both the CM and the UM groups were not different to those of the CC group (Fig. 3a).
Plasma fHb and LDH levels in children presenting with SMA were not different to those of other malaria groups in the validation cohort
The levels of both fHb and LDH through convalescence to recovery in the SMA group in the validation cohort do not appear to be higher than those of the UM and CM groups (Fig. 3b,c). LDH levels returned to normal at recovery in all malaria groups; levels at PD28 were not different to the level in the CC group (Fig. 3c).
Plasma HP and fHb levels correlate differently with malaria parasite density
The plasma HP levels significantly inversely correlated with parasite density in the UM group (Supplementary Table 1). On the other hand, there was no correlation between fHb and parasite density in the UM group (Supplementary Table 1). No correlations exist between plasma HP and parasite density in the CM and SMA groups (Supplementary Table 1). A significant direct correlation (p = 0.03) between fHb and parasite density was detected in the SMA group, not reciprocated in the CM group (Supplementary Table 1).
HP isoform phenotype does not show association with SMA or plasma HP level in the HTS cohort
We found no association between HP isoform phenotype and SMA (Fig. 4a). The low HP levels observed in the SMA are equally independent of HP isoform (Fig. 4a,b). The plasma levels of the different isoform phenotypes within a disease group are not different (Fig. 4b).
The HP1-1 isoform is associated with CM (p = 0.038) with an estimated OR of 2.1 (1.04–4.4); there is a higher frequency of HP1-1 in the CM group (0.46) compared to the CC (0.29), UM (0.35) and SMA (0.29) groups (Fig. 4a). The observed association between CM and HP1-1 does not appear to be related to plasma HP level within the isoforms (Fig. 4b).
A Single Nucleotide Polymorphism (rs12162087) (g.-1203G > A) is associated with low HP levels and SMA disease in the HTS cohort
Twelve variants with MAF ≥ 10% found in the HP gene, all located 3.4 kb upstream of the translation start site, were analyzed (Table 2) as summarized in Fig. 5. The HP plasma level of healthy malaria-negative CC group was used to establish the association with the rs12162087 variant as HP plasma level at PD0 in the malaria-positive disease clinical groups might not actually reflect their actual congenital level (Fig. 5). We used ANOVA and linear regression analysis to detect a SNP that correlates with HP circulatory level in the healthy subjects, the CC group, and found rs12162087 (g.-1203G > A; 16:72087307) to be significantly associated (p = 8.5 × 10−6) (Fig. 5 and Table 2). The GG genotype is associated with very low HP plasma level; the AA genotype has the highest level of plasma HP while the plasma HP level in the GA genotype is intermediate (Fig. 5).
To evaluate whether rs12162087 is associated with malaria syndromes, we performed a genetic association study. The analysis revealed a significant association between SMA and rs12162087 (p = 0.03). The (Homo-Ref: GG) genotype frequency is higher in SMA (19.8%) compared to CC (8.2%) with an OR of 2.74 (95% CI = 1.11–6.78). All together our findings in the Nigerian cohort show that the genotype HP rs12162087 GG is more common in SMA and is associated with lower HP levels (Fig. 5).
Discussion
Since the discovery of haptoglobin in 193851, its levels have been used as a marker of hemolysis20. The causes and consequences of low plasma HP in clinical malaria are difficult to dissect. Genetic factors, inflammatory acute phase responses and hemolysis-induced anemia have been suggested to play a role in the low level of HP observed in life-threatening childhood SMA28,44,45, a severe malaria syndrome that requires urgent blood transfusion. A common assumption has been that low HP level in SMA results from parasite-induced hemolysis31,46. However, by measuring the levels of HP, fHb and LDH on hospital admission, during convalescence to recovery, our study shows that low HP levels are associated with susceptibility to SMA.
Our study contributes to the understanding of why some children go onto developing life-threatening SMA while other children the symptoms are restricted to UM. In the present study, HP levels at acute onset were lowest in children with SMA (one order of magnitude lower) and also lower than normal in the CM group, an observation that appears consistent with the hemolysis-induced hypo-haptoglobinemia hypothesis18,20,21,22. However, while the plasma HP levels recovered in children with UM and CM, most children presenting with SMA had a low HP level that remained low during convalescence to recovery, suggesting that hypo-haptoglobinemia was already present in these patients prior to the malaria episode and the acute onset of SMA.
It is intractable to carry-out a large clinical study where HP levels are measured at the initial day of infection, especially in holoendemic high-transmission malaria settings such as ours. To tackle this challenge, we focused on the disease trajectory of the malaria-positive clinical groups from day of diagnosis (PD0) through convalescence (PD7, PD14) to recovery (PD28) in order to establish a possible genetic etiology of the observed hypo-haptoglobinemia. In our study, children recovery at four weeks (PD28) post-diagnosis is strictly defined by clinical and laboratory parameters such as return to normal values of PCV, fHb, LDH and reticulocyte counts. Our definition of recovery at PD28 is also consistent with previous studies that show that hemoglobin levels as low as 5.0 g/dL in SMA returns to normal 2–3 weeks after commencement of treatment52. Similarly, other studies have reported that as parasite disappears from the blood, either by antimalarial therapy or host mechanism, both hemoglobin level and bone marrow morphology returns to normal53 2–3 weeks which is within the convalescence phase54. Furthermore, in acquired hypo-haptoglobinemia, induced by malaria or hemolytic disease, HP levels return to normal 2 weeks after the commencement of antimalarial treatment32,47. In line with these previous findings and coupled with the clinical and laboratory evidence of recovery provided in our study, the PD28 recovery time-point is sufficient for the HP level and other clinical parameters to normalize as it happens in the UM and CM clinical groups. By measuring HP levels at these time-points we have been able to study the role of congenital hypo-haptoglobinemia over that of other etiologic causes. Although hypo-haptoglobinemia can be induced by hemolysis, a genetic contribution to this phenomenon can also be equally important as our data suggest. Our study provides evidence that the etiology of hypo-haptoglobinemia observed in children with acute-onset SMA could be due to genetic factors as HP levels fail to rise after the underlying cause of pathology has been successfully treated and the children recovered four weeks after diagnosis.
The evidence that the rs12162087 (g.-1203G > A) genetic variant, with an observed frequency of 0.67 in this predominantly Yoruba Ibadan’s cohort, is associated with plasma HP levels in the malaria-negative CC group is fairly compelling. First, the association is determined within a single cohort, the malaria-negative well-children cohort, reducing the possibility that confounders play a role. Second, this variant is the only common variant that has such strong evidence suggesting little confounding acting across SNPs. Third, the variant is not out of Hardy-Weinberg equilibrium, suggesting that it is well genotyped by our HTS protocol and there is not stratification bias in the CC group. We show that children with rs12162087 GG genotype showed a risk of 2.74 to develop SMA compared to those with the rs12162087 GA and AA genotypes.
This is the first time a variant in the distal upstream region of the HP gene is reported to be associated with both circulatory HP level and the disease, SMA29,55. The rs12162087 (g.-1203G > A) variant has not been associated with another trait before. The variant is upstream of the transcriptional start site of the HP gene, raising the likelihood that it might be a regulatory variant affecting gene expression. Furthermore, in HaploReg this variant is highlighted as enhancer histone marks and it is associated with eQTL. We suggest that there is a possible interaction between the distal upstream region and the proximal upstream (promoter) region of the HP gene through DNA looping that could affect the expression of the protein.
Aside the hypo-haptoglobinemia observed in SMA patients, we also showed that plasma HP level is independent of HP isoform phenotype. Replicating the findings of a previous study in Ghana56, we found that the Hp1-1 isoform is significantly more prevalent in children presenting with CM than in the CC group in our Nigerian cohort. However, our larger sample size than the previous study56 did not detect any significant differences between Hp1-1 isoform in the SMA when compared to CC. Consistent to a study carried out in Kenya57, we found that the Hp2-1 isoform does not confer protection against SMA. In the Kenyan cohort57, the Hp2-2 isoform is associated with the greatest risk of CM and Hp2-1 with greatest protection against CM. In our west-Africa CM cohorts we found that Hp1-1 was more common in the CM patients, implying a potential for greater risk factor, whilst Hp2-1 and Hp2-2 seem to be associated with protection. Further multi-site studies are needed to dissect the differences between these west- and east- African cohorts and to investigate if there is a correlation between HP isoform and the reported rs12162087 variant.
The reported long heterozygous and homozygous deletions of the HP gene known to be associated with hypo-haptoglobinemia28,36 are rare in this population. This deletion was found in only 1 out of 401 subjects whose HP genes were sequenced. These deletions appear not to be the major cause of hypo-haptoglobinemia in this predominantly Yoruba population.
Our data shows that the degree of intravascular hemolysis in the SMA group is not different to that of other malaria positive groups. In acute-onset life-threatening childhood SMA rapid PCV decrease58 is predominantly driven by increased clearance of uninfected erythrocytes by the reticulo-endothelial system49 as intravascular hemolysis of infected erythrocytes due to schizogony cannot explain by itself this dramatic change. In line with our previous SMA study49, we suggest that, in the cohorts with baseline low plasma HP and Hb, particularly the SMA group, clearance of non-infected erythrocytes is driven by the reticulo-endothelial system in the spleen through an as yet unknown CD8(+) T-cell-dependent mechanism49. This splenic clearance of erythrocytes could be responsible for the low Hb level in the SMA group. This further indicates that extravascular hemolysis is implicated more strongly in the pathology of SMA rather than intravascular hemolysis.
It is unknown why some children develop severe forms of malaria such as SMA whilst in others the infection is restricted to the mild form of the disease. Our study suggests that a low circulatory HP level is a risk factor for the occurrence of SMA in contrast to the other malaria syndromes studied. Children bearing the genotype at risk rs12162087 g.-1203G/G had one order of magnitude lower HP levels which is consistent also with plasma HP levels observed to be low in SMA children at disease onset and at recovery.
Although multi-site studies within Africa and outside Africa, with populations under different malaria burden, are needed to further dissect the relationship between of HP genotypes with hypo-haptoglobinemia, our findings provide evidence that the hypo-haptoglobinemia phenotype and the presence of the rs12162087 g.-1203G/G genotype could be used as predictors for acute onset SMA. We expect that our findings will prompt these multi-site studies, not only to replicate association, but to dissect what other characteristics of other clinical settings can reinforce the significance of genetic control of HP plasma levels in determining acute onset severe anemia phenotypes including that induced by malaria.
Finally, our study contributes to the development of risk assessment algorithms to identify children with higher risk to develop SMA, which is lethal without urgent access to safe blood transfusion, this being the case in all (urban, peri-urban and rural) areas in high-transmission malaria holoendemic regions in sub-Saharan Africa. Improved risk assessment algorithms will also have a substantial impact on the use and management of health-care resources such as blood banks across low- and middle-income regions of the World under malaria burden.
References
World Health Organization. World Malaria Report 2017. doi:CC BY-NC-SA 3.0 IGO (World Health Organization, 2017).
World Health Organization. World Malaria Report 2016. World Malaria Report doi:CC BY-NC-SA 3.0 IGO (World Health Organization, 2016).
Bamiselu, O. F. et al. Adherence to malaria diagnosis and treatment guidelines among healthcare workers in Ogun State, Nigeria. BMC Public Health, https://doi.org/10.1186/s12889-016-3495-x (2016).
Ajetunmobi, W. A. et al. Haemoglobinuria among children with severe malaria attending tertiary care in Ibadan, Nigeria. Malar. J. 11 (2012).
Burte, F. et al. Severe childhood malaria syndromes defined by plasma proteome profiles. PLoS One 7, e49778 (2012).
Ferreira, A., Balla, J., Jeney, V., Balla, G. & Soares, M. P. A central role for free heme in the pathogenesis of severe malaria: The missing link? Journal of Molecular Medicine https://doi.org/10.1007/s00109-008-0368-5 (2008).
Perkins, D. J. et al. Severe malarial anemia: Innate immunity and pathogenesis. International Journal of Biological Sciences, https://doi.org/10.7150/ijbs.7.1427 (2011).
Mendonca, V. R. et al. Association between the haptoglobin and heme oxygenase 1 genetic profiles and soluble CD163 in susceptibility to and severity of human malaria. Infect. Immun. 80, 1445–1454 (2012).
Han, T. H., Hyduke, D. R., Vaughn, M. W., Fukuto, J. M. & Liao, J. C. Nitric oxide reaction with red blood cells and hemoglobin under heterogeneous conditions. Proc Natl Acad Sci USA 99, 7763–7768 (2002).
Schaer, D. J., Buehler, P. W., Alayash, A. I., Belcher, J. D. & Vercellotti, G. M. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121, 1276–1284 (2013).
Quaye, I. K. Haptoglobin, inflammation and disease. Trans R Soc Trop Med Hyg 102, 735–742 (2008).
Sertorio, J. T. et al. Haptoglobin polymorphism affects nitric oxide bioavailability in preeclampsia. J Hum Hypertens 27, 349–354 (2013).
Smithies, O. Zone electrophoresis in starch gels: group variations in the serum proteins of normal human adults. Biochem. J. 61, 629–641 (1955).
Smithies, O. & Walker, N. F. Notation for serum-protein groups and the genes controlling their inheritance. Nature 178, 694–695 (1956).
Robson, E. B., Polani, P. E., Dart, S. J., Jacobs, P. A. & Renwick, J. H. Probable assignment of the alpha locus of haptoglobin to chromome 16 in man. Nature 223, 1163–1165 (1969).
Nielsen, M. J. & Moestrup, S. K. Receptor targeting of hemoglobin mediated by the haptoglobins: roles beyond heme scavenging. Blood 114, 764–771 (2009).
Schaer, C. A., Schoedon, G., Imhof, A., Kurrer, M. O. & Schaer, D. J. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res 99, 943–950 (2006).
Delanghe, J., Langlois, M. & De Buyzere, M. Congenital anhaptoglobinemia versus acquired hypohaptoglobinemia. Blood 91, 3524 (1998).
Delanghe, J. R. & Langlois, M. R. Hemopexin: a review of biological aspects and the role in laboratory medicine. Clin Chim Acta 312, 13–23 (2001).
Gupta, S., Ahern, K., Nakhl, F. & Forte, F. Clinical usefulness of haptoglobin levels to evaluate hemolysis in recently transfused patients. Adv Hematol 2011, 389854 (2011).
Imrie, H. et al. Haptoglobin levels are associated with haptoglobin genotype and alpha+ -Thalassemia in a malaria-endemic area. Am. J. Trop. Med. Hyg. 74, 965–971 (2006).
Kim, H. et al. Anaphylactic transfusion reaction in a patient with anhaptoglobinemia: the first case in Korea. Ann Lab Med 32, 304–306 (2012).
Ko, D. H. et al. A review of haptoglobin typing methods for disease association study and preventing anaphylactic transfusion reaction. Biomed Res Int 2013, 390630 (2013).
Ballas, S. K. Lactate dehydrogenase and hemolysis in sickle cell disease. Blood 121, 243–244 (2013).
Shah, P., Mehta, V. M., Cowger, J. A., Aaronson, K. D. & Pagani, F. D. Diagnosis of hemolysis and device thrombosis with lactate dehydrogenase during left ventricular assist device support. J Hear. Lung Transpl. 33, 102–104 (2014).
Kato, G. J. et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood 107, 2279–2285 (2006).
Tabbara, I. A. Hemolytic anemias. Diagnosis and management. Med Clin North Am 76, 649–668 (1992).
Koda, Y., Soejima, M., Yoshioka, N. & Kimura, H. The haptoglobin-gene deletion responsible for anhaptoglobinemia. Am. J. Hum. Genet. 62, 245–252 (1998).
Teye, K. et al. A novel I247T missense mutation in the haptoglobin 2 beta-chain decreases the expression of the protein and is associated with ahaptoglobinemia. Hum. Genet. 114, 499–502 (2004).
Rougemont, A. et al. Hypohaptoglobinaemia as an epidemiological and clinical indicator for malaria. Results of two studies in a hyperendemic region in West Africa. Lancet 2, 709–712 (1988).
Langlois, M. R. & Delanghe, J. R. Biological and clinical significance of haptoglobin polymorphism in humans. Clin. Chem. 42, 1589–1600 (1996).
Saad, H. A. & Geoffery, P. Malaria: A Hematological Perspective. Definition, Epidemiology, Diagnosis and Management of the Anemia of Malaria Tropical M, (Imperial College Press, 2004).
Carter, K. & Worwood, M. Haptoglobin: a review of the major allele frequencies worldwide and their association with diseases. Int J Lab Hematol 29, 92–110 (2007).
Allison, A. C., Blumberg, B. S. & Ap, R. Haptoglobin types in British, Spanish Basque and Nigerian African populations. Nature 181, 824–825 (1958).
Constans, J., Viau, M., Gouaillard, C. & Clerc, A. Haptoglobin polymorphism among Saharian and West African groups. Haptoglobin phenotype determination by radioimmunoelectrophoresis on Hp O samples. Am. J. Hum. Genet. 33, 606–616 (1981).
Koda, Y. et al. Simple PCR detection of haptoglobin gene deletion in anhaptoglobinemic patients with antihaptoglobin antibody that causes anaphylactic transfusion reactions. Blood 95, 1138–1143 (2000).
Oliviero, S., Morrone, G. & Cortese, R. The human haptoglobin gene: transcriptional regulation during development and acute phase induction. EMBO J (1987).
Orimadegun, A. E., Fawole, O., Okereke, J. O., Akinbami, F. O. & Sodeinde, O. Increasing burden of childhood severe malaria in a Nigerian tertiary hospital: implication for control. J Trop Pediatr 53, 185–189 (2007).
Bachmann, J. et al. Affinity Proteomics Reveals Elevated Muscle Proteins in Plasma of Children with Cerebral Malaria. PLoS Pathog. 10 (2014).
Burte, F. et al. Circulatory hepcidin is associated with the anti-inflammatory response but not with iron or anemic status in childhood malaria. Blood 121, 3016–3022 (2013).
Njuguna, P. & Newton, C. Management of severe falciparum malaria. J Postgr. Med 50, 45–50 (2004).
Trampuz, A., Jereb, M., Muzlovic, I. & Prabhu, R. M. Clinical review: Severe malaria. Crit Care 7, 315–323 (2003).
World Health Organization. Treatment of Severe Malaria. Guidel. Treat. Malar, https://doi.org/10.1016/0035-9203(91)90261-V (2015).
Casals-Pascual, C. & Roberts, D. J. Severe malarial anaemia. Curr Mol Med 6, 155–168 (2006).
Koda, Y., Soejima, M. & Kimura, H. Anhaptoglobinemia and hypohaptoglobinemia. Ryoikibetsu Shokogun Shirizu 676–678 (1998).
Boreham, P. F., Lenahan, J. K., Port, G. R. & McGregor, I. A. Haptoglobin polymorphism and its relationship to malaria infections in The Gambia. Trans R Soc Trop Med Hyg 75, 193–200 (1981).
Trape, J. F. et al. Malaria, cause of ahaptoglobinaemia in Africans. Trans R Soc Trop Med Hyg 79, 430–434 (1985).
Reyburn, H. et al. Association of transmission intensity and age with clinical manifestations and case fatality of severe Plasmodium falciparum malaria. JAMA, https://doi.org/10.1001/jama.293.12.1461 (2005).
Safeukui, I. et al. Malaria induces anemia through CD8+ T Cell-dependent parasite clearance and erythrocyte removal in the spleen. MBio 6 (2015).
Orimadegun, A. E. & Sodeinde, O. Glucose-6-phosphate dehydrogenase status and severity of malarial anaemia in Nigerian children. J. Infect. Dev. Ctries, https://doi.org/10.3855/jidc.1837 (2011).
Wobeto, V. P., Zaccariotto, T. R. & Sonati, M. Polymorphism of human haptoglobin and its clinical importance. Genet. Mol. Biol. 31, 602–620 (2008).
Raffray, L. et al. Severe delayed autoimmune haemolytic anaemia following artesunate administration in severe malaria: A case report. Malar. J, https://doi.org/10.1186/1475-2875-13-398 (2014).
Abdalla, S. H. Hematopoiesis in human malaria. Blood Cells (1990).
Phillips, R. E. et al. The importance of anaemia in cerebral and uncomplicated falciparum malaria: Role of complications, dyserythropoiesis and iron sequestration. QJM, https://doi.org/10.1093/oxfordjournals.qjmed.a067960 (1986).
Teye, K. et al. A-61C and C-101G Hp gene promoter polymorphisms are, respectively, associated with ahaptoglobinaemia and hypohaptoglobinaemia in Ghana. Clin Genet 64, 439–443 (2003).
Quaye, I. et al. Haptoglobin 1-1 is associated with susceptibility to severe Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg 94, 216–219 (2000).
Atkinson, S. H. et al. Epistasis between the haptoglobin common variant and α+ thalassemia influences risk of severe malaria in Kenyan children. Blood, https://doi.org/10.1182/blood-2013-10-533489 (2014).
Ray, S. et al. Proteomic analysis of Plasmodium falciparum induced alterations in humans from different endemic regions of India to decipher malaria pathogenesis and identify surrogate markers of severity. J Proteomics, https://doi.org/10.1016/j.jprot.2015.04.032 (2015).
Acknowledgements
The authors thank all the children, guardians and parents who participated in this study. We thank the consultants, clinical registrars, nurses, clinical laboratory and administrative staff at the Departments of Paediatrics and Haematology of the College of Medicine of the University of Ibadan, University College Hospital Ibadan, Nigeria for all the support they provided for the present study. We thank Mr. Abdul Sesay at the Crick Institute Advanced Sequencing Platform for help and advice for DNA sequencing. We thank the research and administrative staff who provided support at the College of Medicine of the University of Ibadan, Nigeria; the Crick Institute, UK; the European Network of Malaria Research Excellence; the Aix-Marseille Université, INSERM, Marseille, France and the Faculty of Engineering, University College London, United Kingdom. This work was supported by the College of Medicine of the University of Ibadan, Ibadan, Nigeria; the UK Medical Research Council (MC_U117585869); the European Network of Malaria Research Excellence (EviMalaR); the Aix-Marseille Université, INSERM, Marseille, France; the Francis Crick Institute, London, UK (FC001097); the Department of Computer Science, Faculty of Engineering Sciences of University College London, United Kingdom; the UK Engineering and Physical Sciences Research Council (EP/P028608/1) and the UK Global Challenges Research Fund (GCRF). The Childhood Malaria Research Group (CMRG) is a joint research and innovation equal-partnership between the College of Medicine of University of Ibadan, Nigeria and University College London, London, UK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
D.F.-R., B.J.B. and O.S. designed the study. S.E.A., F.B., S.M., B.J.B., S.O., I.L., W.A.S., O.S., M.W., A.A.H., A.J.D. and D.F.-R. carried out the study. F.A., B.J.B., N.K.A., G.O., S.O., I.L., W.A.S., O.S. and D.F.-R. carried patient recruitment and executed the case-control study at the CMRG, Ibadan, Nigeria. S.E.A., F.B., S.M., N.K.A., H.D., L.A., B.N., L.H., G.E., O.S. and D.F.-R. carried out laboratory experimental work. S.E.A., F.B., S.M., B.J.B., A.A.H., G.E., B.N., L.H., A.J.D., O.S. and D.F.-R. analyzed the data. S.E.A., F.B., S.M., B.J.B., O.S., A.A.H. and D.F.-R. wrote the manuscript, which was approved by all authors. D.F.-R. is project lead and senior corresponding author.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abah, S.E., Burté, F., Marquet, S. et al. Low plasma haptoglobin is a risk factor for life-threatening childhood severe malarial anemia and not an exclusive consequence of hemolysis. Sci Rep 8, 17527 (2018). https://doi.org/10.1038/s41598-018-35944-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35944-w
Keywords
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
