
Abstract
Multiple Sclerosis (MS) is an inflammatory and neurodegenerative disease of the central nervous system. The inflammatory process in MS is driven by both T and B cells and current therapies are targeted to each of these cell types. Epigenetic mechanisms may provide a valuable link between genes and environment. DNA methylation is the best studied epigenetic mechanism and is recognized as a potential contributor to MS risk. The objective of this study was to identify DNA methylation changes associated with MS in CD19+ B-cells. We performed an epigenome-wide association analysis of DNA methylation in the CD19+ B-cells from 24 patients with relapsing-remitting MS on various treatments and 24 healthy controls using Illumina 450 K arrays. A large differentially methylated region (DMR) was observed at the lymphotoxin alpha (LTA) locus. This region was hypermethylated and contains 19 differentially methylated positions (DMPs) spanning 860 bp, all of which are located within the transcriptional start site. We also observed smaller DMRs at 4 MS-associated genes: SLC44A2, LTBR, CARD11 and CXCR5. These preliminary findings suggest that B-cell specific DNA-methylation may be associated with MS risk or response to therapy, specifically at the LTA locus. Development of B-cell specific epigenetic therapies is an attractive new avenue of research in MS treatment. Further studies are now required to validate these findings and understand their functional significance.
Similar content being viewed by others


Introduction
Multiple Sclerosis is an inflammatory and neurogenerative disease leading to demyelination and axonal loss. Risk of developing MS is thought to be influenced by both genetic and environmental factors. The primary environmental factors that influence disease pathology are sunlight exposure, Epstein-Barr virus (EBV) infection and smoking1. Genome wide association studies (GWAS) have identified 149 genes associated with MS risk with approximately one third coming from variations in the major histocompatibility complex (MHC)2,3. Despite this, there still remains a large element of unexplained heritability in terms of disease pathology.
Epigenetic mechanisms are capable of modifying the genome without changes to the DNA sequence and can be inherited. One well-studied epigenetic mechanism is DNA methylation, which is the addition of a methyl group to CpG dinucleotides. We, and others, have used genome-wide DNA methylation technologies to identify differentially methylated positions (DMPs) in the T-cells of MS patients compared to healthy controls4,5,6,7,8. In two independent studies of CD4+ T-cells, we found a striking differentially methylated region (DMR) located within the major histocompatibility complex (MHC) region, with a major peak at HLA-DRB1 and RNF394,6. Using the same cohort of patients we assessed DMPs in CD8+ T-cells and found 79 DMPs, all of which showed minor association with MS but none of which overlapped with any of the DMPs found in CD4+ T-cells5. A study by Bos et al. also found little overlap between the methylation profiles of CD4+ and CD8+ T-cells, highlighting the importance of investigating individual cell subtypes.
There is evidence to suggest that T-cells may have a role in MS pathology (reviewed in Martin et al.9). However, it is becoming increasingly clear that B-cells may also play a substantial role in helping to drive disease. Activated B-cells may contribute to MS pathology as antibody producing cells, antigen presenting cells or as a source of pro-inflammatory cytokines (reviewed in Lehmann-Horn et al.10). Evidence for this is in the success of B-cell depleting monoclonal antibodies, such as rituximab11 and ocrelizumab12 as MS therapies. Additionally, many currently approved MS therapies, for example fingolimod and dimethyl fumarate, also have an impact on B cells through reduced numbers or a shift in phenotype towards a more anti-inflammatory cytokine profile (reviewed in Lehmann-Horn et al.10).
In an effort to identify B-cell specific DMPs associated in MS, we performed genome-wide DNA methylation study of CD19+ B-cells from MS patients and healthy controls. We used the same cohort and data analysis techniques as our previous studies so that the results could be compared to those from the CD4+ and CD8+ T-cells.
Methods and Materials
Ethics Statement
The Hunter New England Health Research Ethics Committee and University of Newcastle Ethics committee approved this study (05/04/13.09 and H-505-0607 respectively), and methods were carried out in accordance with institutional guidelines on human subject experiments. Written and informed consent was obtained from all patient and control subjects. MS patients gave written and verbal consent. The Australian Red Cross Blood Service ethics committee approved the use of blood from healthy donors.
Sample Processing
We performed an epigenome-wide association study (EWAS) of CD19+ B-cells using the same patient cohort, work flow and data analysis as described in our previous study4. Briefly, whole blood was collected from 24 RRMS patients and 24 healthy controls. All patients were diagnosed with RRMS according to the McDonald criteria13. PBMCs were isolated from 45 mL of whole blood by density gradient centrifugation on lymphoprep (StemCell Technologies, Canada). CD19+ B cells were isolated using positive selection, magnetic separation kits (Stem Cell Technologies, Canada) according to the manufacturer’s protocols. Purity was assessed using FITC conjugated anti-CD19 antibody (clone H1B19, catalog #60005FL.1, StemCell Technologies, Canada) and the FACS CantoII flow cytometer (BD Biosciences, USA) and analyzed using the FACSDiva software (BD Biosciences, USA). All samples met a minimum purity threshold of ≥90%. DNA was extracted using the QiaAMP DNA micro kit (Qiagen, USA). DNA was then bisulphite converted and hybridized to Illumina 450 K arrays (service provided by the Australian Genome Research Facility).
Data analysis
Raw fluorescence data were processed using a combination of R/Bioconductor and custom scripts. Raw data was parsed into the Bioconductor MINFI package. Methylation data was background corrected and quantile normalized according to MINFI routines. Data was cleaned by removing control probes, probes which map multiple times to the genome, cross-reactive probes and failed probes for which the intensity of both the methylated and the unmethylated probes was <1000 units across all samples. A threshold of 1000 units was selected based on the profile of the available negative control probes. Y chromosome probes were filtered out. All probe sequences were mapped to the human genome (buildHg19) using BOWTIE14 to identify potential hybridization anomalies. We chose to retain probes containing single nucleotide polymorphisms (SNPs) and filter these out post hoc where appropriate (see results section).
Measures of methylation (β values) were produced for each probe and ranged from completely unmethylated (β = 0) to completely methylated (β = 1). To identify differentially methylated positions (DMPs) associated with MS subtypes in this cohort, we first calculated the difference in median β value by subtracting the median β value of controls (mediancont) from the median β value for cases (mediancase). This produced a Δmeth score ranging from −1 (hypomethylated) to 1 (hypermethylated). A two-sample Kolmogorov-Smirnov test (K-S test) was used to determine if Δmeth was statistically significant. A K-S test was chosen over the F test because of the marked variation in the distribution of the β values among the probes. Rather than base our CpG selection strictly on statistical significance (P-values) of the K-S test, which is overly limiting due to the small sample size and could miss important signal, we used a selection strategy based on a combination of P-value and effect size (ie. Δmeth score). We have used this approach successfully in previous studies to implicate differential methylation at HLA in CD4+ cells with regard to MS4,5,6. A CpG was considered a DMP if the P-value was <0.05 and the absolute β value was >±0.1. Differentially methylated regions (DMRs) were called if at least two DMPs were found within a 500 base pair (bp) span of each other and were altered in the same direction (either all hypermethylated or all hypomethylated).
Over-Representation Analysis (ORA): To assess the biological relevance of DMPs in terms of MS pathology we conducted an ORA on resultant the DMP list using the WebGestalt engine (www.webgestalt.org) incorporating the KEGG pathways database.
Results
DMP and DMR analyses
Table 1 shows the patient demographic for 24 MS patients and 24 healthy controls (Table 1). A total of 7618 CpGs met the criteria for a DMP (Table S1). Figure 1 shows the genome-wide distribution of differential methylation (Δmeth) for all DMPs. Amongst the DMPs, we observe an overall hypomethylation in MS cases, with 4731 (62%) of DMPs being hypomethylated and 2887 (38%) being hypermethylated in MS patients versus controls. When we considered genomic features for all DMPs we found 1869 (24.5%) map to intergenic regions, 3226 (42.3%) within the gene body, 1254 at the transcriptional start site (TSS1500 or TSS200) (16.5%), 699 in the 5′ untranslated region (UTR) (9.2%), 211 map to the 1st exon (2.8%) and 359 in the 3′UTR (4.7%) (Fig. 2).

A genome-wide differential methylation plot. Data points outside the circle (red) represent increased methylation (i.e. ∆meth), in multiple sclerosis (MS) patients compared to controls whereas points inside the circle (blue) represent decreased methylation in MS patients compared to healthy controls.

Distribution of DMPs over each of the genomic regions Y-axis represents proportion of total DMPs (7618) in each category (shown as percentage).
DMPs were ranked by Δmeth values. The two top ranked DMPs were located within the lymphotoxin alpha (LTA) gene (alias: tumor necrosis factor beta, TNFβ - hereafter referred to as LTA). These two sites had Δmeth values of 0.504 and 0.486 (50.4% and 48.6% hypermethylated respectively) in the MS patient group compared to the control group (P < 0.0001). Of the CpGs that met the criteria for a DMP, 19 are found in the LTA locus within a region of 860 bp. All sites are hypermethylated with Δmeth scores between 0.15 and 0.5 (between 15% and 50%) and are located within the TSS/5′UTR (Table 2).
Genetic influence at the LTA locus
One technical limitation of array technology is the influence that SNPs may have on the calculated methylation levels (β values). Of the 19 DMPs identified at the LTA TSS, 13 of the corresponding probes contain an adjacent SNP which may potentially influence the methylation profile (Table 2). Rather than remove these sites from our analysis, we assessed the genetic influence on the methylation signal by visualizing the distribution of β values.
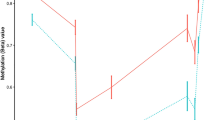
Figure 3A shows an example of a CpG site whose methylation signal is known to be influenced by a SNP located at this probe. This example shows that the β values cluster into 3 distinct regions representing the 3 possible genotypes (homozygous allele 1, homozygous allele 2 or heterozygous). Figure 3B shows the influence of SNPs on the top LTA CpG site in our DMR. The β values form a uniform spread, providing support that the SNP is not influencing the methylation signal at this particular CpG site. A similar result is seen in all DMPs within the LTA cluster (data not show). In addition to this, we compared the profiles of LTA in CD4+ and CD8+ T-cells from our previous data sets with the same cohort. We found that hypermethylation at the LTA TSS appears to be specific to CD19+ B-cells, providing further support that the methylation effects observed in this cohort are at least partially exclusive from the underlying genotype (Fig. 4).

Tukey box plot showing distribution of beta values for (A) a probe where the SNP is driving the methylation values and (B) the top LTA site from this study. The box plot shows the data within the interquartile range and the median is represented by a solid black line. Whiskers show maximum and minimum values. Grey bars indicate region for each genotype (homozygous allele 1 (1/1), heterozygous (1/2), and homozygous allele 2 (2/2)). Each point represents either an individual control (blue) or MS patients (red). Y axis shows β values.

Differential methylation at other genes within the MHC locus
Our previous study in CD4+ T-cells identified a peak of differential methylation on chromosome 6 that mapped to the MHC region4. Specifically, we found a differentially methylated region that spanned 11 sites at the well-established MS risk gene, HLA-DRB1 that was unique to CD4+ T-cells4,5. To determine if there is any overlap of DMPs in the MHC region between CD4+ T-cells and CD19+ B-cells, we performed a closer analysis of the MHC region. Figure 5 shows that although a similar distinct peak is present at the MHC region, it corresponds primarily to the DMR at LTA and to a lesser extent HLA-DRB1. However, there are 4 DMPs in CD19+ B-cells that overlap with the sites found in CD4+ T-cells (Table 3). These sites correspond to probes cg04985482, cg06032479, cg17416722, and cg24147543. The first site maps to the MHC class I polypeptide related sequence A (MICA) locus and the remaining three sites all map to sites within HLA-DRB1. All sites are altered in the same direction (hypo- or hypermethylated) and have a similar differential methylation value in both cell subsets (Table 3).

Methylation at the MHC locus in CD19+ B cells Manhattan plot showing methylation level (Δmeth) for all probes that fall within the MHC locus (Chr6: 29054321-32978719). Points above 0 represent hypermethylated sites, points below 0 represent hypomethylated sites. Grey dotted line indicates 10% change in methylation.
Differential methylation at sites outside the MHC locus
To explore the importance of methylation outside the MHC region, we filtered DMPs outside the MHC to include only those contained within the TSS or 5′UTR (1953 DMPs). We chose the TSS and 5′UTR as an initial filtering step because DNA methylation that occurs in the promoter regions is generally associated with transcriptional repression, but its role elsewhere in the genome is more complex and less well understood15. We then further filtered the list to include only DMRs. A DMR was considered if i) there were 2 or more DMPs, ii) these DMPs fell within a 500 bp span iii) the DMPs were altered in the same direction. This generated a list of 276 genes which contained a DMR in their 5′UTR or TSS.
A comparison of this list to genes with a known association to MS2,3,16,17,18 revealed 4 DMRs (Table 4). Choline transporter-like 2 (SLC44A2) and Lymphotoxin β receptor (LTBR) each have 2 hypomethylated DMPs within their TSS/5′UTR which are 72 and 113 bp apart, respectively. Caspase recruitment domain-containing protein 11 (CARD11) has 2 DMPs at the 5′UTR. Both of which are hypermethylated and 101 bp apart. There is a third DMP which is hypermethylated at the 5′UTR of CARD11; however, it is located >62,000 bp downstream of the DMR so it did not fulfil the criteria to be part of the DMR. At the TSS of the CXC chemokine receptor 5 (CXCR5), there are 2 DMRS that are 9266 bp apart. The first of these has 5 hypermethylated DMPs within an 86 bp span. These DMPs are between 20.7% and 31.1% hypermethylated. The second contains 3 DMPs within 42 bp of each other that are 17.7%, 13.9% and 18% hypermethylated.
Further analysis revealed several other DMRs outside the MHC region which reside in genes that may have biological significance to MS pathology. Of interest, there are 5 DMPs which lie within a 269 bp span at the cluster of differentiation 19 (CD19) locus. All are found within the TSS or 5′UTR and are hypermethylated by 22.4–30.7%. There is also a DMR at interleukin 21 receptor (IL21R) where 4 hypermethylated DMPs lie within a 264 bp span at the TSS.
Over-Representation Analysis (ORA)
The 7618 DMPs identified in the methylation analysis were located in 2899 genes. To assess the biological relevance of this gene set in terms of MS pathology we conducted a ORA using the WebGestalt engine to identify potential pathways associated with the 2899 gene set. Pathway analysis revealed significant alignment to innate immune system (293 genes, P = 4.08E-09), B-cell receptor signaling pathway (28 genes, P = 3.31E-04), cytokine signaling in Immune system (166 genes, 4.14E-04), and signaling by interleukins (119 genes, 1.46E-03). Table 5 shows the top 10 pathways identified.
Discussion
B-cells are gaining recognition in MS as potential regulators of disease pathology. In this study, we are the first to describe changes in the global DNA methylation profile in the CD19+ B-cells of MS patients compared to healthy controls. We find a slight overall hypomethylation and enrichment of genes involved in innate immunity and B-cell receptor and cytokine signaling pathways. We have identified a large, hypermethylated DMR in the TSS of LTA that is unique to the B-cell population. In addition, we identified four smaller DMRs at genes which contain known MS-associated SNPs, SLC44A2, LTBR, CXCR5, and CARD11.
The large DMR at LTA is of interest due to its longstanding, strong associations with MS. LTA encodes for the pro-inflammatory cytokine lymphotoxin-alpha (LT-α). LTA is over-expressed in CD4+ T-cells, CD8+ T-cells and CD19+ B-cells of RRMS patients19. Furthermore, in RRMS patients the LTA CSF/PBMC expression ratios are increased, and positively correlate with CD19 expression in CSF cells19. LTA producing cells have been found in the immediate vicinity of the demyelinating process in MS patients20 and expression is present in acute and chronic active brain lesions in MS patients21. One inconsistency is that we have shown hypermethylation in the TSS, which implies potential downregulation of transcription (as opposed to over-expression). The most likely explanation for this inconsistency is the presence of hydroxymethylation. Bisulfite conversion does not distinguish between hydroxymethylated and methylated sites; thus, both are considered methylated by the methods used in this study. Unlike methylation, which negatively correlates with transcription, hydroxymethylation has been found to positively correlate with active transcription22,23,24,25. Therefore, it is plausible that the methylation changes at LTA are due to changes in hydroxymethylation which would result in the overexpression seen in previous studies.
Another explanation for the inconsistency between our findings and previous studies could be due to transcript variants. LTA is known to have eight transcript variants, with multiple start sites26. Thus, the hypermethylation seen in our study may be related to an alternate transcriptional variant to that identified in previous studies. Alternately, previous studies were conducted primarily in treatment naïve patients, whereas our cohort only contains 1 treatment naïve sample. Therefore, hypermethylation and decreased LTA expression may be a result of treatment effects, or simply be reflective of disease stabilization.
Although we find DMRs at four other genes previously associated with MS, the functional significance of these DMRs is unclear. SLC44A2 is found in the peripheral tissues and has been associated with thrombosis and autoimmune hearing loss but not MS27. CXCR5 is used as the defining marker for follicular B helper T-cells (TFH) but its expression has not been demonstrated in B-cells28.
A recent study found LTBR expression levels increased in the animal model of MS, experimental autoimmune encephalitis (EAE), and that blockage of this receptor ameliorated disease in mice29. The same study investigated LTBR expression in RRMS patients and found increased transcript levels in patients who were resistant to interferon beta (IFNβ) therapy29. Hypomethylation in the TSS may be correlated with increased transcription of LTBR; however, the study by Inoue and colleagues used PBMCs, which contain a mixture of T-cells and B-cells, so it remains to be elucidated if increased expression is occurring in B-cells.
Although CARD11 does not yet have a demonstrated, functional role in MS, it is an essential scaffolding platform for the CARD11/ BCL10/MALT1 (CBM) complex30. NFκB governs the BCR-induced (B cell receptor) NFκB activation through a complex series of phosphorylation events that results in destruction of the NFκB inhibitor, IκB30. One known mechanism of action of the common MS therapy, dimethyl fumarate, is inhibition of the NFκB transcription factor; therefore, an intriguing possibility is that dysregulation of this pathway may play a role in MS pathology31.
Although not part of the MHC locus or previously linked to MS, IL21R is involved in other autoimmune conditions such as systemic lupus erythematosus (SLE)32 and arthritis33. It has been linked to B-cell proliferation and survival as well as B-cell apoptosis which suggesting a role in immune cell function34,35.
Autoimmune diseases often have overlapping aetiological and genetic backgrounds36. In our previous studies, we found that there is also overlap in epigenetic profiles of CD4+ T-cells from SLE and MS patients6. Recently, Julià et al.37 assessed the DNA methylation profiles of B-cells from rheumatoid arthritis (RA) patients and performed a comparison with SLE patients37. To determine if there is overlap in the epigenetic profiles of CD19+ B-cells we compared our results to this study. Of the ten probes identified in their study, five also show differential methylation in the same direction (all hypermethylated) as in our study (Table 6). This suggests a common epigenetic precursor or epigenetic effect among related autoimmune diseases.
One important consideration for our study is that the patients tested were taking various medications at the time of recruitment including interferons, glatiramer acetate, natalizumab and fingolimod. Only one patient was treatment naïve and 4 had been off treatment for more than 6 months. Although this study controlled for age, sex and treatment effects (as much as possible), due to our limited size we cannot control for changes associated with various environmental factors. Additionally, we were unable to control for B cell subtype compositions. As a third of the patients were taking fingolimod, this may have caused a significant change in the circulating cells.
This study adds to our knowledge of epigenetic factors in MS and further highlights the need to investigate individual cell subtypes when assessing DNA methylation in disease. It also raises several new and important questions including i) are these changes due to treatment effects ii) is the change in methylation at LTA due to hydroxymethylation iii) what role do environmental factors play on methylation changes iv) are the methylation effects due to changes in B cell subtypes and v) are DNA methylation changes a pre-disposing factor for MS or are they a result of disease pathology? Further studies are required using larger, treatment naïve cohorts that include epidemiological data will help extract if these results are due to treatment effects and allow the addition of environmental factors such as vitamin D, EBV virus infection and smoking as covariates in the analysis. Additionally, further studies should attempt to extrapolate the relative effect of methylation versus hydroxymethylation, possibly using a more targeted approach such as next generation sequencing. Overall, our results suggest that B-cell specific epigenetics may play a role in MS pathology. B-cell specific epigenetic therapies which target LTA expression would therefore be an attractive new avenue of research in MS treatments.
Availability of Data and Material
The datasets generated or analyzed during the current study are included in this published article (Supplementary Table 1). Raw data files are available from Assoc. Prof. Rodney A. Lea.
References
Amato, M. P. et al. Environmental modifiable risk factors for multiple sclerosis: Report from the 2016 ECTRIMS focused workshop. Mult Scler, 1352458516686847, https://doi.org/10.1177/1352458516686847 (2017).
International Multiple Sclerosis Genetics, C IL12A, MPHOSPH9/CDK2AP1 and RGS1 are novel multiple sclerosis susceptibility loci. Genes Immun 11, 397–405, https://doi.org/10.1038/gene.2010.28 (2010).
International Multiple Sclerosis Genetics, C. et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 45, 1353–1360, https://doi.org/10.1038/ng.2770 (2013).
Graves, M. et al. Methylation differences at the HLA-DRB1 locus in CD4+ T-Cells are associated with multiple sclerosis. Mult Scler 20, 1033–1041, https://doi.org/10.1177/1352458513516529 (2013).
Maltby, V. E. et al. Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin Epigenetics 7, 118, https://doi.org/10.1186/s13148-015-0152-7 (2015).
Maltby, V. E. et al. Differential methylation at MHC in CD4+ T cells is associated with multiple sclerosis independently of HLA-DRB1. Clin Epigenetics 9, 71, https://doi.org/10.1186/s13148-017-0371-1 (2017).
Bos, S. D. et al. Genome-wide DNA methylation profiles indicate CD8+ T cell hypermethylation in multiple sclerosis. PLoS One 10, e0117403, https://doi.org/10.1371/journal.pone.0117403 (2015).
Baranzini, S. E. et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 464, 1351–1356, https://doi.org/10.1038/nature08990 (2010).
Martin, R., Sospedra, M., Rosito, M. & Engelhardt, B. Current multiple sclerosis treatments have improved our understanding of MS autoimmune pathogenesis. Eur J Immunol 46, 2078–2090, https://doi.org/10.1002/eji.201646485 (2016).
Lehmann-Horn, K., Kinzel, S. & Weber, M. S. Deciphering the Role of B Cells in Multiple Sclerosis-Towards Specific Targeting of Pathogenic Function. Int J Mol Sci 18, https://doi.org/10.3390/ijms18102048 (2017).
Hauser, S. L. et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 358, 676–688, https://doi.org/10.1056/NEJMoa0706383 (2008).
Kappos, L. et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 378, 1779–1787, https://doi.org/10.1016/S0140-6736(11)61649-8 (2011).
Thompson, A. J. et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol, https://doi.org/10.1016/S1474-4422(17)30470-2 (2017).
Liu, Y. & Schmidt, B. Long read alignment based on maximal exact match seeds. Bioinformatics 28, i318–i324, https://doi.org/10.1093/bioinformatics/bts414 (2012).
Schubeler, D. Function and information content of DNA methylation. Nature 517, 321–326, https://doi.org/10.1038/nature14192 (2015).
Patsopoulos, N. A. et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet 9, e1003926, https://doi.org/10.1371/journal.pgen.1003926 (2013).
International Multiple Sclerosis Genetics, C. et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 357, 851–862, https://doi.org/10.1056/NEJMoa073493 (2007).
International Multiple Sclerosis Genetics, C. et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219, https://doi.org/10.1038/nature10251 (2011).
Romme Christensen, J. et al. Cellular sources of dysregulated cytokines in relapsing-remitting multiple sclerosis. J Neuroinflammation 9, 215, https://doi.org/10.1186/1742-2094-9-215 (2012).
Matusevicius, D. et al. Multiple sclerosis: the proinflammatory cytokines lymphotoxin-alpha and tumour necrosis factor-alpha are upregulated in cerebrospinal fluid mononuclear cells. J Neuroimmunol 66, 115–123 (1996).
Selmaj, K., Raine, C. S., Cannella, B. & Brosnan, C. F. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J Clin Invest 87, 949–954, https://doi.org/10.1172/JCI115102 (1991).
Nestor, C. E. et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res 22, 467–477, https://doi.org/10.1101/gr.126417.111 (2012).
Pastor, W. A. et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473, 394–397, https://doi.org/10.1038/nature10102 (2011).
Ficz, G. et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473, 398–402, https://doi.org/10.1038/nature10008 (2011).
Jin, S. G., Wu, X., Li, A. X. & Pfeifer, G. P. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res 39, 5015–5024, https://doi.org/10.1093/nar/gkr120 (2011).
Yokley, B. H., Selby, S. T. & Posch, P. E. A stimulation-dependent alternate core promoter links lymphotoxin alpha expression with TGF-beta1 and fibroblast growth factor-7 signaling in primary human T cells. J Immunol 190, 4573–4584, https://doi.org/10.4049/jimmunol.1201068 (2013).
Traiffort, E., O’Regan, S. & Ruat, M. The choline transporter-like family SLC44: properties and roles in human diseases. Mol Aspects Med 34, 646–654, https://doi.org/10.1016/j.mam.2012.10.011 (2013).
Moser, B. CXCR5, the Defining Marker for Follicular B Helper T (TFH) Cells. Front Immunol 6, 296, https://doi.org/10.3389/fimmu.2015.00296 (2015).
Inoue, M. et al. An interferon-beta-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat Neurosci 19, 1599–1609, https://doi.org/10.1038/nn.4421 (2016).
Sommer, K. et al. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity 23, 561–574, https://doi.org/10.1016/j.immuni.2005.09.014 (2005).
Pistono, C. et al. What’s new about oral treatments in Multiple Sclerosis? Immunogenetics still under question. Pharmacol Res 120, 279–293, https://doi.org/10.1016/j.phrs.2017.03.025 (2017).
Herber, D. et al. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol 178, 3822–3830 (2007).
Jungel, A. et al. Expression of interleukin-21 receptor, but not interleukin-21, in synovial fibroblasts and synovial macrophages of patients with rheumatoid arthritis. Arthritis Rheum 50, 1468–1476, https://doi.org/10.1002/art.20218 (2004).
Ozaki, K. et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol 173, 5361–5371 (2004).
Mehta, D. S. et al. IL-21 induces the apoptosis of resting and activated primary B cells. J Immunol 170, 4111–4118 (2003).
Parkes, M., Cortes, A., van Heel, D. A. & Brown, M. A. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet 14, 661–673, https://doi.org/10.1038/nrg3502 (2013).
Julia, A. et al. Epigenome-wide association study of rheumatoid arthritis identifies differentially methylated loci in B cells. Hum Mol Genet 26, 2803–2811, https://doi.org/10.1093/hmg/ddx177 (2017).
Acknowledgements
This study was supported by the John Hunter Charitable Trust. R.A.L., V.E.M. and K.A.S. are supported by fellowships from Multiple Sclerosis Research Australia. K.A.S. was supported by a scholarship from the Trish foundation. V.E.M. is supported by a fellowship from the Canadian Institutes of Health Research. We would like to thank the MS patients and clinical team at the John Hunter Hospital MS clinic who participated in this study and the Australia Red Cross Blood Service for providing healthy control samples. We also acknowledge the Analytical Biomolecular Research Facility at the University of Newcastle for flow cytometry support and the Australian Genome Research Facility for performing the bisulfite conversions and hybridizations to the Illumina 450 K arrays.
Author information
Authors and Affiliations
Contributions
V.E.M. performed experiments, was involved in interpretation of the data, wrote the manuscript and revised all versions of the manuscript. R.A.L. and M.C.B. performed data analysis, interpreted the data, and critically reviewed the manuscript. M.C.G. contributed to the original study design, performed experiments, and critically reviewed the manuscript. K.A.S. performed experiments and critically reviewed the manuscript. L.T. contributed to initial study design and critically reviewed the manuscript. J.L.S. and R.J.S. initiated and designed the original study, they critically reviewed the manuscript and are responsible for the infrastructure in which in the study was conducted. J.L.S. supervised all aspects of the study.
Corresponding author
Ethics declarations
Competing Interests
Dr. Lechner-Scott’s institution receives non-directed funding as well as honoraria for presentations and membership on advisory boards from Sanofi Aventis, Biogen Idec, Bayer Health Care, Merck Serono, Teva and Novartis Australia.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maltby, V.E., Lea, R.A., Graves, M.C. et al. Genome-wide DNA methylation changes in CD19+ B cells from relapsing-remitting multiple sclerosis patients. Sci Rep 8, 17418 (2018). https://doi.org/10.1038/s41598-018-35603-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35603-0
Keywords
This article is cited by
-
Identification and protective role of CD34+ stromal cells/telocytes in experimental autoimmune encephalomyelitis (EAE) mouse spleen
Histochemistry and Cell Biology (2023)
-
Epigenetic regulation of B cells and its role in autoimmune pathogenesis
Cellular & Molecular Immunology (2022)
-
Epigenome-wide association studies: current knowledge, strategies and recommendations
Clinical Epigenetics (2021)
-
Immune Memory in Aging: a Wide Perspective Covering Microbiota, Brain, Metabolism, and Epigenetics
Clinical Reviews in Allergy & Immunology (2021)
-
DNA methylation changes in metabolic and immune-regulatory pathways in blood and lymph node CD4 + T cells in response to SIV infections
Clinical Epigenetics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
