
Abstract
Chronic obstructive pulmonary disease (COPD) comprises multiple phenotypes such as airflow obstruction, emphysema, and frequent episodes of acute worsening of respiratory symptoms, known as exacerbations. The goal of this pilot study was to test the usefulness of unbiased metabolomics and transcriptomics approaches to delineate biological pathways associated with COPD phenotypes and outcomes. Blood was collected from 149 current or former smokers with or without COPD and separated into peripheral blood mononuclear cells (PBMC) and plasma. PBMCs and plasma were analyzed using microarray and liquid chromatography mass spectrometry, respectively. Statistically significant transcripts and compounds were mapped to pathways using IMPaLA. Results showed that glycerophospholipid metabolism was associated with worse airflow obstruction and more COPD exacerbations. Sphingolipid metabolism was associated with worse lung function outcomes and exacerbation severity requiring hospitalizations. The strongest associations between a pathway and a certain COPD outcome were: fat digestion and absorption and T cell receptor signaling with lung function outcomes; antigen processing with exacerbation frequency; arginine and proline metabolism with exacerbation severity; and oxidative phosphorylation with emphysema. Overlaying transcriptomic and metabolomics datasets across pathways enabled outcome and phenotypic differences to be determined. Findings are relevant for identifying molecular targets for animal intervention studies and early intervention markers in human cohorts.
Similar content being viewed by others


Introduction
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death in the United States1. In 2015, the economic burden of COPD in the United States through 2020 was estimated to be ~$90 billion2, while in the European Union a recent 2017 release estimated the economic burden to be ~ €48.4 billion3. COPD occurs predominately in smokers; however, only 10–50% of smokers develop COPD4. Current therapy is focused on treating symptoms such as chronic cough and excessive sputum production, and preventing exacerbations. There are no therapies that reduce the progression of disease or mortality. Diagnosis and treatment are complicated because COPD may manifest as multiple phenotypes including airway obstruction, emphysema (destruction of alveoli), and frequent exacerbations. Identifying biomarkers and pathways that distinguish COPD phenotypes can potentially lead to more specific treatments.
In addition to clinical challenges, there remain significant gaps in understanding the pathways and mechanisms involved in the pathogenesis of COPD phenotypes. Moreover, there are inherent challenges in clinical omics-based research that make studying COPD difficult, including (1) complexity of patient stratification across multiple phenotypes5, (2) obtaining suitable numbers of study subjects across phenotypes, (3) lack of validated informatics tools and strategies for large clinical datasets6,7, and (4) limitations within the omics technological platforms. The latter includes challenges in integrating clinical metadata with multiple datasets from functional genomics, proteomics, and/or metabolomics sources; i.e. a true “systems approach”.
Some progress has been made in the fields of functional genomics and metabolomics; however, no discovery-based approach has yet resulted in validated clinical biomarkers. For example, genetic studies previously identified alpha-1 antitrypsin in 1964 as a cause of COPD8; however alpha-1 antitrypsin deficiency accounts for only 1% of COPD cases9. Most recently, proteomic approaches have identified new biomarkers such as plasma sRAGE for the presence and progression of emphysema10,11. Metabolomics approaches have identified potential markers of disease severity or therapeutic candidates such as purines12, sphingolipids13, glycerophospholipids14, and amino acids which differentiate patients with or without emphysema and/or cachexia15. While these omic-centric studies have added to the knowledge base, only limited information is obtained. A systems- or pathways-based approach could further pinpoint potential mechanisms by integrating datasets and thereby increasing statistical power. This approach has been used in plant and bacterial studies and has recently been applied to human disease. For example, integrating functional genomics and metabolomics identified markers associated with poor outcome in human neuroendocrine cancers16, determined predictors of asthma control17, and determined novel therapeutic targets in lung cancer18.
While single ‘omic studies have helped to address the knowledge gap, to date no study has integrated transcriptomics and metabolomics from a COPD cohort to determine if specific pathways are associated with individual COPD outcomes. This could be a useful strategy since functional genomics measures the association between genes, transcripts and clinical phenotype19 and metabolomics provides a readout of the physiological state of the human body20. Therefore, we used metabolomics and transcriptomics to identify markers associated with six COPD health outcomes, then mapped the significant results to pathways for each outcome. The overall goal is to identify dysregulated pathways that distinguish outcomes.
The novelty and strength of our study lies in the uniqueness of the highly phenotyped and matched samples across several stages of COPD severity. Using transcriptomics and metabolomics datasets provides information at a molecular level that is not available when only a single profiling technology is used. Determining how the molecular basis of disease phenotypes vary will improve treatment options and clinical outcomes since unique therapies can be adapted to each patient based on their individual phenotype.
Results
Cohort description
Clinical characteristics are provided in Table 1. Exacerbation frequency is the total number of exacerbations (0–6) in the year prior to enrollment. Exacerbation severity is the total number of severe exacerbations (0–3) in the year prior to enrollment. Age was uniform across Global Initiative for Chronic Obstructive Lung Disease (GOLD) Stages. Gender was balanced across males (n = 79) and females (n = 70). Clinical phenotypes were associated with GOLD stage, smoking pack-years, asthma, and current smoking status.
Approximately 10–15% of the current and former smoker population falls into the Preserved Ratio, Intact Spirometry (PRISm)21 category. Studies in COPDGene have found that these subjects are not as healthy as control subjects21 and it is not clear where these patients fit in the spectrum of COPD. We therefore included PRISm subjects in the transcriptomics analyses since patients within that category presented with outcomes such as exacerbations or emphysema. None of the PRISm subjects overlapped with the metabolomics dataset and were therefore not included in the metabolomics analysis.
Individual outcome associations
We evaluated 12,531 transcripts and 2,999 metabolites for associations with COPD outcomes (Fig. 1). The highest number of significant transcript differences was found in FEV1% predicted (1,652) while no transcript differences were associated with exacerbation severity. FEV1/FVC had the most significant metabolites (269) while emphysema and BDR had none. There was minimal overlap in the significant transcripts (Fig. 2A) and metabolites (Fig. 2B) associated with each outcome.

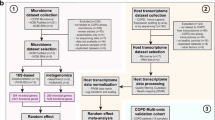
Flow chart summarizing the omics methods and analysis. Peripheral blood mononuclear cells (PBMC) (n = 136 human subjects) and matched plasma (n = 131) were prepared and analyzed using functional genomics and metabolomics, respectively. Metabolomics samples were analyzed randomly in triplicate. Regression model fitting in R resulted in a list of statistically significant transcript probes and metabolites associated with each outcome. These significant transcript probes and metabolites were mapped to pathways using IMPaLA. Pathway p-values for metabolites and transcripts were combined by a meta-analysis using Fisher’s method to obtain a single p-value and FDR for each pathway. In the outcome tables, ‘# sig’ refers to the number of statistically significant transcripts probes and metabolites. BMI: body mass index, FEV1: forced expiratory volume in 1 second, FVC: forced vital capacity.

Venn diagrams. (A) Edwards’ Venn diagram showing the overlap of significant transcript probes (p ≤ 0.015, FDR ≤ 0.1) across COPD outcomes. (B) Edwards’ Venn diagram showing the overlap of statistically significant metabolites (p ≤ 0.05, FDR ≤ 0.15) across COPD outcomes. (C) Classic Venn diagram showing the overlap of statistically significant pathways (p ≤ 0.05, FDR ≤ 0.2, ≥3 hits). The overlapping pathways are shown in red arrows. The unique pathways are listed in Table 4. %Emphy: % emphysema, Exac Freq: exacerbation frequency, Exac Sever: exacerbation severity, BDR: bronchodilator response, FEV1: Forced expiratory volume in 1 second, FVC: Forced vital capacity, FEV1%Pred: FEV1% predicted.
The top gene transcripts for each outcome based on smallest p-value and FDR are listed in Table 2. ACTG1, a cytoplasmic protein found in all cell types22, was associated with seven transcripts, all with increased expression with increasing exacerbation frequency. For emphysema, the top gene transcripts were all associated with a decrease with worsening emphysema. These genes are associated with apoptosis (BCL10, SUMO2), cancer (CCDC186, RECQL), regulation of dietary iron absorption (CYBRD1), lipid binding (OSBPL8), protein transport (TMCO3), protein binding (TOPORS), and the mitochondria (SLC25A43, TFAM)22. The gene interactions among these significant associations were investigated, with FEV1/FVC having the most interactions (Supplementary Document S1).
For the metabolomics analysis, only significant compounds that were confidently identified are shown in Table 3. A large number of amino acids were associated with exacerbation frequency while both amino acids and carbohydrates associate with exacerbation severity. Predominantly glycerophospholipids are associated with FEV1/FVC, and several lipid classes associate with FEV1% predicted. Carnitine(C14:2) was increased with increasing exacerbation frequency, acetylcarnitine* was decreased with increasing exacerbation severity, and octanoylcarnitine** was decreased with COPD for FEV1/FVC. The metabolite interactions for the significant associations are shown in Supplementary Document S1. Interactions were present for exacerbations and absent for lung function.
Specific metabolites were associated with disease progression based on CT scan and FEV1
Clinical data from quantitative computerized tomography (CT) scans and lung function measurements from each patient was used to quantify disease progression. This clinical data was incorporated with metabolomics to identify small molecule changes associated with disease severity. There were 90 significant compounds in the emphysema progression analysis compared to 142 significant compounds in FEV1 progression analysis. Three compounds overlapped between the two comparisons. These were PC(36:4)* (% emphysema p = 0.0016, FEV1% predicted = 0.034), PE-Cer(d16:1/18:0) (% emphysema p = 0.0272, FEV1% predicted = 0.019), and an unannotated compound at mass 771.5351 Da (% emphysema, p = 0.0215, FEV1% predicted = 0.0489). PC(36:4)* and PE-Cer(d16:1/18:0) are negatively correlated with emphysema progression and positively correlated with FEV1 progression, while the unannotated compound was negatively correlated with both emphysema and FEV1 progression.
Pathway associations
Fifteen pathways were significantly associated with FEV1/FVC, 22 pathways with FEV1% predicted, 7 with exacerbation frequency, and 15 with exacerbation severity (Table 4). The transcript expressions for the significant pathways for BDR were all increased (p ≤ 0.0013, FDR ≤ 0.0954) post-BDR treatment; for phagosome these were ATP6V0B, ATP6V1F, FCGR2A, CYBB, HLA-DMA, RAC1, SCARB1, TUBA1B, TUBA1C, and for lysosome these were ATP6V0B, NPC2, NAGA, GAA, GUSB, HEXA, PSAP.
Three pathways were unique to FEV1/FVC (Fig. 2C), with fat digestion and absorption being the most significant. Nine pathways were unique to FEV1% predicted, with T cell receptor signaling as the most significant. There was one unique pathway associated with exacerbation frequency (antigen processing and presentation). There were eight unique pathways related to exacerbation severity, with arginine and proline metabolism as the most significant. Sphingolipid metabolism was significant in exacerbation severity with ganglioside GD3 (d18:0/23:0), lactosylceramide (d18:1/14:0), and the glycosphingolipid Fucα1–4GlcNAcβ1-3Galβ1-3GlcNAcβ1-3Galβ1-4Glcβ-Cer(d18:1/22:0) positively associated (FDR < 0.0001) with disease severity. Sphingolipid metabolism was also significant in the lung function outcomes (FDR < 0.0001), with two glycosphingolipids positively associated with FEV1% predicted (lung size), and three glycosphingolipids and four ceramides positively associated with FEV1/FVC (airflow obstruction). Emphysema was related to two unique pathways (oxidative phosphorylation and mRNA surveillance pathway). There were no unique pathways associated with BDR. Individual pathway entities are available in Supplementary Document S2.
Transcriptome and metabolome pathways
Oxidative phosphorylation was uniquely associated with emphysema and therefore examined more closely. All gene transcripts were decreased (Fig. 3A) and are located in complex I, II, and IV. Likewise, antigen processing and presentation (Fig. 3B) was uniquely associated with exacerbation frequency. This pathway comprises two sub-pathways (MHC I and MHC II), however only MHC I was significantly perturbed with an overall decrease. Eight unique metabolome pathways were associated with exacerbation frequency, two of which are amino acid-related metabolic pathways; arginine and proline metabolism was decreased (Fig. 4A) and glycine, serine and threonine metabolism was increased (Fig. 4B).

Transcriptomics pathways. (A) Oxidative phosphorylation in emphysema. (B) Antigen processing and presentation for exacerbation frequency. Blue stars indicate: associated with a decrease with worsening outcome. Purple stars represent both an increase or decrease in expression if multiple transcripts are mapped to a single gene. Pathway image is modified from KEGG85,86,87.

Metabolomics pathways. (A) Arginine and proline metabolism in exacerbation severity. (B) Glycine, serine and threonine metabolism in exacerbation severity. Red boxes indicate associated with an increase with worsening outcome and blue boxes indicate associated with a decrease with worsening outcome.
Energy and cellular degradation pathways
Pathways associated with exacerbation severity and exacerbation frequency differed to some extent; therefore, HumanCyc23 was used to determine what was responsible for the differences. Exacerbation severity comprised eight degradation pathways and three energy pathways, while exacerbation frequency comprised three degradation pathways and no perturbed energy pathways (Supplementary Document S3).
Integrated transcriptome/metabolome analysis
Three pathways were examined to determine association between gene transcripts and metabolites. Glycerophospholipid metabolism was based on FEV1% predicted (Fig. 5A). As disease severity increases from GOLD 0-GOLD 4, eight gene transcripts (CDS2, CHKB, CRLS1, DGKA, DGKE, GNPAT, LPIN1, PLA2G6) were decreased (p ≤ 0.015, FDR ≤ 0.1) and five gene transcripts (CEPT1, ETNK1, GPCPD1, LPCAT2, PISD) were increased (p ≤ 0.015, FDR ≤ 0.1). Lipids that decreased with disease severity were LysoPE(16:0)*, DG(36:2)*, PC(36:2)*, PE(P-16:0), PI(41:1) (FDR ≤ 0.05), and LysoPC(20:0)* (p = 0.0018, FDR = 0.15). Conversely, PG(29:2), PS(39:7), and TG(42:0) increased (FDR ≤ 0.05) with disease severity.

Integrated transcriptomics and metabolomics pathway diagrams. (A) Glycerophospholipid metabolism in FEV1% predicted. (B) Fat digestion and absorption in FEV1/FVC modified from KEGG85,86,87. FA: fatty acid, BA: bile acid, PA: phosphatidic acid, PL: phospholipids, MAG: monoglycerides, DAG: diglycerides, TAG: triglycerides, CE: cholesterol ester, CL: cholesterol. Red font and red boxes indicate associated with an increase with worsening disease. Blue font and blue boxes indicate associated with a decrease with worsening disease. Black font or uncolored boxes indicate no statistical significance.
Fat digestion and absorption (Fig. 5B) was uniquely significant in FEV1/FVC with an associated increase with disease of triglycerides, cholestrol esters, phospholipids, and bile acids, and a decrease in monoglycerides, diglycerides, and docosahexaenoic acid (DHA) with disease. The SCARB1 gene located in the brush border-membrane, and ABCA1 located in the basolateral membrane, were associated with an increase in expression in COPD subjects compared to healthy controls. The protein encoded by SCARB1 is a plasma membrane receptor for high density lipoprotein cholesterol (HDL)24. Lastly, glycerolipid metabolism, also uniquely significant in FEV1/FVC revealed an increased association with phosphatidic acid and the AGPAT9 gene that encodes it (Supplementary Document S4).
Pathway relationships
To determine whether one or multiple pathways were driving the outcome perturbations, pathways were plotted based on KEGG25. An integrated pathway map (Fig. 6) demonstrates that MAPK and PI3K-AKT signaling pathways, although not statistically significant, were each connected to eight pathways. In comparison, lysosome and T cell receptor signaling pathway each had six connections while purine metabolism and glycine, serine and threonine metabolism each had five connections.

Overview of outcome perturbations and pathway relationships. The connections between the pathways were mapped using KEGG. White boxes indicate no statistical significance, while colored boxes indicate significance based on outcome as described in the Legend.
Discussion
Overlaying the significant transcripts and compounds using Venn diagrams allowed us to view the differences amongst the various outcomes in a discernable manner. For example, while a larger overlap between FEV1/FVC and FEV1% predicted might be expected, we observed minimal overlap. This is however not surprising because although these two outcomes are related, especially in a COPD cohort, their measures are different. FEV1/FVC ratio is a measure of obstruction and is required in the COPD diagnosis, while FEV1% is a measure of lung size. Therefore, a patient can have small lungs (FEV1%) and still have no obstruction and occasionally is the reverse (for mild COPD). Also, not surprising is the minimal overlap across all the tested outcomes since COPD is a heterogeneous disease. This approach was important in allowing us to distinguish phenotypes and outcomes.
Overall, a metabolomic signature differentiated the COPD outcomes, which was supported in the transcriptomics analysis for corresponding pathways. Both omics datasets demonstrated that specific COPD phenotypes and outcomes are associated with different transcriptome and metabolome pathways. This supports the premise that distinct COPD phenotypes have distinct mechanisms. There were also differences in the types of omics associations. For example, emphysema appears to have a larger number of gene associations compared to exacerbation severity, which has more metabolic associations. Therefore, we chose to focus the discussion on the most significant and/or unique pathways for each outcome, as a means of narrowing down the large amount of data generated from this study.
We first interpreted exacerbations as a group and observed that for exacerbation frequency, there are no significant differences in the energy pathways and very few associations in the degradation pathways. Conversely, for exacerbation severity, the energy pathways are more dysregulated and accompanied by an increase in the number of degradation pathways, particularly carbohydrate degradation, nucleoside degradation, fatty acid degradation, and amino acid degradation. This is supported by the energy association in degradation pathways; carbohydrate breakdown provides a source of cellular energy for other syntheses such as protein transport, nucleosides act as signaling molecules or provide chemical energy to cells, and fatty acids store energy in the cell26. We then observed that sphingolipid metabolism was significant in exacerbation severity. In our previous studies, we observed that trihexosylceramides, a subclass of sphingolipids, were positively associated with severe exacerbations13.
Interestingly we also observed for exacerbations that while antigen processing and presentation was unique to exacerbation frequency, only the MHC I pathway (leading to CD8 T cell activation) and not the MHC II pathway (leading to CD4 T cell activation) is affected. The activation of CD4 T cells is beneficial to sustaining memory of CD8 T cells following acute infection27. Since COPD exacerbations are associated with viral28,29 and bacterial30,31 infections, the perturbation of CD8 but not CD4 in our study suggests that a crucial connection is not being made, and this may predispose patients to frequent exacerbations as their immune system is weakened. An alternative explanation is that CD4 T cells were activated and CD8 T cells are now destroying the viral or bacterial pathogen in the more frequent exacerbations. Interestingly, an outcome-pathway shift is observed where the perturbed genes within the MHC I pathway for exacerbation frequency end at the T cell receptor signaling pathway which is uniquely perturbed in FEV1% predicted.
We then examined the emphysema phenotype. Oxidative phosphorylation was unique to and significant in the emphysema outcome. Cigarette smoking (CS) is a major cause of emphysematous COPD. CS is known to produce reactive oxygen species (ROS). Excessive production of ROS can lead to oxidative damage, leading to cell apoptosis32. In a recent study, exposure of human lung fibroblasts to various doses of nicotine and e-cigarette condensate inhibited myofibroblast differentiation and inhibited oxidative phosphorylation complex III33. Therefore, in spite of adjusting for smoking status, our results are not unexpected as our cohort is comprised of current and former smokers.
Next, we focused on the BDR outcome. Unlike the other outcomes, BDR was associated with two cytoplasmic pathways, lysosome and phagosome. Lysosomes break down proteins, nucleic acids, carbohydrates and lipids26. Phagosomes fuse with lysosomes allowing the ingested material to be digested; some of the material is recycled to the plasma membrane26. The perturbed transcripts in these pathways showed increased expression and may serve as markers of airflow reversibility.
We then examined FEV1% predicted outcome. Glycerophospholipid metabolism, although significant in many of the other outcomes, was highly significant in this lung function outcome. These lipids play a role in respiratory infections34 and in asthma and COPD35 since they constitute lung surfactant36,37,38. Surfactant, a complex combination of proteins and ~90% lipids39, is secreted by the airway epithelial cells into the airspace to reduce surface tension and provides a barrier to pathogens. In the FEV1% predicted outcome, the lipids classes LysoPE and PG, decreased and increased respectively, and have been previously suggested as markers of lung remodeling40. Their associated genes PLA2G6 and LPCAT2 were also decreased and increased respectively and could serve as targets in future studies to repair disrupted surfactant composition.
Sphingolipids were also highly associated with this outcome and FEV1/FVC. This is congruent with our recent publications that discuss in depth the significance of sphingolipids to COPD pathogenesis, including signaling related to structural cell fate, innate immune responses, and lymphocyte trafficking41,42. Other pathways uniquely and significantly associated with the FEV1/FVC was involved in fat digestion and absorption, typicaly reflecting perturbations between the intestine and blood. SCARB1 may be driving the cholesterol (CL) and cholesterol ester (CE) transport and regulation while the phospholipids (PL) may be driving ABCA1. Since bile acids and triglycerides were increased with disease, and the omega-3 fatty acid DHA was decreased with disease, this pathway suggests a diet or nutrition component associated with disease. This is supported by a recent publication on a cohort of 34,739 women where long-term consumption of fruits was inversely assocated with COPD incidence43. Diet has been suggested to play a role as a risk factor for many chronic diseases including COPD44,45, and suggestions have been made on the role of nutritional supplement therapy in the treatment of COPD46.
Finally, we attempted to deconstruct the results into a pathway map where relations could be observed among the outcomes and pathways. Based on the overview pathway map, MAPK and PI3K-AKT signaling pathways appeared to be the unifying links among many of the perturbed pathways. In a recent study exposing human bronchial epithelial cells to urban particulate matter, airway inflammation was triggered via activation of MAPK and NF-κB signaling pathways47. Although MAPK signaling was not significant in our study, NF-κB signaling was significant in FEV1% predicted. MAPK and PI3K-AKT signaling pathways may therefore be the driving force for downstream events. Researchers have already begun exploring these targets48,49,50,51.
A limitation of our study is the use of plasma and PBMCs as these may not directly translate to the lung environment. However, our previous studies, both published and unpublished, have observed a high degree of congruency for compounds in lung lavage fluid, lung tissue and plasma in both human and animal studies52,53. Also non-fasting samples may affect results as food intake affects the plasma metabolite profile54, We therefore used stringent statistics to filter out many of the diet-associated compounds. Lastly, patients in this study were taking medications, which is unavoidable with this type of cohort. However, we did our best to filter those medications from the final dataset prior to analysis. Future work would incorporate lavage fluid and increase sample numbers to gain more power for discriminating metabolite-transcript-phenotype differences. Targeted quantitative analysis of pathway-specific molecules could provide increased power and more quantitative results. Although these results need to be validated in a larger cohort, they point to therapeutic targets to be tested in animal experiments, and the most significant species from each outcome could serve as non-invasive markers of disease outcomes.
Despite these limitations, we were able to determine unique pathways that can be leveraged for treatment targets. For example, oxidative phosphorylation was decreased with worsening emphysema. It is perturbed due to cigarette smoke exposure33; cigarette smoking is a major cause of emphysema and produces reactive oxygen species, leading to oxidative damage. Interventions such as rotenone, antimycin A, or oligomycin can be used to inhibit oxidative phosphorylation33,55,56, while other small molecule interventions can be used to activate the pathway. Autophagy was highly significant for FEV1/FVC and can be inhibited using, 3-methyladenine57,58. Another pathway that can be leveraged is antigen processing and presentation. This pathway, decreased in exacerbation frequency and is dysregulated in a rat model of COPD59. Therefore, activating these pathways using either small molecules or gene interventions in animal models could be beneficial in developing phenotype-specific COPD treatments to reduce or halt lung damage or viral load. These pathway interventions in mouse models have the potential to lead to phenotype-specific treatment targets. Other follow up studies could include targeted analysis in COPD or smoking cohorts such as COSYCONET60, Birmingham61, SPIROMICS62, or KORA63,64.
Conclusion
Our pilot study has shown that using a pathway enrichment approach to analyze transcriptomics and metabolomics data from matched samples, identified perturbed biological pathways associated with COPD outcomes and identified pathways that distinguish between outcomes. In addition, our results show that the various phenotypes and outcomes have distinct pathways associated with each one, in spite of COPD being a heterogenous disease. Investigators may choose to select the strongest pathways (those with low p-values) for follow up analyses using either mouse intervention studies via knockouts, mouse treatment studies, or using targeted analysis in other COPD or smoking cohorts. The long term clinical applicability of this work would be in the use of blood-based markers where a simple blood panel could identify patient outcomes and indicate patient-targeted treatment. This work could help in the management of COPD.
Methods
Ethics statement
All methods were performed in accordance with the relevant guidelines and regulations. Human subjects were from the COPDGene cohort65 which is a National Institute of Health–sponsored multicenter study of the genetic epidemiology of COPD. COPDGene was approved by the institutional review board at each participating center; all subjects were enrolled from January 2008 to April 2011 and provided written informed consent. The current analysis was approved by the National Jewish Health Institutional Review Board.
Study population
The COPDGene study enrolled 10,192 non-Hispanic White and Black individuals, aged 45–80 years old with at least a 10 pack-year history of smoking, who had not had an exacerbation of COPD for at least the previous 30 days. Additional information on the COPDGene study and the collection of clinical data has been previously described65. There were ten Preserved Ratio, Intact Spirometry (PRISm)21 subjects included in the transcriptomics analysis since these subjects presented with exacerbations or emphysema but are considered an unclassified. Patients who were pregnant or with co-morbidities such as a history of other lung disease (except asthma), active cancer under treatment, and other cardiac hospitalizations, were excluded from the study65. Non-fasting blood was collected from a random sampling of subjects (n = 149) from a single clinical center (National Jewish Health) that represented the entire cohort and used for omics analysis. Peripheral blood mononuclear cells (PBMC) from 136 subjects were used for functional genomics and fresh frozen plasma from 131 subjects were used for metabolomics, with an overlap of 118 subjects for both omics technologies. The cohort characteristics are shown in Table 1.
Clinical data collection and definitions of COPD outcomes
The clinical data included forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC), FEV1 percent (%) predicted, percent (%) emphysema on CT scan10, exacerbation frequency, exacerbation severity, and bronchodilator response (BDR). COPD was defined as post bronchodilator ratio of FEV1/FVC < 0.70. Current or former smokers without spirometry evidence of airflow obstruction (FEV1/FVC ≥ 0.70) were classified as controls66. Subjects with without airflow obstruction but FEV1 < 80% of predicted where classified as (PRISm)21. Emphysema was measured using quantitative high resolution CT as previously described67 and quantified as the percent of lung attenuation lung voxels (LAA) ≤−950 HU on the inspiratory images for the whole lung. Emphysema was further categorized as none (LAA ≤ 5%), mild (5% < LAA ≤ 10%), moderate (10% < LAA ≤ 20%), or severe (20% < LAA). Exacerbations (flare ups) of COPD are characterized by acutely worse cough, sputum, and dyspnea. Only moderate exacerbations (treated by corticosteroids or antibiotics) or severe exacerbations (causing an admission to the hospital) were evaluated. Patients had no exacerbations for at least 30 days prior to sample collection. BDR determined the reversibility of airway obstruction by measuring FEV1 before and after the administration of bronchodilator medication. A significant response was defined as an increase in FEV1 of 12% and 200 mL increase68.
Sample collection
Blood was drawn into a BD Vacutainer Cell Preparation Tube for peripheral blood mononuclear cells (PBMC) and plasma as previously described65. PBMCs were used for gene expression profiling using the Affymetrix Human Genome U133 plus 2.0 Gene Array, (GEO accession number GSE42057)69. Plasma was used for untargeted liquid-chromatography mass spectrometry (LC-MS)-based metabolomics.
Chemicals, standards and reagents
All solvents were LC-MS grade. Water and isopropyl alcohol were purchased from Honeywell Burdick & Jackson (Muskegon, Michigan); chloroform, acetonitrile, methanol, acetic acid, low retention microcentrifuge tubes, serological pipettes were purchased from Fisher Scientific (Fair Lawn, New Jersey); plastic pipette tips were purchased from USA Scientific (Orlando, Florida); methyl tert-butyl ether was purchased from J.T. Baker (Central City, Pennsylvania); internal standards were purchased from Avanti Polar lipids Inc. and Sigma Aldrich (St. Louis, MO); pyrex glass culture tubes were purchased from Corning Incorporated (Corning, New York).
Sample preparation
Blood samples for functional genomics analysis were prepared as previously described69. Briefly, blood was collected from subjects, processed within 30 minutes and peripheral blood mononuclear cells (PBMC) were isolated from the supernatant for RNA isolation as previously described69. Fresh frozen plasma was isolated using P100 tubes as previously described70 and stored at −80 °C until sample preparation for untargeted liquid-chromatography mass spectrometry (LC-MS)-based metabolomics. For LC-MS, 100 µL of each sample underwent protein precipitation using methanol, followed by liquid-liquid extraction using methyl-tert butyl ether as previously described71,72 to obtain an aqueous fraction and a lipid fraction. All samples were prepared randomly to avoid batch effects from phenotype, gender, or GOLD stage.
Liquid chromatography mass spectrometry
The samples from the lipid fraction were analyzed randomly and each sample vial was run randomly in triplicate using an Agilent 1290 series pump with an Agilent Zorbax Rapid Resolution HD (RRHD) SB-C18, 1.8 micron (2.1 × 100 mm) analytical column and an Agilent Zorbax SB-C18, 1.8 micron (2.1 × 5 mm) guard column. The autosampler tray temperature was set at 4 °C, column temperature was set at 60 °C, and the sample injection volume was 4 µL. The flow rate was 0.7 mL/min with the following mobile phases: mobile phase A was water with 0.1% formic acid, and mobile phase B was 60:36:4 isopropyl alcohol:acetonitrile:water with 0.1% formic acid. Gradient elution was as follows: 0–0.5 minutes 30–70% B, 0.5–7.42 minutes 70–100% B, 7.42–9.9 minutes 100% B, 9.9–10.0 minutes 100–30% B, followed by column re-equilibration. The lipid fraction mass spectrometry conditions were as follows: Agilent 6210 Time-of-Flight mass spectrometer (TOF-MS) in positive ionization mode with dual electrospray (ESI) source, mass range 60–1600 m/z, scan rate 2.03, gas temperature 300 °C, gas flow 12.0 L/min, nebulizer 30 psi, skimmer 60 V, capillary voltage 4000 V, fragmentor 120 V, reference masses 121.050873 and 922.009798 (Agilent reference mix).
The samples from the aqueous fraction were analyzed randomly with each sample vial run randomly in triplicate using an Agilent 1200 series pump using a Phenomenex Kinetex HILIC, 2.6 µm, 100 Å (2.1 × 50 mm) analytical column and an Agilent Zorbax Eclipse Plus-C8 5 µm (2.1 × 12.5 mm) narrow bore guard column. The autosampler tray temperature was set at 4 °C, column temperature was set at 20 °C, and the sample injection volume was 1 µL. The flow rate of 0.6 mL/min with the following mobile phases: mobile phase A was 50% ACN with pH 5.8 ammonium acetate, and mobile phase B was 90% ACN with pH 5.8 ammonium acetate. Gradient elution was as follows: 0.2 minutes 100% B, 0.2–2.1 minutes 100–90% B, 2.1–8.6 minutes 90–50% B, 8.6–8.7 minutes 50–0% B, 8.7–14.7 minutes 0% B, 14.7–14.8 minutes 0–100% B, followed by column re-equilibration. The aqueous fraction mass spectrometry conditions were as follows: Agilent 6520 Quadrupole Time-of-Flight mass spectrometer (Q-TOF-MS) in positive ionization mode with ESI source, mass range 50–1700 m/z, scan rate 2.21, gas temperature 300 °C, gas flow 10.0 L/min, nebulizer 30 psi, skimmer 60 V, capillary voltage 4000 V, fragmentor 120 V, reference masses 121.050873 and 922.009798 (Agilent reference mix).
Tandem mass spectrometry (MSMS)
The HILIC and C18 chromatographic methods discussed above were replicated for LC-MS/MS analysis using 10, 20, and 40 eV collision energies on a 6520 Q-TOF-MS (Agilent) with a 500 ms/spectra acquisition time, 4 m/z isolation width, and 1 minute delta retention time.
Metabolomics quality control
Samples were randomly selected from 2000 subjects from the COPDGene cohort comprising both healthy subjects and COPD patients to create aliquots of pooled QC plasma samples. At least 20 of the 131 subjects in this study were included in the pooled sample. Samples were prepared as described above and pooled for use as quality control (QC) samples to monitor instrument reproducibility across multiple days. These pooled QC samples were injected after every five samples, followed by a solvent blank. Total ion chromatograms of all samples were evaluated for retention time reproducibility and intensity overlap. Instrument QC samples were analyzed to ensure that peak areas of spiked internal standards in the plasma samples were reproducible with coefficient of variations ≤10%.
Metabolomics spectral peaks extraction
A final list of compounds was obtained by collapsing features (e.g. ions, adducts) into compounds as follows: Spectral peaks were extracted using the following parameters in MassHunter software B.07 (Agilent Technologies): Find by Molecular Feature algorithm, single charge, proton, sodium, potassium, ammonium adducts in positive ionization mode. Data were imported into Mass Profiler Professional software 14.5 (MPP, Agilent Technologies) for mass (15 ppm) and retention time alignment (0.2 minutes). Data from sample preparation blanks and instrument blanks were background subtracted to eliminate noise from contaminants. Because LC-MS data can result in missing values, data was further processed using the ‘Find by Formula’ algorithm parameters (+H, +Na, +K, +NH4 adducts for positive ionization mode, charge states limited to 2, and absolute height >3000 counts). The ‘Find by Formula’ algorithm merged multiple features such as ions, adducts and dimers into a single compound. The final data set was then re-imported into MPP for metabolite annotation.
Metabolite annotation and identification
For the first round of identification, ID Browser within the Mass Profiler Professional software v14.5 was used to annotate metabolites with putative identifications. This software utilizes an in-house database comprising data from METabolite LINk (METLIN), Human Metabolome Database (HMDB), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Lipid Maps and applies isotope ratios, accurate mass, chemical formulas, and scores to provide preliminary identifications. A database score ≥70 out of a possible 100 was considered acceptable for annotation confidence. Chosen elements for molecular formula generation were: C, H, N, O, S, and P. An error window of ≤10 ppm was used with a neutral mass range up to 2500 Da and positive ions selected as H, Na, K, and NH4. The database identifications were limited to the top 10 best matches based on score, and charge state was limited to a maximum of 2. There were 1,633 out of 2,999 compounds that were putatively matched to a name in a database.
For improved confidence, only the statistically significant compounds were fragmented as described in the Tandem MS section above, and searched using either an in-house mass, retention time, and MSMS library built from purchased standards, or their MSMS fragments were matched to reference standards from the NIST14 and NIST17 MSMS spectral library73. For compounds where their fragments were not present in a reference spectral library, MetFrag74,75 was used for in silico fragmentation. A list of the confirmed and putative IDs is available in Supplementary Documents S5 and S6.
Reporting level of confidence
Within the manuscript and in Table 3, the symbol * indicates that the compound name was confirmed by accurate mass (<10 ppm) and matching MSMS fragments to the NIST spectral library. The symbol **indicates that the compound name was confirmed by accurate mass (<10 ppm), retention time, and MSMS match to purchased standards. These are MSI 1. All other compound names are MSI 2 and are based on accurate mass (<10 ppm) and isotopic abundance and distribution.
Data processing
Metabolomics
Processing of metabolomics data was performed using the R package MSPrep76. Only compounds identified in at least 2 of 3 sample vial injection triplicates were summarized to obtain one measurement per compound per subject, all others were filtered out. A maximum coefficient of variation of 0.5 was set as a threshold for utilizing the mean of the triplicates or the median of the triplicates. This threshold was selected to account for when the extraction software may have missed a peak, thus resulting in one replicate having low to no signal and therefore causing a larger CV across replicates. The data was then filtered for compounds that were found in at least 80 percent of subjects. Missing data was then imputed using Bayesian Principal Component Analysis (BPCA), which uses linear combinations of principal axis vectors to estimate the missing value and is not sensitive to the level of missing data77. The data was then normalized using the Cross-Contribution Compensating Multiple Standard Normalization (CRMN) method to remove unwanted batch variation78. Following filtering, imputation, and normalization the numbers of metabolites decreased from 1,813 to 662 and 6,183 to 2,337 compounds in the aqueous and lipid fractions respectively, for a total of 2,999 compounds. Many of the compounds that were removed from further analysis were attributed to instrument noise, software extraction artefacts, and single hits from individual subjects.
Functional Genomics
Transcript expression was measured using Affymetrix Human Genome U133 plus 2.0 Gene Array (GEO accession number GSE42057; Affymetrix, Santa Clara, CA). Quality control, batch correction, normalization, filtering, and log transformation was performed as previously described69. Following filtering, transcripts were reduced from 54,675 to 12,531. Subsequent statistical analysis on the reduced transcript list is described below.
Statistical analysis
To assess the significance of transcript and metabolite levels in COPD phenotypes, a series of regression models were fit for each outcome and the form of the model depended on the distribution of the outcome. For FEV1% predicted, a linear regression was used. For FEV1/FVC and % emphysema beta regressions were used. Number of exacerbations was modeled as a negative binomial; since many subjects had no exacerbations, a zero-inflation portion was added. Severity of exacerbations used logistic regression. Since BDR was constrained between 0 (no response) and 1 (positive response), logistic regression was used for BDR. For all models, covariates were included. Covariates for FEV1% predicted were age, BMI, gender, smoking status, parents have COPD, and ATS Pack-years. Covariates for FEV1/FVC were age, gender, asthma, and smoking status. Covariates for % emphysema were age, gender, BMI, FEV1% predicted, and smoking status. Covariates for exacerbation frequency and exacerbation severity were FEV1% predicted, gastro esophageal reflux, St. George’s Respiratory Questionnaire (SGRQ) score, and gender. Covariates for BDR were age, gender, smoking status and asthma. Significant differences within the cohort, such as age and smoking, were controlled for in subsequent analyses.
Then, to assess the significance of various metabolites and transcripts, the log-transformed metabolite or transcript levels were added to the models as the independent variable, and the significance of the effect of these metabolite or transcript levels were reported. For the transcripts, p ≤ 0.015 was considered significant with FDR ≤ 0.1 adjustment. For the metabolites, p ≤ 0.05 was considered significant with FDR ≤ 0.15 adjustment. All analyses were performed in R79.
Pathway enrichment analysis
The significant transcript probes were converted to official gene symbols using DAVID Bioinformatics Database version 6.880 ID conversion tool. The significant compounds were converted to KEGG IDs using Chemical Translation Service81. The lists of statistically significant transcripts and metabolites within each COPD outcome were searched using IMPaLA82 to obtain pathways relevant to each outcome. In IMPaLA, the gene transcripts and associated values were uploaded using ‘gene symbol’, the metabolites and associated values were uploaded in the ‘KEGG’ identifier, and pathway over-representation analysis was performed. A meta-analysis using Fisher’s method provided a combined p-value and a corrected p-value for the pathways. The results were filtered as follows: KEGG pathways, p ≤ 0.05, FDR ≤ 0.2 and ≥3 pathway hits. A more lenient FDR ≤ 0.2 cut-off was used since the individual samples within each group for each outcome or phenotype were small. Figure 1 highlights the experimental procedure used to generate results from the two datasets.
Visualization
Venn diagrams were created to show the overlap of statistically significant transcript probes, compounds, and pathways for the COPD outcomes using the publicly available jvenn program83.
Cellular function relationships for each outcome were visualized using the ‘Omics Dashboard’ of HumanCyc23 (https://humancyc.org/dashboard/dashboard-intro.shtml) using ‘Analysis’. The gene symbols for the significant transcript probes and their coefficients were uploaded in tab-delimited format for expression analysis. The KEGG IDs for the significant compounds and their coefficients were uploaded in tab-delimited format for metabolomics analysis. The interactions between the top gene transcripts and top metabolites were explored using ConsensusPathDB (http://consensuspathdb.org/)84 and selecting ‘over-representation analysis’. The selected identifiers were ‘gene symbol’ and ‘ChEBI’ to determine gene interactions and pathways interactions for the gene transcripts and metabolites, respectively. For the transcriptomics data, protein, genetic, biochemical, and gene regulatory interactions were considered; for protein interactions, only binary protein interactions with high and medium confidence were allowed, while complex interactions were ignored. Intermediate nodes were allowed for all interactions. For the metabolomics data, only KEGG pathways with a minimum overlap of 2 and a p-value cut-off of 0.05 was allowed. Finally, outcome and pathway relationships were visualized using KEGG85 to plot pathways for each outcome.
Data Availability
The mass spectrometry data from this publication has been deposited to the Metabolomics Workbench database http://www.metabolomicsworkbench.org/ and assigned the identifier PR000438. The microarray data from this publication is available in the Gene Expression Omnibus (GEO) database https://www.ncbi.nlm.nih.gov/geo and assigned the identifier GSE4205769.
References
CDC. Chronic obstructive pulmonary disease among adults—United States, 2011. Morb. Mortal. Weekly Rep. 61, 938–943 (2012).
Ford, E. S. et al. Total and state-specific medical and absenteeism costs of COPD among adults aged >/= 18 years in the United States for 2010 and projections through 2020. Chest 147, 31–45, https://doi.org/10.1378/chest.14-0972 (2015).
GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. The Lancet 5, 691–706, https://doi.org/10.1016/s2213-2600(17)30293-x (2017).
Goldklang, M. P., Marks, S. M. & D’Armiento, J. M. Second hand smoke andCOPD: lessons from animal studies. Frontiers in Physiology 4, 30, https://doi.org/10.3389/fphys.2013.00030 (2013).
Friedlander, A. L., Lynch, D., Dyar, L. A. & Bowler, R. P. Phenotypes of Chronic Obstructive Pulmonary Disease. COPD: Journal of Chronic Obstructive Pulmonary Disease 4, 355–384, https://doi.org/10.1080/15412550701629663 (2007).
Gomez-Cabrero, D. et al. Data integration in the era of omics: current and future challenges. BMC Systems Biology 8, https://doi.org/10.1186/1752-0509-8-S2-I1 (2014).
Wanichthanarak, K., Fahrmann, J. F. & Grapov, D. Genomic, Proteomic, and Metabolomic Data Integration Strategies. Biomarker Insights 7, 1–6, https://doi.org/10.4137/BMI.S29511 (2015).
Kueppers, F., Briscoe, W. A. & Bearn, A. G. Hereditary Deficiency of Serum α1-Antitrypsin. Science 146, 1678–1679, https://doi.org/10.1126/science.146.3652.1678 (1964).
Berndt, A., Leme, A. S. & Shapiro, S. D. Emerging genetics of COPD. EMBO Molecular Medicine 4, 1144–1155, https://doi.org/10.1002/emmm.201100627 (2012).
Carolan, B. J. et al. The association of plasma biomarkers with computed tomography-assessed emphysema phenotypes. Respir Res 15, 127, https://doi.org/10.1186/s12931-014-0127-9 (2014).
Yonchuk, J. G. et al. Circulating soluble receptor for advanced glycation end products (sRAGE) as a biomarker of emphysema and the RAGE axis in the lung. Am. J. Respir. Crit. Care Med. 192, 785–792, https://doi.org/10.1164/rccm.201501-0137PP (2015).
Esther, C. R. Jr., Lazaar, A. L., Bordonali, E., Qaqish, B. & Boucher, R. C. Elevated Airway Purines in COPD. Chest 140, 954–960 (2011).
Bowler, R. P. et al. Plasma Sphingolipids Associated with Chronic Obstructive Pulmonary Disease Phenotypes. Am. J. Respir. Crit. Care Med. 191, 275–284, https://doi.org/10.1164/rccm.201410-1771OC (2015).
Chen, Q. et al. Serum Metabolite Biomarkers Discriminate Healthy Smokers from COPD Smokers. PloS one 10, e0143937, https://doi.org/10.1371/journal.pone.0143937 (2015).
Ubhi, B. K. et al. Targeted metabolomics identifies perturbations in amino acid metabolism that sub-classify patients with COPD. Molecular BioSystems 8, 3125–3133 (2012).
Ippolito, J. E. et al. An integrated functional genomics and metabolomics approach for defining poor prognosis in human neuroendocrine cancers. Proc. Natl. Acad. Sci. USA 102, 9901–9906, https://doi.org/10.1073/pnas.0500756102 (2005).
McGeachie, M. J. et al. The metabolomics of asthma control: a promising link between genetics and disease. Immunity, Inflammation and Disease 3, 224–238, https://doi.org/10.1002/iid3.61 (2015).
Liu, Y. et al. Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer discovery 3, 870–879, https://doi.org/10.1158/2159-8290.cd-13-0015 (2013).
Bino, R. J. et al. Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 9, 418–425, https://doi.org/10.1016/j.tplants.2004.07.004 (2004).
Gieger, C. et al. Genetics Meets Metabolomics: A Genome-Wide Association Study of Metabolite Profiles in Human Serum. PLoS Genet. 4, e1000282 (2008).
Wan, E. S. et al. Epidemiology, genetics, and subtyping of preserved ratio impaired spirometry (PRISm) in COPDGene. Respir Res 15, 89, https://doi.org/10.1186/s12931-014-0089-y (2014).
(Gene [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004 – [cited 2017 Jan 02]. Available from, https://www.ncbi.nlm.nih.gov/gene/.
Romero, P. et al. Computational prediction of human metabolic pathways from the complete human genome. Genome biology 6, R2, https://doi.org/10.1186/gb-2004-6-1-r2 (2005).
Stelzer, G. et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Current protocols in bioinformatics 54, 1.30.31–31.30.33, https://doi.org/10.1002/cpbi.5 (2016).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114, https://doi.org/10.1093/nar/gkr988 (2012).
Cooper, G. M. & Sunderland, M. A. In The Cell: A Molecular Approach (Sinauer Associates, 2000).
Sun, J. C., Williams, M. A. & Bevan, M. J. CD4(+) T cells are required for the maintenance, not programming, of memory CD8(+) T cells after acute infection. Nat. Immunol. 5, 927–933, https://doi.org/10.1038/ni1105 (2004).
Alfredo, P. et al. Pathophysiology of viral-induced exacerbations of COPD. International Journal of Chronic Obstructive Pulmonary Disease 2, 477–483 (2007).
McKendry, R. T. et al. Dysregulation of Antiviral Function of CD8(+) T Cells in the Chronic Obstructive Pulmonary Disease Lung. Role of the PD-1–PD-L1 Axis. Am. J. Respir. Crit. Care Med. 193, 642–651, https://doi.org/10.1164/rccm.201504-0782OC (2016).
Sethi, S. et al. Airway bacterial concentrations and exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 176, 356–361, https://doi.org/10.1164/rccm.200703-417OC (2007).
Erkan, L. et al. Role of bacteria in acute exacerbations of chronic obstructive pulmonary disease. International Journal of Chronic Obstructive Pulmonary Disease 3, 463–467 (2008).
Li, X. et al. An acetyl-L-carnitine switch on mitochondrial dysfunction and rescue in the metabolomics study on aluminum oxide nanoparticles. Particle and fibre toxicology 13, 4, https://doi.org/10.1186/s12989-016-0115-y (2016).
Lei, W., Lerner, C., Sundar, I. K. & Rahman, I. Myofibroblast differentiation and its functional properties are inhibited by nicotine and e-cigarette via mitochondrial OXPHOS complex III. Sci Rep 7, 43213, https://doi.org/10.1038/srep43213 (2017).
Mander, A., Langton-Hewer, S., Bernhard, W., Warner, J. O. & Postle, A. D. Altered Phospholipid Composition and Aggregate Structure of Lung Surfactant Is Associated with Impaired Lung Function in Young Children with Respiratory Infections. American Journal of Respiratory Cell and Molecular Biology 27, 714–721, https://doi.org/10.1165/rcmb.4746 (2002).
Pniewska, E. & Pawliczak, R. The Involvement of Phospholipases A2 in Asthma and Chronic Obstructive Pulmonary Disease. Mediators of Inflammation 2013, 12 pages, https://doi.org/10.1155/2013/793505 (2013).
Berry, K. A. et al. MALDI imaging MS of phospholipids in the mouse lung. J. Lipid Res. 52, 1551–1560, https://doi.org/10.1194/jlr.M015750 (2011).
Schürch, S., Lee, M. & Gehr, P. Pulmonary surfactant: surface properties and function of alveolar and airway surfactant. Pure and Applied Chemistry 64, 1745–1750 (1992).
Goerke, J. Pulmonary surfactant: functions and molecular composition. Biochimica et biophysica acta 1408, 79–89 (1998).
Scott, J. E. The Pulmonary Surfactant: Impact of Tobacco Smoke and Related Compounds on Surfactant and Lung Development. Tobacco Induced Diseases 2, 3–25, https://doi.org/10.1186/1617-9625-2-1-3 (2004).
Hishikawa, D., Hashidate, T., Shimizu, T. & Shindou, H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J. Lipid Res. 55, 799–807, https://doi.org/10.1194/jlr.R046094 (2014).
Alberg, A. J. et al. Plasma Sphingolipids and Lung Cancer: A Population-Based, Nested Case–Control Study. Cancer Epidemiol. Biomarkers Prev. 22, 1374–1382, https://doi.org/10.1158/1055-9965.EPI-12-1424 (2013).
Petrache, I. & Petrusca, D. N. The Involvement of Sphingolipids in Chronic Obstructive PulmonaryDiseases. Handbook of Experimental Pharmacology 216, 247–264, https://doi.org/10.1007/978-3-7091-1511-4_12 (2013).
Kaluza, J., Harris, H. R., Linden, A. & Wolk, A. Long-term consumption of fruits and vegetables and risk of chronic obstructive pulmonary disease: a prospective cohort study of women. Int. J. Epidemiol., https://doi.org/10.1093/ije/dyy178 (2018).
Hanson, C. et al. The Relationship between Dietary Fiber Intake and Lung Function in the National Health and Nutrition Examination Surveys. Annals of the American Thoracic Society 13, 643–650, https://doi.org/10.1513/AnnalsATS.201509-609OC (2016).
Hanson, C., Rutten, E. P., Wouters, E. F. & Rennard, S. Influence of diet and obesity on COPD development and outcomes. Int J Chron Obstruct Pulmon Dis 9, 723–733, https://doi.org/10.2147/copd.S50111 (2014).
Itoh, M., Tsuji, T., Nemoto, K., Nakamura, H. & Aoshiba, K. Undernutrition in patients with COPD and its treatment. Nutrients 5, 1316–1335, https://doi.org/10.3390/nu5041316 (2013).
Wang, J. et al. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-kappaB signaling pathway. Journal of thoracic disease 9, 4398–4412, https://doi.org/10.21037/jtd.2017.09.135 (2017).
Bewley, M. A. et al. Differential Effects ofp38, MAPK, PI3K or Rho Kinase Inhibitors on Bacterial Phagocytosis and Efferocytosis by Macrophages in COPD. PLoS one 11, e0163139, https://doi.org/10.1371/journal.pone.0163139 (2016).
Liu, X., Bao, H., Zeng, X. & Wei, J. Effects of resveratrol and genistein on nuclear factor-κB, tumor necrosis factor-α and matrix metalloproteinase-9 in patients with chronic obstructive pulmonary disease. Molecular medicine reports 13, 4266–4272, https://doi.org/10.3892/mmr.2016.5057 (2016).
Leus, N. G. et al. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-kappaB p65 transcriptional activity. Biochem. Pharmacol. 108, 58–74, https://doi.org/10.1016/j.bcp.2016.03.010 (2016).
Mitani, A., Ito, K., Vuppusetty, C., Barnes, P. J. & Mercado, N. Restoration of Corticosteroid Sensitivity in Chronic Obstructive Pulmonary Disease by Inhibition of Mammalian Target of Rapamycin. Am. J. Respir. Crit. Care Med. 193, 143–153, https://doi.org/10.1164/rccm.201503-0593OC (2016).
Cruickshank-Quinn, C. et al. Metabolomic similarities between bronchoalveolar lavage fluid and plasma in humans and mice. Sci Rep 7, https://doi.org/10.1038/s41598-017-05374-1 (2017).
Miller, M. et al. Gene and metabolite time-course response to cigarette smoking in mouse lung and plasma. PLoS ONE 12, e0178281, https://doi.org/10.1371/journal.pone.0178281 (2017).
Barton, S. et al. Targeted plasma metabolome response to variations in dietary glycemic load in a randomized, controlled, crossover feeding trial in healthy adults. Food & function 6, 2949–2956, https://doi.org/10.1039/c5fo00287g (2015).
Cannon, D. T., Liu, J., Sakurai, R., Rossiter, H. B. & Rehan, V. K. Impaired Lung Mitochondrial Respiration Following Perinatal Nicotine Exposure in Rats. Lung 194, 325–328, https://doi.org/10.1007/s00408-016-9859-2 (2016).
Fan, J. et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 9, https://doi.org/10.1038/msb.2013.65 (2013).
Wu, Y. et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 285, 10850–10861, https://doi.org/10.1074/jbc.M109.080796 (2010).
Seglen, P. O. & Gordon, P. B. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proceedings of the National Academy of Sciences of the United States of America 79, 1889–1892 (1982).
Li, J. et al. Integrating 3-omics data analyze rat lung tissue of COPD states and medical intervention by delineation of molecular and pathway alterations. Bioscience reports 37, https://doi.org/10.1042/bsr20170042 (2017).
Karch, A. et al. The German COPD cohort COSYCONET: Aims, methods and descriptive analysis of the study population at baseline. Respir. Med. 114, 27–37, https://doi.org/10.1016/j.rmed.2016.03.008 (2016).
Adab, P. et al. Cohort Profile: The Birmingham Chronic Obstructive Pulmonary Disease (COPD) Cohort Study. Int. J. Epidemiol. 46, 12, https://doi.org/10.1093/ije/dyv350 (2016).
Couper, D. et al. Design of the Subpopulations and Intermediate Outcomes in COPD Study (SPIROMICS). Thorax 69, 491–494, https://doi.org/10.1136/thoraxjnl-2013-203897 (2014).
Holle, R., Happich, M., Lowel, H. & Wichmann, H. E. KORA–a research platform for population based health research. Gesundheitswesen (Bundesverband der Arzte des Offentlichen Gesundheitsdienstes (Germany)) 67(Suppl 1), S19–25, https://doi.org/10.1055/s-2005-858235 (2005).
Wichmann, H. E., Gieger, C. & Illig, T. KORA-gen–resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen (Bundesverband der Arzte des Offentlichen Gesundheitsdienstes (Germany)) 67(Suppl 1), S26–30, https://doi.org/10.1055/s-2005-858226 (2005).
Regan, E. A. et al. Genetic epidemiology of COPD (COPDGene) study design. COPD 7, 32–43, https://doi.org/10.3109/15412550903499522 (2010).
Pauwels, R. A. et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: National Heart, Lung, and Blood Institute and World Health Organization Global Initiative for Chronic Obstructive Lung Disease (GOLD): executive summary. Respir Care 46, 798–825 (2001).
Carolan, B. J. et al. The association of adiponectin with computed tomography phenotypes in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 188, 561–566, https://doi.org/10.1164/rccm.201212-2299OC (2013).
Pellegrino, R. et al. Interpretative strategies for lung function tests. The European respiratory journal 26, 948–968, https://doi.org/10.1183/09031936.05.00035205 (2005).
Bahr, T. M. et al. Peripheral blood mononuclear cell gene expression in chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology 49, 316–323, https://doi.org/10.1165/rcmb.2012-0230OC (2013).
Sun, W. et al. Common Genetic Polymorphisms Influence Blood Biomarker Measurements in COPD. PLoS Genet. 12, e1006011, https://doi.org/10.1371/journal.pgen.1006011 (2016).
Yang, Y. et al. New sample preparation approach for mass spectrometry-based profiling of plasma results in improved coverage of metabolome. J. Chromatogr. 1300, 217–226, https://doi.org/10.1016/j.chroma.2013.04.030 (2013).
Cruickshank-Quinn, C. et al. Multi-step preparation technique to recover multiple metabolite compound classes for in-depth and informative metabolomic analysis. Journal of Visualized Experiments 89, e51670, https://doi.org/10.3791/51670 (2014).
NIST. NIST/EPA/NIH Mass Spectral Library with Search Program (Data Version: NIST 14, Software Version 2.2g), http://www.nist.gov/srd/nist1a.cfm (2014).
Ruttkies, C., Schymanski, E. L., Wolf, S., Hollender, J. & Neumann, S. MetFrag relaunched: incorporating strategies beyond in silico fragmentation. J Cheminform 8, 3, https://doi.org/10.1186/s13321-016-0115-9 (2016).
Wolf, S., Schmidt, S., Muller-Hannemann, M. & Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics 11, 148, https://doi.org/10.1186/1471-2105-11-148 (2010).
Hughes, G. et al. MSPrep—Summarization, normalization and diagnostics for processing of mass spectrometry–based metabolomic data. Bioinformatics 30, 133–134, https://doi.org/10.1093/bioinformatics/btt589 (2014).
Oba, S. et al. A Bayesian missing value estimation method for gene expression profile data. Bioinformatics 19, 2088–2096, https://doi.org/10.1093/bioinformatics/btg287 (2003).
Redestig, H. et al. Compensation for Systematic Cross-Contribution Improves Normalization of Mass Spectrometry Based Metabolomics Data. Analytical chemistry 81, 7974–7980, https://doi.org/10.1021/ac901143w (2009).
R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, http://www.R-project.org/.
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44–57, https://doi.org/10.1038/nprot.2008.211 (2009).
Wohlgemuth, G., Haldiya, P. K., Willighagen, E., Kind, T. & Fiehn, O. The Chemical Translation Service–a web-based tool to improve standardization of metabolomic reports. Bioinformatics (Oxford, England) 26, 2647–2648, https://doi.org/10.1093/bioinformatics/btq476 (2010).
Kamburov, A., Cavill, R., Ebbels, T. M. D., Herwig, R. & Keun, H. C. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 27, 2917–2918, https://doi.org/10.1093/bioinformatics/btr499 (2011).
Bardou, P., Mariette, J., Escudie, F., Djemiel, C. & Klopp, C. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 15, 293, https://doi.org/10.1186/1471-2105-15-293 (2014).
Kamburov, A. et al. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 39, D712–D717, https://doi.org/10.1093/nar/gkq1156 (2011).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–d361, https://doi.org/10.1093/nar/gkw1092 (2017).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–462, https://doi.org/10.1093/nar/gkv1070 (2016).
Acknowledgements
This study was supported by National Heart, Lung and Blood Institute (NHLBI R01 HL 095432, P20 HL113445, U01 HL089856, U01 HL089897); and NCRR/NIH UL1 RR025780.
Author information
Authors and Affiliations
Contributions
N.R., R.B. and I.P. conceived and designed the experiments; C.C.Q. and R.P. performed the experiments; S.J., K.K., G.H. and C.C.Q. analyzed the data; C.C.Q. and N.R. wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cruickshank-Quinn, C.I., Jacobson, S., Hughes, G. et al. Metabolomics and transcriptomics pathway approach reveals outcome-specific perturbations in COPD. Sci Rep 8, 17132 (2018). https://doi.org/10.1038/s41598-018-35372-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35372-w
Keywords
This article is cited by
-
An integrated metabo-lipidomics profile of induced sputum for the identification of novel biomarkers in the differential diagnosis of asthma and COPD
Journal of Translational Medicine (2024)
-
Plasma metabolomics and quantitative interstitial abnormalities in ever-smokers
Respiratory Research (2023)
-
A metabolome-wide case-control study of african american breast cancer patients
BMC Cancer (2023)
-
Phenomic Imaging
Phenomics (2023)
-
Mechanism-aware imputation: a two-step approach in handling missing values in metabolomics
BMC Bioinformatics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
