
Abstract
Isopods of the genus Idotea have an unusual ability to feed on algae containing high amounts of chemical defense molecules, such as species of the genera Fucus and Ulva. In this study, we compared gene expression patterns of Idotea balthica individuals fed with Fucus vesiculosus to individuals fed with Ulva lactuca. We generated the first-ever transcriptome assembly for this species, and found 3,233 differentially expressed genes across feeding regimes. However, only a handful of biological functions were enriched with regard to differentially expressed genes, the most notable being “alkaloid metabolic process”. Within this category, we found eight differentially expressed cytochrome P450 (CYP) unigenes, all of which had a higher expression in the U. lactuca diet treatment. A phylogenetic analysis showed that the differentially expressed CYP genes are closely related to a CYP gene described from the hepatopancreas of the spiny lobster Panulirus argus, and we hypothesize that these transcripts are involved in metabolite detoxification. This is a first step in the understanding of this algae-grazer interaction, and will form a basis for future work to characterize cytochrome P450 functioning in marine crustaceans.
Similar content being viewed by others
Introduction
Isopod crustaceans of the globally distributed genus Idotea are common in coastal marine areas. These species are generalist grazers on macroalgae and sea grasses, and can be ecologically important in structuring algal communities1. For example, in some areas in the Baltic Sea (Northern Europe), densities of Idotea balthica can rise to astonishing numbers (>80 individuals/100 g wet weight of the brown alga Fucus vesiculosus), which may temporarily cause a complete loss of F. vesiculosus in the area2. In areas with strong grazing pressure, algal genotypes with defensive strategies against grazing are favored by natural selection3. One common defense strategy among macrophytes is synthesis of secondary metabolites, which are bioactive compounds not directly related to growth or reproduction. In algae, these are often involved in grazer deterrence by decreasing the palatability of the tissue4. As some algal secondary metabolites have been found to have pharmaceutical applications, much research has been undertaken to gain a better understanding of these compounds5,6,7,8,9. As a result, a whole range of bioactive compounds have now been discovered in seaweeds10.
However, the mechanism involved in grazer deterrence has been more difficult to conclusively elucidate to date. It has been suggested that the main grazer-deterring compounds in brown algae are polyphenolic compounds which have been named “phlorotannins”11,12. These compounds have the ability to bind to proteins and thus reduce the digestible nutrient content of the algae, while at the same time degrading the chemical environment within the gut of the grazers. Jormalainen et al.13 observed that phlorotannin extracts reduced the assimilation efficiency of I. balthica, but this did not deter the isopods from feeding. Other feeding trials with algae of varying phlorotannin concentrations have yielded mixed results, in some cases deterring grazing while in some cases actually stimulating feeding14. In green algae, such as the “sea lettuce” Ulva lactuca, dimethylsulfoniopropionate (DMSP) and acrylic acids15, as well as compounds producing reactive oxygen species (ROS)16 are induced under grazing, and have been suggested to have a role in grazer deterrence and toxicity. ROS ingestion could cause oxidative damage in the guts of grazers, as has been shown in insects feeding of plant tissue17. However, while chemical defense compounds found in Ulva species caused sea urchin deterrence, they had the opposite effect on Idotea wosnesenskii15. In common periwinkles (Littorina spp.), a recent study found that the deterrence effects of Ulva lactuca were species-specific, which the authors hypothesized could be due to differences in feeding strategies (generalist vs specialist)18.
As a consequence of the evolution of chemical defense compounds in the algae, it is expected that the grazers would evolve mechanisms for tolerance or detoxification of the algal compounds. A tolerance mechanism could act in different ways. For example, a highly efficient general detoxification mechanism for toxic compounds can be fine-tuned based on the diet. This would allow them to be generalists while still feeding on algal species with high quantities of secondary metabolites such as F. vesiculosus and U. lactuca. Common to all animals, the Cytochrome P450 (CYP) system is one of the major players in metabolism and detoxification of compounds produced by other organisms19,20,21. Cytochrome P450 is a superfamily of enzymes, consisting of several hundreds of different types which are classified into different families22. Some of these have an important role in general metabolism and in development, while others are more involved in detoxification23. For example, CYP enzymes are induced in caterpillars after being fed with nicotine-containing material24. In the marine environment, it has been shown that some variants of the CYP2 family, involved in detoxification of xenobiotic compounds, are present in the hepatopancreas of crustaceans25,26. While very few comparative studies have been conducted to date on crustacean CYP variants, it has been suggested that generalist algal grazers would benefit from the evolution of a high diversity of CYP genes27,28, as has been observed in the genome sequence of the purple urchin (Strongylocentrotus purpuratus)29. Each of these could then evolve an ability to metabolize a specific group of compounds, generating a finely-tuned diet-based detoxification machinery. In addition, it is possible that the isopods have acquired specialized gut microbiota, which assist them in digesting toxic compounds before they are able to diffuse from the gut into the tissues of the animal. In terrestrial isopods with diets containing hard-to-digest compounds such as cellulose, it is well-known that the gut microbiota plays an important role in digestion30,31. The functional roles of gut microbes in species of the genus Idotea have not yet been investigated, but there have been some studies of the overall microbiota indicating that isopods from different geographic areas and with different diets possess distinct microbiomes31,32. It is also possible that some of the functions usually performed by gut bacteria could have been transferred to the isopods themselves. For example, the lignocellulose metabolic machinery otherwise only present in bacteria and fungi, can be found also in the genomes of isopods of the wood-boring genus Limnoria33. In particular, the glycosyl hydrolase family 7 (GH7) has 3 representatives which are all expressed in the gut transcriptome of Limnoria quadripunctata34 suggesting horizontal gene transfer from symbiont to host33.
We here present a study investigating the detoxification mechanism of algal defense compounds in I. balthica using an RNA sequencing approach. We conducted an experiment where individuals of I. balthica were fed with different algal diets - one brown alga (F. vesiculosus) which is a common diet item for these isopods, and one green alga (U. lactuca) which is not as common in the isopods’ habitat. Representing two evolutionarily distant groups of algae, it is likely that there are considerably differences in metabolite structure among the two. Subsequently, we examined differences in gene expression among individuals from the different diet treatments supported by a reference transcriptome assembled from a combination of all tissue types from one adult male I. balthica. Our hypothesis was that the I. balthica individuals would induce expression of diet-specific detoxification enzymes, either through isopod-specific detoxification compounds, or as a response within the Cytochrome P450 pathway. If the latter was the case, we predicted a diversity of different Cytochrome P450 forms in the transcriptome sequence of I. balthica, of which many would be differentially expressed in different diet treatments, and that these would be similar in sequence to variants of Cytochrome P450 found in other marine algal grazers.
Results
Transcriptome Assembly and Annotation
The total number of reads amounted to 296,173,922, out of which 262,020,548 remained after quality-trimming. Rcorrector was able to correct 46,535,134 bases pre-assembly, and the in silico normalization yielded a final set of 10,099,252 read pairs which were used as input for the de novo assembly (Table 1).
The raw assembly was filtered using read evidence and presence of ORFs using TransRate, with a resulting output assembly of 40,122 transcripts in 30,147 unigenes, an N50 of 1,710 bp and a total length of 47,523,534 bp (Table 1). Mapping the reads of the reference individual back to the filtered assembly, 67% of the reads mapped (58% with high quality). TransDecoder could find 21,439 putative peptides encoded by the filtered assembly (Table 1). The BUSCO analysis searched for 2,675 Arthropod single-copy orthologs in the assembly, and found 1,645 (61.5%). Only 46.3% of these were single-copy and complete, and the remainders were fragments (19.5%), duplicates (23.0%) or triplicates (11.2%) (Table 1), resulting from either assembly fragmentation or separate assembly of the two alleles.
The PANNZER annotation, using an arthropod database, yielded annotations for 10,655 out of the 40,122 transcripts, while a separate BLASTx to NCBI’s non-redundant protein database (nr) yielded annotations for 19,498 transcripts (Table 1; Supplementary Table 1). Filtering out putative, hypothetical and predicted hits, this number was reduced to 6,572 BLASTx-annotated transcripts. Finally, BLAST hits from the most common micro-organismal genera were filtered out, leaving 5,875 transcripts annotated by BLAST. In total, 12,925 transcripts were annotated by BLAST and PANNZER.
The sequences of the Limnorea quadripunctata lignocellulose digestion genes originating from microbes (GH7 and GH9) had no BLAST matches in the I. balthica transcriptome.
Feeding Trials
The sequencing depth ranged between 21,541,785 and 48,180,703 reads per individual (mean = 38,037,887 reads, st.dev. = 5,937,275 reads) (Supplementary Table 2). The proportion of reads that aligned to the reference transcriptome was between 61.1% and 88.6% (mean = 81.3%, st.dev. = 8.73%). Out of these, between 55.1% and 73.9% mapped to multiple locations in the assembly (at the transcript level) (mean = 65.1%, st.dev. 6.37%).
No reads were found to map to I. chelipes and I. granulosa COI sequences, whereas many (mean = 74,581, st.dev = 59,750) mapped to I. balthica COI, showing that all experimental individuals were indeed I. balthica.
The RSEM analysis of gene expression extracted expected count data, averaged by transcript/isoform for each unigene (Supplementary Table 3). The expected counts were rounded to the nearest integer and analysed in DESeq2, resulting in 3,233 genes being differentially expressed between feeding treatments (Fucus or Ulva) after multiple test adjustment (p < 0.05) (Supplementary Table 4). Out of these, 1,145 could be annotated (924 by PANNZER, 547 by BLASTx to nr) (Supplementary Table 5). Of these, 15 could potentially be microbial (or have isoforms within the “gene” which were microbial), as suggested by the BLASTx to nr taxon-filtering, although 2 out of the 15 did have a valid non-microbial PANNZER annotation (TRINITY_DN73620 (TEM beta-lactamase) and TRINITY_DN93555 (T-complex protein 1 subunit delta)) (Supplementary Table 5).
The multiple-test adjusted p-values from the differential expression analysis were used as input for GO enrichment Gene Score Resampling analysis. Despite the large number of differentially expressed genes (3,233), only 2 functional categories were significantly enriched for differential expression. Those were “nicotine metabolic process” (GO:0018933, adjusted p-value 2.01 * 10−9) and “alkaloid metabolic process” (GO:0009820, corrected p-value 4.02 * 10−9) (Supplementary Table 6). Nicotine metabolic process is nested within alkaloid metabolic process, and all genes in the former are also present in the latter. There are 16 genes responsible for the enrichment (Table 2), most of which are various forms of Cytochrome P450. All of the significantly differentially expressed genes in the list were down-regulated in the F. vesiculosus diet treatment. Quantitative PCR of three representative Cytochrome P450 genes confirmed this result (Supplementary Figure 1).
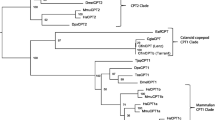
The differentially expressed Cytochrome P450 isoforms were merged by sequence identity into 8 different sequences, and a BLASTp search using the ORF protein sequences showed that all belong to the CYP2 family. The results of the Bayesian phylogeny inference showed that all 8 sequences form a monophyletic clade together with Cytochrome P450 2L1, described from the hepatopancreas of the spiny lobster Panulirus argus, with strong statistical support (Q27712.1; Fig. 1). In addition, one of the contigs with Cytochrome P450 domains also contained components with ADP-ribosylation domains (TRINITY_DN91434_c7). There was only one differentially expressed unigene in the GO category (TRINITY_DN91758) which was not annotated as a Cytochrome P450, but rather as a nicotinamide phosphoribosyltransferase by BLAST to the nt database (Table 2).

Phylogenetic tree of eight I. balthica CYP450 transcripts (DN######), together with 100 CYP450 sequences downloaded from GenBank (See Supplementary Table 1 for a list of Accession Numbers). Node support values (Bayesian posterior probabilities) are denoted by the coloured circles. The clade grouping all I. balthica transcripts together with a Panulirus argus transcript (Q27712.1) is highlighted in yellow.
Discussion
The Cytochrome P450 family 2 is specifically involved in detoxification of xenobiotic compounds by mono-oxygenation or hydroxylation, thereby inactivating potential bioactive compounds which have been ingested35. Usually, CYP detoxification involves additional binding proteins such as NADP+, which can act as electron receivers36. The CYP2 transcripts found in this study are most closely related to those described from the hepatopancreas of spiny lobsters (CYP2L25). However, to the best of our knowledge, no work has been done on the actual mode of detoxification within crustaceans, and it is not known which specific compounds these CYP2 proteins might be metabolizing. Further work is thus essential here to investigate these genes and their functions.
Nicotinamide phosphoribosyltransferase (NAMPT) was the only non-CYP2 unigene within the functionally enriched categories in our experiment. NAMPT plays a critical role in production of NAD+37, which has an important role in metabolism via the electron transport chain. It has recently been shown that a knockout of NAMPT has a cascading effect on the cytochrome P450 pathway and inducing mortality in mice38, suggesting that NAMPT is needed for the CYP2 proteins to function.
Interestingly, all of the CYP2 transcripts were more highly expressed in I. balthica fed with U. lactuca than in isopods fed with F. vesiculosus. Several recent studies have shown that Ulva produces a range of secondary metabolites which deter both gastropods18 and sea urchins39. F. vesiculosus also possesses a range of secondary metabolites, but as F. vesiculosus is the natural diet of I. balthica, we can speculate that they might be able to detoxify F. vesiculosus compounds through a separate, taxon-specific, pathway. As support of this hypothesis, our results indicate that there are many unannotated transcripts which are upregulated in the F. vesiculosus diet treatment. Some of these could be involved in detoxification. An alternative explanation could perhaps also be found in that F. vesiculosus possesses a structural defense in its much more robust and leathery thallus, which might relax the need for production of secondary metabolites aimed at grazer deterrence, when compared to U. lactuca which has a thin and soft thallus.
In isopods of the genus Limnoria, lignocellulose metabolic genes (primarily GH7) have been horizontally transferred from bacteria to the isopod, allowing them to digest plant (wood) material without symbiotic gut microbes33. In the I. balthica transcriptome assembled in this study, we found no signs of GH7 homologs. Perhaps this is not surprising, as genes involved specifically in lignocellulose metabolism might be irrelevant to grazers of lignocellulose-lacking algae, such as I. balthica.
For future work, proteomic and metabolomic tools could help us understand how metabolite concentrations change with varying transcript levels. The link between transcript expression and physiological performance is not always straightforward due to e.g. non-linear relationships between transcript and protein expression and/or between protein expression and activity40, so gene expression studies such as this one can only elucidate one piece of a more complex puzzle.
In conclusion, our data shows that expression of the Cytochrome P450 family 2 is influenced by the species of macroalgae that I. balthica is feeding on. As this is a gene family involved in detoxification, this suggests that these genes may be important in the isopod detoxification of secondary metabolites produced by the algae, and that there is a diet-specific regulation at the individual transcript level. It would be of great interest to further investigate CYP gene family evolution in I. balthica. However, in order to do this, a reliable genomic reference sequence will be required. Until then, we can conclude that the picture is complex and more work will certainly be needed before the mechanisms of metabolite detoxification in marine grazers can be fully understood.
Materials and Methods
The analysis followed a standard RNA-seq pipeline, similar to the one published by De Wit et al.41 (Fig. 2). Below are detailed Methods for each of the steps listed in the flowchart.

Flowchart of the RNA-Seq analysis undertaken in this study.
Transcriptome Assembly and Annotation
The male I. balthica individual from which the transcriptome was assembled was sampled in September, 2014 at Kristineberg, Sweden (58°14.869′N;11°26.883′E). Total RNA was extracted using a TRIzol (Thermo Fisher Scientific) protocol and cleaned with a Zymo RNA Clean and Concentrator Kit (Zymo Research). RNA concentration was measured using a QuBit RNA assay (Thermo Fisher Scientific) and RNA integrity was assessed using a MOPS denaturing agarose gel. cDNA libraries were prepared at the National Genomics Infrastructure in Stockholm, Sweden using a TruSeq RNA Library Preparation Kit v2 (Illumina). This protocol includes poly-A selection of mRNA, cDNA synthesis, Illumina adapter ligation and a 12-cycle PCR reaction. A single lane of an Illumina HiSeq2500 sequencer (Illumina) was then used to paired-end sequence 125 base pair (bp) long reads.
The ‘best practice’ transcriptome assembly pipeline by De Wit et al.42 was followed to generate a total male transcriptome. All bioinformatic analyses were run on the CSC - IT Center for Science’s (Espoo, Finland; www.csc.fi) Taito super-cluster. Briefly, raw reads were assessed for quality using FASTQC (v0.11.2; www.bioinformatics.babraham.ac.uk/projects/fastqc), after which TRIMMOMATIC (v0.33)43 was used to trim adapters and trimming read ends for low-quality bases. Also, a sliding window of four bases was used for trimming reads of average base quality <15. Finally, sequences that were less than 36 bases long were dropped.
Pre-assembly error correcting was done using RCORRECTOR (v2015-11-0644) with a k-mer size of 20 to reduce the number of substitution errors incorporated into the assembly. TRINITY (v2.1.0) was used for in silico normalization45,46 with k-mer size 25 and a coverage limit of 50. The reads were then de novo assembled using TRINITY with a k-mer size of 25.
The assembly was quality evaluated and filtered using TRANSRATE (v1.0.147). The normalized reads were used for read-mapping. Only contigs with read evidence and containing ORFs were kept. The filtered assembly was assessed for the presence of putative peptides by TransDecoder (available at http://transdecoder.github.io/). The raw data used was submitted to the NCBI Sequence Read Archive (SRA) and the final version of the assembly was submitted to the Transcriptome Shotgun Assemblies (TSA) database (BioSample SAMN08960750).
A BUSCO analysis was run to evaluate the completeness of the transcriptome assembly with respect to the number of known core single-copy orthologous genes identified48. In this study, BUSCO was run in transcriptome mode using the arthropod lineage dataset as a reference. In cases where orthologues were found to be duplicated in the assembly, the exact number of times they were found was determined using a custom script.
Annotation was conducted in two separate ways. Firstly, functional annotation was conducted using PANNZER49. PANNZER searches the UniProtKB/Swiss (http://www.uniprot.org) database and uses a Z-score for a more reliable automated annotation49. The query taxon id 82763 (Idotea balthica) was specified to the software; the other settings were left as default. In addition, annotations were found using BLASTX (v2.2.31+50) against the NCBI non-redundant protein sequence database (nr; v2015-12-01) using an e-value cutoff of 10−5. Results were filtered to separate out uninformative terms (i.e. hypothetical, putative, predicted and unknown) using a custom script. The term-filtered output was then grouped by taxon using a custom script, after which bacterial and protozoan genera were filtered out. All scripts used can be found on GitHub (https://github.com/The-Bioinformatics-Group/Idotea_balthica_ transcriptome_project/tree/master/Annotation/blastx2nr/scripts).
Finally, in order to investigate the potential of horizontal gene transfer of lignocellulose digestion genes from microbes, as previously found in Limnoria quadripunctata, the glycosyl hydrolase (GH) family 7 and 9 members described in L. quadripunctata (GenBank Acc.Nos FJ940756– FJ940761) were BLASTed with a standard nucleotide BLAST against the I. balthica transcriptome assembly, using default settings.
Feeding Trial Experiment
To find out which genes were differentially expressed under different feeding regimes, and if specific “detoxification genes” are activated in isopods when digesting algal metabolites, we performed a feeding experiment. Twenty-four individuals of I. balthica were collected at the same time and in the same location as the transcriptome individual (September 2014; 58°14.869′N;11°26.883′E). These isopods were kept in tanks with through-flowing seawater until the start of the experiment in the summer of 2015. Isopods were fed daily with their natural diet, the brown alga Fucus vesiculosus prior to onset of the experiment. At the start of the experiment, all individuals had their diet switched to Ulva lactuca, the “sea lettuce”, a green alga preferred by many marine invertebrates. After two weeks feeding on U. lactuca, 12 individuals were switched back to a F. vesiculosus diet. After another two weeks, all 24 animals were killed by decapitation and placed in RNAlater. (One individual in the F. vesiculosus diet treatment died prior to the end of the experiment and was excluded from further analyses). RNA was extracted and assessed as described above and cDNA libraries were prepared using the TruSeq RNA library prep kit v2 (Illumina). cDNA library fragment length was assessed using an Agilent TapeStation with a D1000 tape (Agilent), after which the fragments were multiplexed equimolarly (2 pools per treatment and 6 individuals per pool, except one pool which had 5 individuals) and were sent for sequencing at the National Genomics Infrastructure’s “SNP & SEQ platform” in Uppsala, using an Illumina HiSeq2500 machine (50 bp reads, single-end).
Sequence data were filtered for Illumina adapter sequences and low-quality ends (Q < 20) using fastx toolkit (available at http://hannonlab.cshl.edu/fastx_toolkit/). Filtered reads were then mapped to the transcriptome assembly using bowtie2 (v. 2.2.7)51, allowing for 5 mismatches, both with duplicate reads (two or more short reads with identical nucleotide sequences) kept and with duplicate reads removed, in order to assess duplication bias. As number of duplicates was highly correlated with expression values, and thus likely of biological relevance, duplicates were kept in downstream analyses.
Reads were also mapped (allowing for 1 mismatch) to a list of cytochrome oxidase subunit I (COI) sequences from all three Idotea species present in Swedish waters52, in order to ensure that all individuals were I. balthica (morphological species identification, in particular of juvenile individuals, can be challenging).
Gene expression values were obtained using the RSEM pipeline (https://github.com/bli25ucb/RSEM_tutorial). Briefly, the “–transcript-to-gene-map” option was first used to create a transcript to gene map of the transcriptome assembly, then raw expression values (expected counts) were extracted at the gene level. The expected counts were used as input for a differential expression analysis using DESeq253, rather than FPKM values, as recommended by the manual. Finally, a GO enrichment Gene Score Resampling (GSR) analysis was performed on the multiple-test corrected p-values from the differential gene expression analysis in ErmineJ54. Genes responsible for GO categories being enriched for differential expression were, in addition to the annotation available for the entire transcriptome, also annotated by tBLASTx to the NCBI nt database.
In order to investigate the evolutionary relationships of differentially expressed Cytochrome P450 unigenes, 100 CYP protein sequences obtained from GenBank (Supplementaly. Table 7) were aligned to the translated I. balthica sequences, using Blosum62 cost matrix, and gap open/extension penalties 12/3 in Geneious 4.855. The resulting protein alignment was examined for the best-fitting model of protein evolution using ProtTest 356, after which the best model (JTT + I + G + F) was implemented in an MCMC phylogeny inference analysis using MrBayes v 3.257. Two separate runs of 4 chains each were run for 11.5 M generations, sampling every 1000 generations and, after examining the output for convergence using RWTY in R58, 5 M generations were discarded as burn-in. Remaining generations were summarized into a majority-rule consensus tree and plotted using FigTree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/). All raw data from the feeding trial experiment was submitted to SRA (BioProject PRJNA451082).
Finally, to validate our gene expression results, we performed quantitative PCR on three of the cytochrome P450 genes, using a subset of six individuals, three from the F. vesiculosus diet (T04, T05 and T06) and three from the U. lactuca diet treatments (C04, C06 and C12). Primers were designed based on consensus sequences from all isoforms of the CYP transcripts DN78647, DN87011 and DN89473, as well as 18S rRNA (DN93465), which was used as endogenous control (Supplementary Table 8). 0.5 µg RNA was converted to cDNA using an iScript select cDNA synthesis kit (Bio-Rad), after which a comparative CT experiment using Fast SYBR Green (Thermo Fisher) was performed with an Applied Biosystems StepOnePlus system, following the default “fast” thermal program. The cDNA was diluted 1:10, and each reaction well contained 4 µl diluted cDNA, 1 µl primer F (10 µM), 1 µl primer R (10 µM), 4 µl nuclease-free water, and 10 µl SYBR Green master mix (x2), for a total reaction volume of 20 µl. Each reaction was replicated three times in a 96-well plate. In addition, each primer combination was also run in three negative control reactions. The sample T06 was arbitrarily set as the reference sample for the relative quantification.
References
Bell, T. M. & Sotka, E. E. Local adaptation in adult feeding preference and juvenile performance in the generalist herbivore Idotea balthica. Oecologia 170, 383–393 (2012).
Engkvist, R., Malm, T. & Tobiasson, S. Density dependent grazing effects of the isopod Idotea baltica Pallas on Fucus vesiculosus L in the Baltic sea. Aquat. Ecol. 34, 253–260 (2000).
Nylund, G. M. N., Pereyra, R. T., Wood, H. L. & Johannesson, K. Increased resistance towards generalist herbivory in the new range of a habitat-forming seaweed. Ecosphere 3, 1–13 (2012).
Toth, G. B. & Pavia, H. Induced herbivore resistance in seaweeds: A meta-analysis. J. Ecol. 95, 425–434 (2007).
Geiselman, J. A. & McConnell, O. J. Polyphenols in brown algae Fucus vesiculosus and Ascophyllum nodosum: Chemical defenses against the marine herbivorous snail, Littorina littorea. J. Chem. Ecol. 7, 1115–1133 (1981).
Fleury, B. G., Kelecom, A., Pereira, R. C. & Teixeira, V. L. Polyphenols, terpenes and sterols in Brazilian dictyotales and fucales (Phaeophyta). Bot. Mar. 37, 457–462 (1994).
Rohde, S., Molis, M. & Wahl, M. Regulation of anti-herbivore defence by Fucus vesiculosus in response to various cues. J. Ecol. 92, 1011–1018 (2004).
Forslund, H., Eriksson, O. & Kautsky, L. Grazing and geographic range of the Baltic seaweed Fucus radicans (Phaeophyceae). Mar. Biol. Res. 8, 322–330 (2012).
Haavisto, F., Koivikko, R. & Jormalainen, V. Defensive role of macroalgal phlorotannins: benefits and trade-offs under natural herbivory. Mar. Ecol. Prog. Ser. 566, 79–90 (2017).
Holdt, S. L. & Kraan, S. Bioactive compounds in seaweed: Functional food applications and legislation. J. Appl. Phycol. 23, 543–597 (2011).
Pavia, H. & Toth, G. B. Inducible Chemical Resistance to Herbivory in the Brown Seaweed Ascophyllum nodosum. Ecology 81, 3212–3225 (2000).
Toth, G. B., Langhamer, O. & Pavia, H. Inducible and constitutive defenses of valuable seaweed tissues: Consequences for herbivore fitness. Ecology 86, 612–618 (2005).
Jormalainen, V., Honkanen, T., Vesakoski, O. & Koivikko, R. Polar extracts of the brown alga Fucus vesiculosus (L.) reduce assimilation efficiency but do not deter the herbivorous isopod Idotea baltica (Pallas). J. Exp. Mar. Bio. Ecol. 317, 143–157 (2005).
Kubanek, J., Lester, S. E., Fenical, W. & Hay, M. E. Ambiguous role of phlorotannins as chemical defenses in the brown alga Fucus vesiculosus. Mar. Ecol. Prog. Ser. 277, 79–93 (2004).
Van Alstyne, K. L., Wolfe, G. V., Freidenburg, T. L., Neill, A. & Hicken, C. Activated defense systems in marine macroalgae: Evidence for an ecological role for DMSP cleavage. Mar Ecol Prog Ser 213, 53–65 (2001).
McDowell, R. E., Amsler, C. D., McClintock, J. B. & Baker, B. J. Reactive oxygen species as a marine grazing defense: H2O2 and wounded Ascoseira mirabilis both inhibit feeding by an amphipod grazer. J. Exp. Mar. Bio. Ecol. 458, 34–38 (2014).
Bi, J. L. & Felton, G. W. Foliar oxidative stress and insect herbivory: Primary compounds, secondary metabolites, and reactive oxygen species as components of induced resistance. J. Chem. Ecol. 21, 1511–1530 (1995).
Peckol, P. & Putnam, A. B. Differential toxic effects of Ulva lactuca (Chlorophyta) on the herbivorous gastropods, Littorina littorea and L. obtusata (Mollusca). J. Phycol. 53, 361–367 (2017).
Guengerich, F. P. Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 21, 70–83 (2008).
Berenbaum, M. R., Favret, C. & Schuler, M. A. On defining ‘key innovations’ in an adaptive radiation: Cytochrome P450S and Papilionidae. Am. Nat. 148, S139–155 (1996).
Sotka, E. E. & Whalen, K. E. Herbivore offense in the sea: the detoxification and transport of secondary metabolites. In Algal chemical ecology (ed. Amsler, C. D.) 203–228 (Springer Verlag, 2008).
Coon, M. J. CYTOCHROME P450: Nature’s Most Versatile Biological Catalyst. Annu. Rev. Pharmacol. Toxicol. 45, 1–25 (2005).
Stegeman, J. J. & Hahn, M. E. In Aquatic Toxicology: Molecular, Biochemical, and Cellular Perspectives (eds Malins, D. C. & Ostrander, G. K.) 87–207 (CRC Press, Inc., 1994).
Snyder, M. J. & Glendinning, J. I. Causal connection between detoxification enzyme activity and consumption of a toxic plant compound. J. Comp. Physiol. A 179, 255–261 (1996).
James, M. O. et al. cDNA and protein sequence of a major form of P450, CYP2L, in the hepatopancreas of the spiny lobster. Panulirus argus. Arch. Biochem. Biophys. 329, 31–38 (1996).
Snyder, M. J. Identification of a new cytochrome P450 family, CYP45, from the lobster, Homarus americanus, and expression following hormone and xenobiotic exposures. Arch. Biochem. Biophys. 358, 271–276 (1998).
Li, X., Schuler, M. A. & Berenbaum, M. R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu. Rev. Entomol. 52, 231–253 (2007).
Li, X., Baudry, J., Berenbaum, M. R. & Schuler, M. A. Structural and functional divergence of insect CYP6B proteins: From specialist to generalist cytochrome P450. Proc. Natl. Acad. Sci. USA 101, 2939–2944 (2004).
Sodergren, E. et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science (80-.). 314, 941–952 (2006).
Zimmer, M. The fate and effects of ingested hydrolyzable tannins in Porcellio scaber. J. Chem. Ecol. 25, 611–628 (1999).
Wang, Y., Brune, A. & Zimmer, M. Bacterial symbionts in the hepatopancreas of isopods: Diversity and environmental transmission. FEMS Microbiol. Ecol. 61, 141–152 (2007).
Mattila, J. M., Zimmer, M., Vesakoski, O. & Jormalainen, V. Habitat-specific gut microbiota of the marine herbivore Idotea balthica (Isopoda). J. Exp. Mar. Bio. Ecol. 455, 22–28 (2014).
Cragg, S. M. et al. Lignocellulose degradation mechanisms across the tree of life. Curr. Opin. Chem. Biol. 29, 108–119 (2015).
King, A. J. et al. Molecular insight into lignocellulose digestion by a marine isopod in the absence of gut microbes. Proc. Natl. Acad. Sci. USA 107, 5345–50 (2010).
Danielsson, P. B. The Cytochrome P450 superfamily: Biochemistry, evolution and drug metabolism in humans. Curr. Drug Metab. 3, 561–597 (2002).
Hanukoglu, I. Electron transfer proteins of Cytochrome P450 systems. Adv. Mol. Cell Biol. 14, 29–56 (1996).
Imai, S.-I. Nicotinamide phosphoribosyltransferase (Nampt): A link between NAD biology, metabolism, and diseases. Curr. Pharm. Des. 15, 20–28 (2009).
Zhang, L. Q. et al. Metabolic and molecular insights into an essential role of nicotinamide phosphoribosyltransferase. Cell Death Dis. 8, e2705–11 (2017).
Erickson, A. A., Paul, V. J., Alstyne, K. L., Van & Kwiatkowski, L. M. Palatability of macroalgae that use different types of chemical defenses. J. Chem. Ecol. 32, 1883–1895 (2006).
Pan, T. C. F., Applebaum, S. L., Lentz, B. A. & Manahan, D. T. Predicting phenotypic variation in growth and metabolism of marine invertebrate larvae. J. Exp. Mar. Bio. Ecol. 483, 64–73 (2016).
De Wit, P. et al. The simple fool’s guide to population genomics via RNA‐Seq: an introduction to high‐throughput sequencing data analysis. Mol. Ecol. Res. 12, 1058–1067 (2012).
De Wit, P., Pespeni, M. H. & Palumbi, S. R. SNP genotyping and population genomics from expressed sequences - Current advances and future possibilities. Mol. Ecol. 24, 2310–2323 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Song, L. & Florea, L. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. Gigascience 4, 48 (2015).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. M. & Kelly, S. TransRate: reference free quality assessment of de-novo transcriptome assemblies. Genome Res. 26, 1134–1144 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Koskinen, P., Törönen, P., Nokso-Koivisto, J. & Holm, L. PANNZER: High-throughput functional annotation of uncharacterized proteins in an error-prone environment. Bioinformatics 31, 1544–1552 (2015).
Camacho, C. et al. BLAST plus: architecture and applications. BMC Bioinformatics 10, 1 (2009).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Panova, M., Nygren, A., Jonsson, P. R. & Leidenberger, S. A molecular phylogeny of the north-east Atlantic species of the genus Idotea (Isopoda) with focus on the Baltic Sea. Zool. Scr. 188–199, https://doi.org/10.1111/zsc.12200 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Gillis, J., Mistry, M. & Pavlidis, P. Gene function analysis in complex data sets using Ermine. J. Nat. Protoc. 5, 1148–1159 (2010).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Warren, D. L., Geneva, A. J., Lanfear, R. & Rosenberg, M. RWTY (R We There Yet): An R package for examining convergence of Bayesian phylogenetic analyses. Mol. Biol. Evol. 34, 1016–1020 (2017).
Acknowledgements
The authors would like to acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure, NGI, and Uppmax for providing assistance in massive parallel sequencing and computational infrastructure. The Centre for Marine Evolutionary Biology (CeMEB), coordinated by the University of Gothenburg kindly provided funds for laboratory materials and DNA sequencing. This work resulting from the BONUS BAMBI project was supported by BONUS (Art 185), funded jointly by the EU and the Swedish research council FORMAS.
Author information
Authors and Affiliations
Contributions
P.D.W. was responsible for experiment planning and execution, labwork, gene expression data analysis and writing the manuscript. K.Y. was responsible for transcriptome assembly and writing parts of the manuscript pertaining to this analysis. M.P. was involved in experimental design, data analysis and manuscript writing, C.A. and K.J. were involved in data analysis, manuscript writing and funding.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
De Wit, P., Yamada, K., Panova, M. et al. Diet-dependent gene expression highlights the importance of Cytochrome P450 in detoxification of algal secondary metabolites in a marine isopod. Sci Rep 8, 16824 (2018). https://doi.org/10.1038/s41598-018-34937-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34937-z
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
