
Abstract
Familial Hypercholesterolemia (FH) is a common genetic disorder caused most often by mutations in the Low Density Lipoprotein Receptor gene (LDLr) leading to high blood cholesterol levels, and ultimately to development of premature coronary heart disease. Genetic analysis and subsequent cascade screening in relatives allow diagnosis of FH at early stage, especially relevant to diagnose children. So far, more than 2300 LDLr variants have been described but only a minority of them have been functionally analysed to evaluate their pathogenicity in FH. Thus, identifying pathogenic mutations in LDLr is a long-standing challenge in the field. In this study, we investigated in vitro the activity p.(Asp47Asn) and p.(Thr62Met) LDLr variants, both in the LR1 region. We used CHO-ldlA7 transfected cells with plasmids carrying p.(Asp47Asn) or p.(Thr62Met) LDLr variants to analyse LDLr expression by FACS and immunoblotting, LDL binding and uptake was determined by FACS and analysis of mutation effects was assessed in silico. The in vitro activity assessment of p.(Asp47Asn) and p.(Thr62Met) LDLr variants shows a fully functional LDL binding and uptake activities. Therefore indicating that the three of them are non-pathogenic LDLr variants. These findings also emphasize the importance of in vitro functional LDLr activity studies to optimize the genetic diagnosis of FH avoiding the report of non-pathogenic variants and possible misdiagnose in relatives if cascade screening is carried out.
Similar content being viewed by others


Introduction
Familial Hypercholesterolemia (FH) (OMIM 143890) is a common genetic disease caused most often by mutations in the Low Density Lipoprotein Receptor gene (LDLr; MIM# 606945). These heterogeneous LDLr variants impair the function of the receptor pathway, leading to high blood cholesterol levels, xanthomas, cholesterol accumulation in peripheral tissues and development of atherosclerosis at an early age, ultimately leading to development of premature coronary heart disease (CHD)1. With a heterozygous prevalence as high as 1 in 200 in some populations2, FH constitutes one of the most common genetic disorders leading to an increased cardiovascular risk3. In recent years, the use of new sequencing technologies such as next-generation sequencing and target exome sequencing has enabled the identification of an increased number of new LDLr variants4,5,6. So far, more than 2300 LDLr variants related to FH have been described (ClinVar database). However, only a minority of these variants have been functionally analysed to evaluate their pathogenicity. An improvement of diagnosis and prognosis of FH patients has been highly recommended by World Health Organization (WHO, 1998). The ability to identify FH patients at the earliest opportunity is both economically and socially beneficial with implications for mortality and morbidity7. In 2015, the American College of Medical Genetics and Genomics (ACMG) published an algorithm with the goal of facilitating diagnosis when there is a lack of functional evidence for variant pathogenicity8. According to the ACMG algorithm, variants can be classified as: (i) pathogenic, (ii) likely pathogenic, (iii) uncertain significance, (iv) likely benign, or (v) benign.
It is important to note that at present, there is insufficient data about most LDLr variants to support a quantitative assignment of variant certainty to any of these five categories given the heterogeneity of the disease. Thus, distinguishing pathogenic mutations in LDLr from non-pathogenic ones is a long-standing challenge in the field9,10. To understand the clinical significance of these variants, in vitro functional validation must be performed to enable a definite genetic diagnosis of FH. An accurate genetic diagnosis of FH is not only beneficial for index cases but also for their relatives, since cascade screening is usually carried out in FH diagnosis enabling earlier management. Currently, FH is still underdiagnosed despite the trend toward increased genetic diagnosis.
Recently, accurate and accessible methodologies have been developed to assess LDLr activity in vitro9,11,12,13. Thus, experimental reproducibility between laboratories all over the world ensures rigorous verification of all functional studies performed for each variant.
In the present study, we aimed to functionally characterize the effects of missense mutations found in FH patients whose effect on LDLr is not established.
Materials and Methods
LDLr Variant Selection
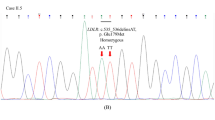
Two LDLr variants - p.(Asp47Asn) and p.(Thr62Met) - were selected to be functionally characterized for three reasons. (1) There was a possibility of an associated LDL binding defect; (2) these variants had been previously documented in FH patients, and (3) the effect of these variants had not been characterized before. Specifically, p.(Asp47Asn) and p.(Thr62Met) were identified through the ClinVar database (https://clinvarminer.genetics.utah.edu) and have been found in 2 and 9 index cases, respectively, by the LIPOchip® platform14 and/or by the SEQPRO LIPO RS® platform from Progenika Biopharma (Derio, Spain), both platforms with the CE mark. Description of the studied variants, conservation and in silico predictions are shown in Table 1.
Construction of p.(Asp47Asn) and p.(Thr62Met) LDLr carrying plasmids
Plasmids containing the p.(Asp47Asn) and p.(Thr62Met) LDLr variants were generated by Innoprot (Derio, Spain). Briefly, variants were introduced into the human LDLr cDNA (NM_000527.4), using the mammalian expression vector pcDNA3 under control of a SV40 promoter by oligonucleotide site-directed mutagenesis using the QuickChange Lightning mutagenesis kit (Agilent) according to the manufacturer’s instructions. The oligonucleotides used to generate the plasmid carrying p.(Asp47Asn) and p.(Thr62Met) LDLr variants was synthesized in vitro and subcloned using the restriction enzymes SacII and EcoRI. The presence of the desired nucleotide alteration was confirmed by PCR and restriction enzyme digestion of the appropriate fragments. The integrity of the remaining LDLr cDNA sequence of the construct was verified by direct sequence analysis.
CHO-ldlA7 Cell culture and transfection
CHO-ldlA7 cells not expressing LDLr kindly provided by M. Krieger (MIT, MA, USA) were maintained in Ham’s F-12 medium containing 10% FBS, 2 mM L-glutamine and antibiotics (100 units/mL penicillin; 100 μg/mL streptomycin). LDLr CHO-ldlA7 cells grown into 6- or 24-well culture plates at 80% confluence were transfected with Lipofectamine® LTX-PlusTM Reagent (Invitrogen) following manufacturer’s indication. LDLr functionality was assessed 48 h after transfection.
Immunodetection of LDLr
Cells were lysed using an ice‐cold buffer containing 50 mM Tris–HCl, pH 7.5, 125 mM NaCl, 1% Nonidet P‐40, 5.3 mM NaF, 1.5 mM NaP, 1 mM orthovanadate, 1 mg/ml complete EDTA-free protease-inhibitor cocktail (Roche), and 0.25 mg/ml Pefabloc, 4‐(2‐aminoethyl)‐benzenesulfonyl fluoride hydrochloride (AEBSF; Roche). Cells were rotated at 4 °C for an hour and centrifuged at 12,000 g during 15 minutes to remove insoluble material. Proteins were fractionated by electrophoresis on non-reducing 8.5% SDS-PAGE for semi-quantitative immunoblotting. Following antibodies were added: rabbit polyclonal anti-LDLr antibody (1:500) (Progen Biotechnik GmbH, Heidelberg, Germany), anti-GAPDH antibody (1:1000) (Nordic Biosite, Täby, Sweden) and horseradish peroxidase-conjugated anti-rabbit antibody (GE Healthcare, Little Chalfont, UK). The primary antibodies were incubated overnight at 4 °C while the secondary antibody incubation was performed at room temperature for an hour. Signals were developed using SuperSignal West Dura Extended Substrate (Pierce Biotechnology, Rockford, IL, USA) in a ChemiDoc XRS (Bio-Rad, Hercules, CA, USA). NIH ImageJ software (http://rsbweb.nih.gov/ij/) was used for band intensity quantification, levels of protein of interest were corrected to GAPDH loading control band intensities. Original blot can be found in Supplementary Fig. S1.
Lipoprotein isolation
Blood plasma used for lipoprotein purification was collected from healthy individuals after 30 min centrifugation, at 12,000 × g at 4 °C. LDL (1.019–1.050 g/mL) was isolated in a sequential ultracentrifugation by adjusting plasma density to 1.21 g/mL by the addition of KBr. Next, a second PBS buffer was added to the top of the solution. Ultracentrifugation was carried out in a TST 41–14 rotor (Kontron, Germany) at 35,400 rpm for 19 h at 4 °C in a Centrikon T-21X0. The white upper band corresponding to VLDL and the intermediate orange band corresponding to LDL were collected and stored at 4 °C. Isolated lipoproteins were used within 2–3 days after purification. This study was approved by the Research Ethics Committee of the University of the Basque Country (Comité de Ética en la investigación y la práctica docente de la Universidad del País Vasco/Euskal Herriko Unibertsitatea; CEIAB/186/2014/MARTÍN PLÁGARO). Methods were carried out according to the approved guidelines. All participants signed the written informed consent. All experiments were carried out according to relevant guidelines and regulations.
Lipoprotein labelling
Lipoproteins (LDL and VLDL) were fluorescently labelled with fluorescein isothiocyanate (FITC) as described previously15. Briefly, LDL and VLDL were loaded in 0.1 M NaHCO3 (pH 9.0) pre-equilibrated Sephadex G-25 column and then incubated with 10 µl/mL FITC (2 mg/mL in DMSO) per lipoprotein millilitre (1 mg/mL) at room temperature under constant gentle agitation for 2 h. After incubation, the non-bounded FITC was eliminated by washing the lipoprotein solution in a Sephadex G-25 column previously balanced in PBS EDTA-free buffer. Protein concentration was determined in all fractions using BSA as standard (Pierce BCA protein assay, Pierce).
Analysis of LDLr expression by
Expression of LDLr at cell membrane was assessed in a FACScalibur using the following antibodies: Mouse anti-human-LDLr (1:100; 2.5 mg/L; Progen Biotechnik GmbH) and Alexa Fluor 488-conjugated goat anti-mouse IgG (1:100; Molecular Probes) as secondary antibody. The inmunostaining was performed as previously described11. Briefly, cells were incubated for 1 hour at room temperature with the primary antibody after consecutive fixing and blocking steps. Cells were finally washed 3 times in PBS-1%BSA and incubated for 1 hour at room temperature with the secondary antibody. Each sample was performed in triplicate and data analysis was acquired with 10,000 events.
Analysis of LDLr activity (lipoprotein binding and uptake) by FACS
Cells were seeded in 24-well plates, at 106 cells/well and transfected as previously described when optimal concentration was reached. 48 hours after transfection, 20 µg/mL FITC-lipoprotein (VLDL or LDL) was added to the cell culture medium and cells were incubated within 4 hours at 37 °C or at 4 °C to determine LDLr activity and its binding to the different lipoproteins respectively. After incubation, cells were rinsed with PBS supplemented with 1% BSA, fixed in 4% paraformaldehyde for 10 minutes and washed again to remove the remaining fixative.
Lipoprotein uptake was determined by adding Trypan blue solution (0.2% final concentration, Sigma-Aldrich, Steinheim, Germany) to the samples. This procedure allows quenching of the extracellular FITC-signal coming from the non-internalized lipoprotein-LDLr complexes. Geometric mean fluorescence intensity of each sample was determined in a FACScalibur Flow cytometer following the manufacturer instructions13. Geometric mean fluorescence intensity of 10,000 events was acquired for each sample. Every assay determination was performed at least three independently times.
Statistical analysis
All measurements were performed at least 3 times, with n = 3 unless otherwise specified, and results represent the mean ± S.D. The differences between p.(Asp47Asn) and p.(Thr62Met) LDLr variants and wt LDLr were tested by a two-tailed Student’s t-test, P-values < 0.05 were considered statistically significant.
Results
In silico prediction analysis of pathogenicity of LDLr variants
Three different software programs were used to predict pathogenicity of p.(Asp47Asn) and p.(Thr62Met) variants. Table 1 shows the obtained results, p.(Asp47Asn) and p.(Thr62Met) LDLr variants.were predicted as pathogenic by SIFT and Mutation taster software and as probably damaging by Polyphen-2 software. These algorithms use mainly amino acid conservation analysis and as the conservation of the two amino acids is very high, the obtained predictions were the expected ones. To confirm these predictions, in vitro functional validation of these LDLr was next assayed.
p.(Asp47Asn) and p.(Thr62Met) LDLr variant expression
Expression of p.(Asp47Asn) and p.(Thr62Met) LDLr variant was analysed by Western blot in CHO-ldlA7 transfected cells as detailed above. Figure 1A (upper panel) shows that expression of p.(Asp47Asn) and p.(Thr62Met) LDLr variants is similar as wt 48 h post-transfection. Immunoblotting was performed to detect GAPDH protein (Fig. 1A, lower panel) in order to confirm equal loading of cytosolic extracts. Then, quantitative densitometry was performed to determine relative levels of LDLr expression (Fig. 1B). These results were further corroborated by assessing LDLr expression by FACS, as shown in Fig. 1C expression of p.(Asp47Asn) and p.(Thr62Met) LDLr variants, determined by IgG-C7 antibody resulted similar to the wt LDLr.

Expression of wt LDLr, p.(Asp47Asn) and p.(Thr62Met) LDLr variants in CHO-ldlA7 transfected cells. Cells were transfected with the corresponding plasmids, LDLr was overexpressed for 48 h and then cells were lysed and analysed by (A) Western blot as described in Materials and Methods section (B) relative band intensity of mature LDLr protein expression was calculated as the ratio of 160 kDa LDLr band intensity to that of GAPDH. (C) LDLr expression of p.(Asp47Asn) and p.(Thr62Met) LDLr variants determined by FACS, two internal controls were used, p.(Trp87)* (a null allele mutant), and Ex3_4del LDLr variant that produces a defective binding LDLr. A representative experiment from three independently performed assays is shown in A. The differences between p.(Asp47Asn) and p.(Thr62Met) LDLr variants and wt LDLr was determined by a two-tailed Student’s t-test, P-values < 0.05 were considered as statistically significant.in (B). The values in (C) represent the mean of triplicate determinations (n = 3); error bars represent ± SD. *P < 0.001 compared to the wt using a Student’s t-test.
p.(Asp47Asn) and p.(Thr62Met) LDLr variant activity
Activity of p.(Asp47Asn) and p.(Thr62Met) LDLr variants was assessed in CHO-ldlA7 cells transfected cells. LDL binding and uptake was determined by FACS using p.(Trp87)* (a null allele mutant), and Ex3_4del LDLr variant that produces a defective binding LDLr as internal controls of the assay16. As shown in Fig. 2A, LDL-LDLr binding activity of p.(Asp47Asn) and p.(Thr62Met) variants resulted similar than wt LDLr (Fig. 2A). As shown in Fig. 2B, uptake of FITC-labelled LDL in cells expressing p.(Asp47Asn) and p.(Thr62Met) variants was similar to that of wt.

LDLr activity of wt, p.(Asp47Asn) and p.(Thr62Met) LDLr variants. (A) LDL-LDLr binding after 4 h incubation at 4 °C; and (B) LDL internalisation efficiency after 4 h incubation at 37 °C. Geometric fluorescence intensity of 10,000 events was acquired in a Facscalibur; extent of LDL binding and uptake was calculated as described in Materials and Methods. The values represent the mean of triplicate determinations (n = 3); error bars represent ± SD. *P < 0.001 compared to the wt using a Student’s t-test.
Discussion
We here report a functional characterization of LDLr activity focused on two LDLr missense variants: p.(Asp47Asn) and p.(Thr62Met), both located at the first cysteine rich domain (LR1) of the ligand binding domain (LBD). The two of them have been previously reported17,18,19,20,21,22,23. They are included in the ClinVar database by different submitters, 4 and 11, respectively. Both variants are classified as “Uncertain significance” by the majority of them but for both variants there is a submitter who classifies them as “pathogenic”. It is remarkable that all of them are predicted by in silico software programs as pathogenic or probably pathogenic, thus ascertaining their activity in vitro allows determining their pathogenicity, which is essential for a genetic diagnosis and appropriate clinical counselling.
The LDLr binding domain contains seven cysteine-rich domains (LR1-LR7)24, each of which contains about 40 amino acids and, each domain conformation is stabilized by three disulphide bridges25. It has been shown that combination of multiple LR’s contributes to lipoprotein recognition and binding, including LR126. As mentioned above, p.(Asp47Asn) and p.(Thr62Met) are located at the LR1 and both are predicted as pathogenic by in silico software programs. Despite being previously described, none of these variants had been functionally characterized. In order to enable diagnoses of patients with these LDLr variants, we assessed the activity of p.(Asp47Asn) and p.(Thr62Met) LDLr varians to gain insight into their role in FH development.
A review of the secondary structure adopted by LR125,27,28,29 shows that Asp47 is involved in a turn formed by residues 46–49 that allow the segment between Cys(IV) and Cys(V) to loop around the side chain of Trp4429. According to our data which show no loss of activity of p.(Asp47Asn) LDLr variant, we surmise that the turn is not affected by the amino acid substitution and that LR1 retains correct folding to allow lipoprotein binding.
It is well established that the six cysteines present in each LR interact via Cys(I)-Cys(III), Cys(II)-Cys(V), and Cys(IV)-Cys(VI) disulfide bonds thus allowing the required structure to coordinate one calcium ion. Given the stringency of these Cys-Cys interactions, it is possible that maintaining the loops that allow proper Cys-Cys bonding requires highly restricted spacing between cysteine pairs. Despite this, spacing between the Cys(V) and Cys(VI) residues in the 7 LRs can vary up to two residues28. It is also remarkable that the Thr residue is only present adjacent to Cys(VI) in LR128 which could suggest a special role for that amino acid in maintaining LR1 conformation, however, and as shown by our results, Thr62 replacement by a Met, does not impair LDLr activity. As shown by western blot and FACS analysis, p.(Thr62Met) LDLr variant is correctly expressed and retains a fully lipoprotein binding and uptake activity. This result can be expected because methionine side chains are relatively flexible, therefore allowing the local chain to adopt the correct LR1 structure. These results indicate that p.(Thr62Met) LDLr variant is a non-pathogenic LDLr variant.
Despite more than 2300 variants have been associated with FH (ClinVar database) and being one of the most common genetic disorders leading increased cardiovascular risk2, clinical diagnosis of FH is still understimated7. The small number of LDLr variants that have been functionally proved to cause FH so far is a good indicator of the importance of functional characterization because functional verification represents the best reference for providing a definite genetic FH diagnosis.
In conclusion, the data presented here puts into the spotlight the relevance of functional validation of LDLr variants to provide a definite clinical diagnosis of FH. We have assessed the activity of p.(Asp47Asn) and p.(Thr62Met) LDLr variants showing that neither of them is pathogenic. This is of special relevance because they are not the cause of the dyslipidemia found in their carriers, so further analysis should be done in these patients to find the real cause underlying FH. This in vitro functional validation that can be used routinely is necessary to identify the pathogenic variants that can enable more personalized treatments and improved prognosis.
References
Goldstein, J. L., Hobbs, H. H. & Brown, M. S. Familial hypercholesterolemia. In: Scriver CR, AB, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York, N. Y.: McGraw-Hill (2001).
Nordestgaard, B. G. et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 34, 3478–3490a, https://doi.org/10.1093/eurheartj/eht273 (2013).
Khera, A. V. et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J Am Coll Cardiol 67, 2578–2589, https://doi.org/10.1016/j.jacc.2016.03.520 (2016).
Chiou, K. R., Charng, M. J. & Chang, H. M. Array-based resequencing for mutations causing familial hypercholesterolemia. Atherosclerosis 216, 383–389, https://doi.org/10.1016/j.atherosclerosis.2011.02.006 (2011).
Maglio, C. et al. Genetic diagnosis of familial hypercholesterolaemia by targeted next-generation sequencing. J Intern Med 276, 396–403, https://doi.org/10.1111/joim.12263 (2014).
Vandrovcova, J. et al. The use of next-generation sequencing in clinical diagnosis of familial hypercholesterolemia. Genet Med 15, 948–957, https://doi.org/10.1038/gim.2013.55 (2013).
Henderson, R., O’Kane, M., McGilligan, V. & Watterson, S. The genetics and screening of familial hypercholesterolaemia. J Biomed Sci 23, 39, https://doi.org/10.1186/s12929-016-0256-1 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424, https://doi.org/10.1038/gim.2015.30 (2015).
Benito-Vicente, A. et al. The importance of an integrated analysis of clinical, molecular, and functional data for the genetic diagnosis of familial hypercholesterolemia. Genet Med 17, 980–988, https://doi.org/10.1038/gim.2015.14 (2015).
Fokkema, I. F., den Dunnen, J. T. & Taschner, P. E. LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum Mutat 26, 63–68, https://doi.org/10.1002/humu.20201 (2005).
Etxebarria, A. et al. Advantages and versatility of fluorescence-based methodology to characterize the functionality of LDLR and class mutation assignment. PLoS One 9, e112677, https://doi.org/10.1371/journal.pone.0112677 (2014).
Etxebarria, A. et al. Functional characterization and classification of frequent low-density lipoprotein receptor variants. Hum Mutat 36, 129–141, https://doi.org/10.1002/humu.22721 (2015).
Etxebarria, A. et al. Functional characterization of splicing and ligand-binding domain variants in the LDL receptor. Hum Mutat 33, 232–243, https://doi.org/10.1002/humu.21630 (2012).
Palacios, L. et al. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis 221, 137–142, https://doi.org/10.1016/j.atherosclerosis.2011.12.021 (2012).
Dardik, R. et al. Homocysteine and oxidized low density lipoprotein enhanced platelet adhesion to endothelial cells under flow conditions: distinct mechanisms of thrombogenic modulation. Thromb Haemost 83, 338–344 (2000).
Esser, V., Limbird, L. E., Brown, M. S., Goldstein, J. L. & Russell, D. W. Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J Biol Chem 263, 13282–13290 (1988).
Bunn, C. F., Lintott, C. J., Scott, R. S. & George, P. M. Comparison of SSCP and DHPLC for the detection of LDLR mutations in a New Zealand cohort. Hum Mutat 19, 311, https://doi.org/10.1002/humu.9021 (2002).
Heath, K. E., Humphries, S. E., Middleton-Price, H. & Boxer, M. A molecular genetic service for diagnosing individuals with familial hypercholesterolaemia (FH) in the United Kingdom. Eur J Hum Genet 9, 244–252, https://doi.org/10.1038/sj.ejhg.5200633 (2001).
Miyake, Y. et al. Update of Japanese common LDLR gene mutations and their phenotypes: Mild type mutation L547V might predominate in the Japanese population. Atherosclerosis 203, 153–160, https://doi.org/10.1016/j.atherosclerosis.2008.07.005 (2009).
Bertolini, S. et al. Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis 227, 342–348, https://doi.org/10.1016/j.atherosclerosis.2013.01.007 (2013).
Fouchier, S. W., Kastelein, J. J. & Defesche, J. C. Update of the molecular basis of familial hypercholesterolemia in The Netherlands. Hum Mutat 26, 550–556, https://doi.org/10.1002/humu.20256 (2005).
Guardamagna, O. et al. The type of LDLR gene mutation predicts cardiovascular risk in children with familial hypercholesterolemia. J Pediatr 155, 199–204 e192, https://doi.org/10.1016/j.jpeds.2009.02.022 (2009).
Thormaehlen, A. S. et al. Systematic cell-based phenotyping of missense alleles empowers rare variant association studies: a case for LDLR and myocardial infarction. PLoS Genet 11, e1004855, https://doi.org/10.1371/journal.pgen.1004855 (2015).
Fass, D., Blacklow, S., Kim, P. S. & Berger, J. M. Molecular basis of familial hypercholesterolaemia from structure of LDL receptor module. Nature 388, 691–693, https://doi.org/10.1038/41798 (1997).
Bieri, S., Djordjevic, J. T., Daly, N. L., Smith, R. & Kroon, P. A. Disulfide bridges of a cysteine-rich repeat of the LDL receptor ligand-binding domain. Biochemistry 34, 13059–13065 (1995).
Iwasaki, T. et al. The important role for betaVLDLs binding at the fourth cysteine of first ligand-binding domain in the low-density lipoprotein receptor. J Hum Genet 49, 622–628, https://doi.org/10.1007/s10038-004-0198-4 (2004).
Bieri, S. et al. Folding, calcium binding, and structural characterization of a concatemer of the first and second ligand-binding modules of the low-density lipoprotein receptor. Biochemistry 37, 10994–11002, https://doi.org/10.1021/bi980452c (1998).
Daly, N. L., Scanlon, M. J., Djordjevic, J. T., Kroon, P. A. & Smith, R. Three-dimensional structure of a cysteine-rich repeat from the low-density lipoprotein receptor. Proc Natl Acad Sci USA 92, 6334–6338 (1995).
Djordjevic, J. T., Bieri, S., Smith, R. & Kroon, P. A. A deletion in the first cysteine-rich repeat of the low-density-lipoprotein receptor leads to the formation of multiple misfolded isomers. Eur J Biochem 239, 214–219 (1996).
Acknowledgements
We thank Rocío Alonso for her excellent technical assistance and Dr. Monty Krieger for kindly providing CHO-ldlA7 cells. This work was supported by ELKARTEK 2016 and and the Basque Government (Grupos Consolidados IT849-13). A.B.-V. and S.J. were supported by a grant PIF (2014–2015) and (2018–2021), Gobierno Vasco respectively.
Author information
Authors and Affiliations
Contributions
A.B.-V., H.S., K.B.U., S.J., U.G.-G., A.L.-S.; performed experiments. M.S. and L.P. Selected the variants. L.P. performed in silico assays. H.O. and C.M. Conceived experiments. C.M. Wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
Progenika Biopharma SA, is an Spanish biotech company founded in 2000 with headquarters in Derio, Bizkaia (SPAIN). Progenika Biopharma SA did not play a role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript and only provided financial support in the form of authors’ salaries to M.S. and L.P. A.B.-V., H.S., K.B.U., S. J., U.G.-G., A.L.-S., H.O. and C.M. declare no competing interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Benito-Vicente, A., Siddiqi, H., Uribe, K.B. et al. p.(Asp47Asn) and p.(Thr62Met): non deleterious LDL receptor missense variants functionally characterized in vitro. Sci Rep 8, 16614 (2018). https://doi.org/10.1038/s41598-018-34715-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34715-x
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
