
Abstract
The Ethylene response factor (ERF) belongs to the APETALA2/ethylene response factor (AP2/ERF) superfamily, located at the end of the ethylene signalling pathway, and has important roles in regulating the ethylene-related response genes. Thus, identifying and charactering this transcription factor would be helpful to elucidate ethylene related fruit ripening regulation in Chinese jujube (Ziziphus jujuba Mill.). In the present study, 119 AP2/ERF genes, including 5 Related to ABI3/VPs (RAV), 17 AP2s, 57 ERFs, 39 dehydration-responsive element-binding (DREB) factors and 1 soloist gene, were identified from the jujube genome sequences. Genome localization, gene duplication, phylogenetic relationships and conserved motifs were simultaneously analysed. Using available transcriptomic data, 85 genes with differential transcripts in the flower, leaf and fruit were detected, suggesting a broad regulation of AP2/ERF genes in the growth and development of jujube. Among them, 44 genes were expressed in the fruit. As assessed by quantitative PCR, 15 up- and 23 downregulated genes corresponding to fruit full maturity were found, while in response to 100 μl l−1 ethylene, 6 up- and 16 downregulated genes were generated. By comparing the output, ZjERF54 and DREB39 were found to be the best candidate genes that positively participated in jujube fruit ripening, while ZjERF25 and ZjERF36, which had an ERF-associated amphiphilic repression (EAR) motif, were ripening repressors. These findings help to gain insights into AP2/ERF gene evolution and provide a useful resource to further understand the ethylene regulatory mechanisms underlying Chinese jujube fruit ripening.
Similar content being viewed by others

Introduction
Chinese jujube (Ziziphus jujuba Mill.) belongs to the Rhamnacease family and is a traditionally popular fruit crop that is native to China1. The fruit has been introduced worldwide due to its immensely nutritional and economic benefits2,3. However, the fresh fruit has a short shelf life; the harvested fruit rots easily, with substantial water loss within 2–3 days under normal ambient conditions4. Knowledge of fruit ripening characterization and its molecular regulation is limited but is urgently required for the development of the jujube industry. Recently, increasing expression levels of ethylene metabolism pathway genes at fruit full maturity have been characterized, suggesting that ethylene-dependent pathways are involved in the ripening of this non-climacteric fruit5. Ethylene response factor (ERF), located at the end of the ethylene signalling pathway, has been found to mediate ethylene-regulated gene expression6. Thus, identification and characterization of the ERF genes in jujube would help understand the ethylene-related ripening regulation, and would also help to improve fruit storage and quality in the long term.
The ERF genes belong to the large superfamily of APETALA2/ethylene response factor (AP2/ERF), which are some of the most important plant transcription factors (TFs). The genes involved in this superfamily commonly share a conserved AP2 domain that consists of approximately 60 amino acid residues and binds to specific DNA sites located in gene promoters, such as the GCC box and the dehydration-responsive element (DRE)7. According to the differences in domain sequence, this superfamily is classified into four families, including Related to ABI3/VP (RAV), AP2, ERF and soloist8. RAV family genes contain an AP2 and a B3 domain, and AP2 family genes usually have multiple repeated AP2 domains, while ERF family genes have a single AP2 domain and are further divided into ERF and the dehydration-responsive element-binding (DREB) subfamily based on the amino acid residue sequence. The remaining genes are named soloist, displaying a low similarity with other family members.
The AP2/ERF superfamily genes have been identified in several plant species, such as 145 types of Arabidopsis9, 167 types of rice10, 121 types of barley11, 146 types of tomato12 and 119 types of kiwifruit13. Increasing research has focused on gene function analyses. For instance, the AP2 family has important roles in regulating flowering time and organ development14,15; the RAV family functions in plant development, abiotic stress responses and disease resistance16,17 and soloist enhances plant tolerance to salt stress and accumulated basal defence against bacterial pathogens18,19. More complex regulatory mechanisms of the ERF and DREB subfamily have been reported, such as involvement in biotic and abiotic stress responses20,21, plant hormone metabolism22 and level of fruit quality23. Recently, the role of the ERF and DREB family in fruit ripening regulation has been recognized. In tomatoes, SlERF.E1, SlERF.E2 and SlERF.E4 were characterized as the main ripening-associated ERF members in ethylene-dependent ripening6. The MaERF11 and MaDREB2 genes acted as a negative regulator in banana fruit ripening24,25,26. These results have suggested the critical effects of AP2/ERF TFs on normal plant growth and fruit ripening processes. However, knowledge related to this superfamily in Chinese jujube is still lacking.
The draft genome of the Chinese jujube has been released27, which has enabled studies on the molecular functional regulation at a genome-wide scale. In the current study, AP2/ERF superfamily genes were identified through the Chinese jujube genome. Gene structure, chromosome localization and gene duplication were simultaneously investigated. Phylogenetic and conserved motif analyses helped to cluster these genes. Tissue-specific expression was also detected with the available transcriptome sequencing dataset. Candidate critical genes associated with five fruit ripening stages were identified, and their expressions in response to exogenous ethylene were determined by quantitative polymerase chain reaction (qPCR). These findings provide insights into the understanding of AP2/ERF gene evolution and further the understanding of the ethylene regulatory network in fruit ripening in Chinese jujube.
Results
Identification and classification of AP2/ERF genes in Chinese jujube
After screening the Chinese jujube genome, a total of 119 genes containing AP2 domain sequences were identified as AP2/ERF superfamily genes (Supplementary File S1). Among the genes, a single gene (Zj.jz031429031), which displayed homology with At4g13040, was classified in the soloist family. According to differences in conserved domains in their encoding proteins (Supplementary File S2), the other 118 genes were classified into three families: 5 genes belonged to the RAV family, containing both an AP2 and a B3 domain; 17 genes belonged to the AP2 family, including 15 genes that had two repeated AP2 domains, 2 genes (ZjAP2.5 and ZjAP2.14) that had only one AP2 domain, and 1 gene (ZjAP2.17) that had four repeated AP2 domains; and the remaining 96 genes belonged to the ERF family, with only one AP2 domain. The deduced amino acid sequences of the AP2 domains in the ERF family were further analysed (Supplementary File S3), and these genes were classified into two subfamilies: 57 members were identified in the ERF subfamily, and 39 members belonged to the DREB subfamily.
A summary of AP2/ERF superfamily genes is listed in Table 1. All of the identified gene lengths ranged from 402 to 6536 bp, and the number of amino acid residues ranged from 133 to 889. The number of introns varied widely among the different families (Table 1, Supplementary File S4). For instance, all AP2 family genes had 3 to 14 introns, and RAV family genes had no introns, while the soloist family had 5 introns. In the ERF or DREB subfamilies, most of the genes had no introns, except ZjERF4, ZjERF42, ZjERF43, ZjERF52, ZjERF53, ZjERF54, ZjDREB19, ZjDREB20, ZjDREB25 and ZjDREB33, which all had one intron.
Phylogenetic relationships and conserved motif analysis
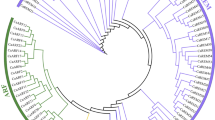
A phylogenetic tree for the AP2/ERF superfamily of jujube and Arabidopsis was constructed based on deduced protein sequences (Supplementary File S5). These genes were significantly classified into three clades as either ERF, DREB, or a mixed clade containing the RAV, AP2, soloist and 13 ERF family genes, which was consistent with the classification described above. When analysed in depth, the ERF family was divided into ten clades (I-X) (Fig. 1), with respect to the previous gene classification in Arabidopsis7. The DREB subfamily contained five clades (I-V), and the remaining five clades (VI-X) belonged to the ERF subfamily.

Phylogenetic relationship of AP2/ERF superfamily genes between Chinese jujube and Arabidopsis. The branch lines for each gene family are coloured consistently. Red, ERF subfamily; Blue, DREB subfamily. The ten clades (I-X) of ERF family genes were divided according to previous classification in Arabidopsis. Each clade is shown in a different coloured circle strip.
The conserved motifs in the ERF family encoding proteins were further investigated. In the ERF subfamily, a total of 19 conserved motifs were detected (Table 2, Fig. 2). Among them, motif 1 and motif 2, the AP2 domain-related motifs, were widely distributed in all subfamily members except for ZjERF56, which lacked this region; while the other motifs were located on different numbers of proteins that varied from 2 to 51. Notably, motif 12, an ERF-associated amphiphilic repression (EAR) motif containing (L/F) DLN (L/P) residues, specifically localized in ZjERF21, ZjERF22, ZjERF23, ZjERF24, ZjERF25, ZjERF36 and ZjERF39. Motif 13, containing the Cys repeat sequence CX2CX4CX2~4C, is likely a zinc-finger motif and is distributed in ZjERF19, ZjERF40, and ZjERF41. In the DREB subfamily, a total of 16 motifs were identified with 39 genes (Table 2, Fig. 3). Motif 1 and motif 2 were commonly shared among these genes, except ZjDREB25. Motif 3 was found in 37 genes, except ZjDREB2 and ZjDREB7. The other motifs were located on different numbers of genes that ranged from 2 to 23. Motif 1 and motif 3 were AP2 domain-related motifs. Motif 2 and motif 4 were identified to have the conserved amino acid residues LNFP and D[IV]QAA, respectively. Detailed information of each motif distribution is listed in Supplementary File S6.

Phylogenetic relationships, conserved motifs and gene structures of the ERF subfamily. The 19 conserved motifs identified by MEME are indicated by coloured rectangles.

Phylogenetic relationships, conserved motifs and gene structures of the DREB subfamily. The 16 conserved motifs identified by MEME are indicated by coloured rectangles.
Chromosome distribution and duplication of AP2/ERF superfamily genes
Among the identified genes, 116 out of 119 were assigned to 12 linkage groups (LGs), which is consistent with the haploid chromosome number of Chinese jujube (Fig. 4). However, three genes, ZjERF1, ZjERF49 and ZjERF51, were not assigned to LGs, but to scaffolds 5919, 219482, and 218390, respectively (Supplementary File S7). The numbers of AP2/ERFs located on each LG ranged widely. LG07 anchored a maximum number of 17 genes, while only two genes were anchored to LG05.

Chromosomal localization and duplication pairs of AP2/ERF genes between Chinese jujube and Arabidopsis. The numbers 1–5 indicate the chromosomes of Arabidopsis, and LG 01-12 indicate the linkage groups of Chinese jujube.
To assess genome duplications, relationship of homologous and paralogous AP2/ERFs genes between jujube and Arabidopsis were analysed. A total of 110 pair relationships were found, containing 41 co-orthologous gene pairs between jujube and Arabidopsis, 18 paralogous pairs in jujube, and 51 paralogous pairs in Arabidopsis (Supplementary File S8). Among the 18 paralogues in jujube, five paralogues (ZjERF45-ZjERF46, ZjERF14-ZjERF15, ZjDREB14-ZjDREB16, ZjDREB15-ZjDREB16, ZjERF30-ZjERF32) were identified as tandem duplication, and the remaining 13 paralogues were classified as the products of segmental duplication.
Specific expression of AP2/ERF superfamily genes in leaf, flower and fruit tissue
Among the 119 AP2/ERF superfamily genes, transcripts of 85 (71.4%) of genes were detected in at least one tissue of leaf, flower or fruit using available transcriptomic data. Heatmap analysis clustered these genes into four districted clades according to differential expression patterns (Fig. 5).

Hierarchical cluster of heatmap for AP2/ERF gene expression in leaf, flower and fruit. The cluster was generated using the Pearson clustering algorithm according to gene expression profiles from the transcriptome data. For each row, blue and red correspond to low and high expression values, respectively, after z-score-normalized transformation. The number for each nod indicates the similarity value.
Clade I (separated by a similarity value of −0.4492) included 8 genes, showing much higher expression in fruit than in leaf and flower; among those genes, ZjDREB12 and ZjDREB17 were specifically expressed in fruit. Clade II (separated by a similarity value of −0.0924) contained 12 genes, exhibiting higher expressions in leaf; among those genes, three genes (ZjAP2.13, ZjDREB11, ZjRAV2) were specifically expressed in leaf. The remaining 65 genes showed higher expression levels in flower, while these genes could be further divided into two different clades. Clade III (separated by a similarity value of 0.6624) contained 17 genes, displaying higher expression levels in flower, which were slightly higher than those in leaf, while their transcripts in fruit were very low. Clade IV (separated by a similarity value of 0.6624) was made up of 48 genes, exhibiting the highest expression in flower, while their transcripts were very low in both fruit and leaf; 17 genes (ZjRAV4; ZjAP2.7; ZjERF16, 17, 29, 30, 31, 41, 43, 44; ZjDREB2, 7, 14, 25, 29, 35, 38) involved in this clade were specifically expressed in the flower. Notably, three genes (ZjAP2.5, ZjERF25, ZjDREB34) involved in this clade displayed the highest expression levels in flower, and their transcripts were slightly higher than those in fruit and relatively low in leaf.
Gene expression associated with jujube fruit ripening
In order to investigate the AP2/ERF gene expressions associated with fruit ripening, five developmental series were selected, including the young fruit (YF), white mature (WM, ripening onset), beginning red (BR), half-red (HR), and fully red (FR), according to the days after full bloom and their peel colour changes. Regardless of the genes not expressed in fruit, the transcripts of 44 (37.0%) genes were detected. The relative expression was visualized by heatmap, in which transcription patterns were distributed into five clades (Fig. 6).

Hierarchical cluster heatmap for AP2/ERF gene expression patterns during fruit development and ripening. The cluster was generated using the Pearson clustering algorithm according to gene expression profile analysis by qPCR. For each row, blue and red correspond to low and high values of gene expression, respectively, after z-score-normalized transformation. The number for each nod indicates the similarity value. YF, young fruit; WM, white-mature fruit; BR, beginning-red fruit; HR, half-red fruit; FR, fully red fruit.
Clade I (separated by a similarity value of −0.2804) contained eight genes. Among those genes, six of them showed preferential expression in YF; their transcripts declined at the WM stage, which represents fruit ripening onset, and they maintained low levels during the ripening process. In addition, the transcripts of ZjDREB17 and ZjERF11 were higher in YF but were also slightly upregulated in HR and FR fruit, respectively. Clade II (separated by a similarity value of −0.1264) included four genes with complicated expression patterns. The transcripts of ZjDREB36 and ZjRAV5 were highly accumulated at the WM stage but were afterward downregulated. The expression of ZjERF25 and ZjERF26 was higher in HR fruit but was downregulated at the FR stage. Clade III (separated by a similarity value of 0.1485) included 16 genes, displaying the highest expression in BR fruit; however, their expression levels declined at fruit full maturity. The remaining 16 genes showed an increased expression along with the fruit ripening process and contained genes from two clades that were separated by a similarity value of 0.5059. Clade IV included 10 genes whose relative transcriptions were high at the HR and FR stage, except ZjDREB32 and ZjERF8, which had slightly lower expression levels in FR fruit. Clade V was made up of six genes, displaying the highest expression in FR fruit, and their transcripts did not highly accumulate before full maturity.
Therefore, most genes (38 out of 44, except 6 genes in clade I) showed a ripening-associated expression pattern with either ripening onset or the dynamic process. Among those genes, 15 of them (ZjAP2.1, 2.4, 2.5; ZjERF11, 23, 24, 27, 28, 35, 37, 42, 53, 54; ZjDREB1, 39) were upregulated and positively associated with full maturity, while the other 23 genes were mostly downregulated and negatively correlated with ripening.
Gene expression in response to exogenous ethylene
To explore the role of AP2/ERF genes in ethylene-dependent ripening, gene expression was investigated upon treatment with 100 μl l−1 exogenous ethylene. The physiological data of fruit responses to ethylene were described in our previous study5, with a slightly induced respiration increase at the first day after treatment (DAT) compared with that of the control, suggesting a positive response of fruit upon exogenous ethylene. Therefore, relative expressions of 44 fruit-expressed genes at DAT 1 were analysed. A variance analysis with a t-test (p < 0.05) was conducted, showing that transcripts of 22 (50%) genes were significantly induced by exogenous ethylene, with 6 up- and 16 downregulated genes (Fig. 7).

Differentially expressed AP2/ERF genes upon 100 μl l−1 ethylene treatment at DAT1. The different letter over the bars represents the significant difference between the mean values. (A) Ethylene up-regulated genes (B) Ethylene downregulated genes.
Gene expression levels that responded to ethylene and associated with ripening were further compared. Among the 15 ripening-upregulated genes, transcripts of 6 genes were ethylene-induced, with two (ZjERF54, ZjDREB39) upregulated and four (ZjAP2.1, 2.4, 2.5; ZjERF53) downregulated. Therefore, ZjERF54 and ZjDREB39 were identified as the best candidate activators of ethylene-regulated fruit ripening in jujube. In contrast, among the 23 ripening-downregulated genes, three genes (ZjRAV5; ZjERF21, 33) were upregulated, and nine genes (ZjAP2.14; ZjERF25, 36; ZjDREB18, 22, 26, 27, 31, 34) were downregulated. Notably, ZjERF25 and ZjERF36 had an EAR motif and were the best putative repressors in ethylene-dependent ripening of jujube.
Discussion
TFs participate in the regulation of plant growth, maturation and senescence, and response to biotic and abiotic stresses, such as drought, salt, and cold. Therefore, studies on TFs are helpful for understanding the plant physiological processes and associated complex regulation networks. Although the AP2/ERF family has been widely reported in several species, the number, gene structure, sequences, and functions of this family were obviously different and diverged from other species20. Thus, the identification and characterization of the AP2/ERF genes in jujube, along with screening the best candidate genes for unique biological events, is an important investigation.
In the present study, comprehensive analyses for the AP2/ERF superfamily were performed across the Chinese jujube genome. In total, 119 genes were identified and classified into four families. The total number of genes for each family was 5, 17, 96 and 1, corresponding to the RAV, AP2, ERF and soloist families, respectively. The total number of Chinese jujube species that carry the AP2/ERF superfamily genes is lower than that in Arabidopsis (145), rice (167), barley (121), tomato (146), grape (132)28, carrot (267)29 and Populus trichocarpa (200)30. This difference has been explained as the result of gene evolution and duplication in plants11,31. Gene duplication has an important role in gene family expansion29 and tandem duplication-produced gene clusters or hot regions, while segmental duplications produce homologous genes, which expand the total gene number32. In total, we identified 18 paralogous pairs, which were produced by genome tandem and segmental duplication in jujube. Previously, the paralogous numbers of AP2/ERF genes in several plant species were reported in rice (41), grape (76), Arabidopsis (51), and carrot (264), all of which were much higher than those in jujube (18). An explanation for this lower gene number could be that fewer genome duplication events occurred in the jujube AP2/ERF superfamily.
An analyses of the phylogenetic relationships and conserved domains helped to cluster the AP2/ERF genes. The existence of AP2 domains in these genes was investigated, displaying a family-specific distribution of conserved domains. However, two genes (ZjAP2.5 and ZjAP2.14) that had only one AP2 domain were classified into the AP2 family due to a close phylogenetic relationship. This classification was similar with that in Arabidopsis, in which four genes involved in the AP2 family contained a single AP2 domain7; in the physic nut, JcAP2-12 contained one AP2 domain33. In addition, multiple alignments among sequences involved in the ERF and DREB subfamilies were generated, which identified the conserved amino acid residues of Ala-33 (A) and Asp-43(D) in the ERF subfamily, and Val-21 (V) and Glu-26 (E) in the DREB subfamily (Supplementary File S3). These amino acid sites were involved in AP2/ERF DNA-binding domains and were considered to be important for distinguishing these subfamilies9.
The number of amino acid residues and introns were also summarized in our study. Notably, the intron number in the AP2 family was larger than that in other families, while no introns were found in the RAV family, and at most one intron was found in the ERF family. This typical pattern of gene structure was consistent with those of previous findings in grape31, peach34 and Medicago truncatula32. Regardless of further investigations, we conjecture that the gene structure is associated with their various functional regulations. In addition, some studies have also suggested that the intron number and distribution are related to plant evolution, while introns of the ERF family genes were probably lost during evolution in higher plants33,35. For example, the number of introns was 61.7% of ERFs and 34.0% of DREBs in moss (Physcomitrella patens), which was markedly higher than that in physic nut (22.2% ERF and 0 DREB) and Arabidopsis (27.7% ERF and only 1 DREB)33. Our results showed that 6 (10.5%) ERFs and 4 (10.2%) DREBs had only one intron, which also supports the previous hypothesis.
Conserved motif analyses provided further insights into gene evolution and potentially functional differences. In the ERF subfamily, 19 motifs distributed around different numbers of genes. Motif 1 and 2 contained a wide region of the AP2 domain, which mainly consisted of three β-sheet regions and one α-helix7. The other motifs were shared among different clades and were associated with specific functions, such as motif 12, which was identified as an EAR motif that displayed repression functions32,36. Motif 13 was a putative zinc-finger motif and may function in DNA binding or protein-protein interactions7. In the DREB subfamily, motif 1 and motif 3 contained the largest region of AP2 domains and were commonly shared among these genes. Motif 2 and 4 contained the conserved residues of LPRP that are involved in CBL-interacting serine/threonine-protein kinase-12 (CIPK12), which is important for plant stress responses37,38. We also compared the identified motifs with those previously found in Arabidopsis and rice, and five motifs (motif 6, motif 8, motif 11, motif 13, and motif 14) were consistent with the CMIV-1, CMIII-3, LWSY, CMI-3, and CMI-2 motif, respectively. However, the functions of these motifs are still unknown, and more work is required for understanding their regulatory functions.
Due to the genome assembly of Chinese jujube that was based on the mapping of scaffolds or contigs by linkage maps27, we were able to anchor the identified AP2/ERF genes onto 12 LGs, which was consistent with haploid chromosome number of Chinese jujube. However, these genes were not uniformly located on each LG, for example, only two genes were mapped to LG 5. A previous study in M. truncatula suggested possible hot regions on chromosomes, such as ten genes that were located on chromosome 6 in a short region32. Accordingly, similar hot regions were found on LG4, LG6 and LG7 in Chinese jujube. Interestingly, tandem duplications of five gene pairs were also detected on these LGs, corroborating the theory that tandem duplication contributed to the occurrence of hot regions or gene clusters32,39. In addition, three genes were not anchored in our results, and we believe that the localization of these genes would be improved with the availability of an improved physical map for Chinese jujube.
Tissue-specific expression analysis showed that AP2/ERF superfamily genes are widely expressed across the leaf, flower and fruit, indicating critical and multiple functional regulations on plant growth and development in Chinese jujube. In total, we detected 85 genes that had transcripts in at least one tissue. The number of genes expressed in leaf, flower and fruit was 64, 80 and 44, respectively. In the RAV family, ZjRAV2 showed higher expression in leaf, and ZjRAV4 and ZjRAV5 highly accumulated in the flower. This family has been suggested to participate in regulating plant growth and abiotic defence40. In the AP2 family, ZjAP2.1 was found to be highly accumulated in the fruit, ZjAP2.3, 2.13, and 2.14 were highly expressed in leaf, and others were markedly accumulated in the flower. This family of genes carries out important roles in regulating floral and leaf organ identity14,41,42 and shows a potential role in fruit ripening regulation, such as SlAP2a functioning as a negative regulator in tomato fruit ripening43. The ZjERF.soloist gene was also highly expressed in the flower. Previous studies have indicated that the soloist family genes could enhance accumulation of salicylic acid and the basal response to stress18,19. In addition, the ERF family genes differentially expressed in tissues, with seven genes highly accumulated in fruit, eight genes expressed highly in leaf, and most of the genes having higher accumulation in the flower. The expression patterns were consistent with their diverse functions in response to hormone accumulation and signalling, biotic and abiotic stress, and plant growth and development20,31,33. Analysis of the tissue-specific expression patterns helped to gain insights into the putative gene functions, and this approach will contribute to further studies on the regulatory mechanisms of biological events in jujube.
The relative expression of AP2/ERF genes during jujube fruit development and ripening processes were visualized by heatmaps. These genes showed differential expression patterns related to each developmental stage, which indicated that the role of AP2/ERF genes were complex in fruit and were not limited to ripening regulation, also including involvement in regulating the fruit quality attributes of colour, texture, and flavour23,44,45; and also involvement in the crosstalk with other plant hormones, such as the VvERF5-1 mediated in auxin-induced upregulation of ethylene biosynthesis in grape46. Although diverse functions of AP2/ERF genes were found, our study aimed to identify genes potentially participating in fruit ripening regulation. The Chinese jujube is a non-climacteric fruit, and an increased expression of genes involved in ethylene metabolism has been found at the FR stage5, indicating the role of ethylene in regulating fruit full ripening is necessary. Therefore, transcript patterns of genes associated with fruit full maturity were identified, along with putative activators and repressors.
We identified 15 genes that were upregulated during ripening, and ZjERF54 and ZjDREB39 were induced by ethylene. Therefore, these two genes were identified to be the best candidate activators in ethylene-related jujube fruit ripening regulation. ZjERF54 belongs to subfamily VII (with respect to the nomenclature in Arabidopsis7) and was ethylene responsive and particularly associated with fruit ripening26,47,48. This subfamily includes tomato LeERF247, apple MdERF149, kiwifruit AdERF4 and AdERF650 and banana MaERF726. All of these genes were upregulated by ripening and could interact with the GCC-box-containing genes. ZjDREB39 belongs to subfamily II, and similarly, a VvERF006 gene, also involved in subfamily II, was found to be upregulated during grape fruit ripening in the flesh tissue, although its function was unknown44. In addition, a MaERF9 gene, which is involved in this subfamily, is upregulated by ethylene, displaying a strong correlation with banana ripening and possibly activating MaACO1 promoter activity26.
In contrast, 23 genes were downregulated by ripening, and 9 genes were simultaneously downregulated by ethylene. We identified two genes (ZjERF25 and ZjERF36) with an EAR motif as the putative repressors of fruit ripening. These two genes belonged to subfamily VIII and had an EAR motif that was related to suppression effects43,51. Interestingly, MaERF11 was also involved in this family, showing downregulated expression during banana fruit ripening and after ethylene treatment. MaERF11 negatively regulated banana fruit ripening via recruiting a histone deacetylase (MaHDA1)24. We also found the CiERF69 and CiERF70, which were involved in subfamily VIII with an EAR motif, showed downregulated expression during citrus fruit development and ripening45. In addition, ZjERF36 was homologous with AT1G28360 (AtERF12), which is a TF that binds to the GCC-box pathogenesis-related promoter element and acts as a transcriptional inhibitor36. These lines of evidence suggest a possible role for the identified candidate genes in jujube fruit ripening. We believe these genes should be considered in further studies on ethylene-related ripening regulations, such as the interactions of transcription factors with promoters of ripening-related genes.
In summary, a total of 119 AP2/ERF superfamily genes were identified and characterized in the Chinese jujube genome sequence. The conserved motif/domain distribution and phylogenetic relationships help classify these genes and provide insights into AP2/ERF gene evolution. The tissue-specific expression patterns reveal a broad functional regulation in the growth and development of the flower, leaf and fruit. The expression profiling of genes during fruit ripening and in response to ethylene resulted in four putative activators or repressors that are involved in jujube fruit ripening. Their functions will be investigated in further studies to better understand fruit ripening regulation in Chinese jujube.
Materials and Methods
Identification of AP2/ERF superfamily genes in Chinese jujube
The database of the gene annotation model of Chinese jujube was downloaded from the website of Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.83fr7)27 and was prepared for a local BLASTP algorithm program. The AP2/ERF superfamily genes and their encoding protein sequences, published in Arabidopsis7 and tomato6, were downloaded from the online database of EnsemblPlants (http://plants.ensembl.org/index.html)52. These proteins were used as query sequences in the local BLASTP program. The BLASTP resultant sequences with the parameters of score (bits) >200 and E-value <0.001 were retrieved and further confirmed for the presence of a conserved AP2 domain using the HMMSCAN online analysis tool (https://www.ebi.ac.uk/Tools/hmmer/search/hmmscan)53.
Phylogenetic analysis of AP2/ERF genes
Multiple sequence alignments of Chinese jujube and Arabidopsis AP2/ERF proteins were performed using Clustal Omega, version 2.1 (https://www.ebi.ac.uk/Tools/msa/clustalo/)54 with default parameters. A phylogenetic tree was subsequently constructed by the neighbour-joining method and was visualized by the Interactive Tree of Life (ITOL, http://itol.embl.de/index.shtml)55. The branches were consistently coloured according to their respective clusters. In addition, phylogenetic trees for the ERF and DREB subfamilies were individually constructed. Their protein sequences were aligned using Clustal Omega and were then visualized using MEGA 7.0 with a bootstrap replicate value of 100043.
Gene structure and conserved motif analyses
Conserved motifs of ERF and DREB subfamily proteins were identified using the online tool Multiple Em for Motif Elicitation (MEME) version 4.12.0 (http://meme-suite.org/tools/meme)56, with the following parameters: (1) the number of occurrences of a single motif distributed among the sequences within the model was set to zero or one per sequence; (2) the maximum number of motifs found was set as 25; (3) the optimum motif width was set to ≥6 and ≤50; and (4) motifs with a matched E-value should be below 0.0532. The resulting motifs, together with the full-length gene sequence data and corresponding CDS regions, were prepared for visualization of the gene structure by Gene Structure Display Server 2.0 (http://gsds.cbi.pku.edu.cn/)57. We integrated the results of the phylogenetic trees, conserved motifs and gene structures.
Genomic localization and duplication analysis of AP2/ERF superfamily genes
Genomic localization of identified genes was retrieved from the reference jujube genome annotation database27. The homology analysis was based on a calculation of the protein sequence similarities in jujube and Arabidopsis using the OrthoMCL program29,58. The default parameters were used, as blast similarities with a percent match less than 50%, and E-value exponents greater than -5 were ignored. The putative duplication events were detected for the AP2/ERF genes. Tandem duplication was identified as two proteins with a similarity of greater than 40% and separated by four or fewer gene loci; others were identified as segmental duplications, separated by more than five genes32,35. These results were visualized using Circos software (http://circos.ca/)59.
Transcriptome data source and bioinformation analysis
Transcriptome sequencing data from six samples, including two plant tissues (leaf and flower) and fruit at four different ripening stages (Z. jujuba ‘Junzao’, YF, WM, HR and FR), were previously generated by our group27. The raw data obtained from Illumina 2000 sequencing were filtered to remove low quality reads through an in-house Perl script. The resulting clean data were then submitted for mapping with the Chinese jujube genome dataset27 using TopHat v2.0.9 software60. Subsequently, the aligned reads were further processed for quantification of gene expression levels by HTSeq v0.6.161. The relative abundance of each gene was normalized as the value of reads per kilobase of exon model per million mapped reads (RPKM)62 and was then prepared for tissue-specific expression analysis.
Plant materials and treatment
The fruit of a cultivar, Z. jujuba ‘Dongzao’, at five developmental stages, YF, WM, BR, HR and FR, were collected from the Jujube Experimental Station of Northwest A&F University (Qingjian, Shaanxi, China; N 37.13, E 110.09) in 2017. The sampling periods were selected according to the days after full bloom and fruit peel colour changes during ripening: 15 DAB, YF; 85 DAB, WM (ripening onset, peel colour turned whitish-green); 100 DAB, BR (<10% red, commercially harvested); 110 DAB, HR (40–60% red); and 115 DAB, FR (100% red, full maturity). For each stage, five fruits were cut into pieces and mixed together, and the samples were immediately frozen in liquid nitrogen. The samples were transferred to a −80 °C freezer for storage until RNA isolation. These samples were prepared for the gene expression analyses of fruit undergoing the ripening processes.
The fresh fruit of ‘Dongzao’ at the WM stage (ripening onset) were harvested by hand and transferred to our lab. The fruit was washed with water and dried for 30 min at room temperature. Then, fruit were randomly divided into two groups and treated with either distilled water or 100 μl l-1 ethylene in a covered plastic container for 16 h. After treatment, the fruit were stored at 20 °C, 70% RH, in darkness. The treated fruit were then cut into pieces, frozen in liquid nitrogen, and used for expression analysis of genes in response to exogenous ethylene.
RNA isolation, cDNA synthesis and qPCR expression analyses
The total RNA was isolated using a plant RNA extraction kit (TaKaRa, Dalian, China) and was digested with DNase I according to the manufacturer’s instructions. The first strand cDNA was synthesized as 200 ng of total RNA using a PrimeScriptTM RT reagent kit with gDNA Eraser (TaKaRa). In addition, RT-qPCR was performed using a SYBR Premix Ex TaqTM II kit (TaKaRa) with a total volume of 10 μL, which contained 1.0 μL of cDNA, 5 μL of SYBR premix solution, 0.4 μM forward/reverse primers and 3.2 μL of dH2O. The PCR thermal program was set as follows: 95 °C for 5 min, followed by 40 cycles of amplification for 5 s at 95 °C, 30 s at 58 °C, 30 s at 72 °C, and a default dissociation stage in a Bio-Rad CFX Connect system. The relative expression was normalized to that of an endogenous reference gene ZjUBQ63 and was finally calculated using the 2−△Ct method64. The primers used for the qPCR analysis are listed in Supplementary File S9.
Gene expression profiling based on transcriptome and qPCR data
Hierarchical clustering of the heatmap for tissue-specific gene expression was performed using the web tool ClustVis (https://biit.cs.ut.ee/clustvis/)65 based on the transcriptome sequencing data. The following default parameters were used: the clustering distance of the Pearson correlation subtracted from 1, a clustering method of average distances of all possible pairs, tree ordering of the tightest cluster first, and number of clusters of one. Notably, the transcripts in the fruit were represented as a mean of the RPKM value at each ripening stage, and gene expression with an RPKM value below 1.0 was considered to be no expression. In addition, gene expression determination in the fruit ripening stages and upon ethylene treatment was performed by qPCR analysis, and the relative gene expressions were calculated as the mean of three biological replicates. The results were then visualized by heatmap analyses in the same methods.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Yao, S. R. Past, present, and future of jujubes- Chinese dates in the United States. Hortscience 48, 672-680 (2013).
Chen, J. P. et al. Chemical and biological assessment of Ziziphus jujuba fruits from China: different geographical sources and developmental stages. J Agric Food Chem 61, 7315–7324, https://doi.org/10.1021/jf402379u (2013).
Gao, Q. H., Wu, C. S. & Wang, M. The jujube (Ziziphus jujuba Mill.) fruit: a review of current knowledge of fruit composition and health benefits. J Agric Food Chem 61, 3351–3363, https://doi.org/10.1021/jf4007032 (2013).
Wu, H., Wang, S., Zhu, J., Meng, X. & Wang, D. Postharvest treatments affecting storage quality of Chinese jujube. In Chinese dates: a traditional functional food (ed Dongheng Liu) Ch. 15, 272–315 (CRC Press, 2016).
Zhang, Z., Huang, J. & Li, X. Transcript analyses of ethylene pathway genes during ripening of Chinese jujube fruit. J Plant Physiol 224-225, 1–10, https://doi.org/10.1016/j.jplph.2018.03.004 (2018).
Liu, M. et al. Comprehensive profiling of ethylene response factor expression identifies ripening-associated ERF genes and their link to key regulators of fruit ripening in tomato. Plant Physiol 170, 1732–1744, https://doi.org/10.1104/pp.15.01859 (2016).
Nakano, T., Suzuki, K., Fujimura, T. & Shinshi, H. Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol 140, 411–432, https://doi.org/10.1104/pp.105.073783 (2006).
Xu, Z. S., Chen, M., Li, L. C. & Ma, Y. Z. Functions and application of the AP2/ERF transcription factor family in crop improvement. J Integr Plant Biol 53, 570–585, https://doi.org/10.1111/j.1744-7909.2011.01062.x (2011).
Sakuma, Y. et al. DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem Biophys Res Commun 290, 998–1009, https://doi.org/10.1006/bbrc.2001.6299 (2002).
Sharoni, A. M. et al. Gene structures, classification and expression models of the AP2/EREBP transcription factor family in rice. Plant Cell Physiol 52, 344–360, https://doi.org/10.1093/pcp/pcq196 (2011).
Guo, B. et al. Genome-wide analysis of APETALA2/ethylene-responsive factor (AP2/ERF) gene family in barley (Hordeum vulgare L.). PLoS One 11, e0161322, https://doi.org/10.1371/journal.pone.0161322 (2016).
Pirrello, J. et al. Functional analysis and binding affinity of tomato ethylene response factors provide insight on the molecular bases of plant differential responses to ethylene. BMC Plant Biol 12, 190, https://doi.org/10.1186/1471-2229-12-190 (2012).
Zhang, A.-d. et al. Isolation, classification and transcription profiles of the ethylene response factors (ERFs) in ripening kiwifruit. Sci Hortic-Amsterdam 199, 209–215, https://doi.org/10.1016/j.scienta.2015.12.055 (2016).
Jofuku, K. D., den Boer, B. G., Van Montagu, M. & Okamuro, J. K. Control of Arabidopsis flower and seed development by the homeotic gene. APETALA2. Plant Cell 6, 1211–1225 (1994).
Huang, Z. et al. APETALA2 antagonizes the transcriptional activity of AGAMOUS in regulating floral stem cells in Arabidopsis thaliana. New Phytol 215, 1197–1209, https://doi.org/10.1111/nph.14151 (2017).
Feng, C. Z. et al. Arabidopsis RAV1 transcription factor, phosphorylated by SnRK2 kinases, regulates the expression of ABI3, ABI4, and ABI5 during seed germination and early seedling development. Plant J 80, 654–668, https://doi.org/10.1111/tpj.12670 (2014).
Wei, Y. et al. RAV transcription factors are essential for disease resistance against cassava bacterial blight via activation of melatonin biosynthesis genes. J Pineal Res, https://doi.org/10.1111/jpi.12454 (2017).
Giri, M. K. et al. The Arabidopsis thaliana At4g13040 gene, a unique member of the AP2/EREBP family, is a positive regulator for salicylic acid accumulation and basal defense against bacterial pathogens. J Plant Physiol 171, 860–867, https://doi.org/10.1016/j.jplph.2013.12.015 (2014).
Sun, Z. M. et al. Overexpression of the Lotus corniculatus soloist gene LcAP2/ERF107 enhances tolerance to salt stress. Protein Pept Lett 23, 442–449 (2016).
Sun, Z. M., Zhou, M. L., Xiao, X. G., Tang, Y. X. & Wu, Y. M. Genome-wide analysis of AP2/ERF family genes from Lotus corniculatus shows LcERF054 enhances salt tolerance. Funct Integr Genomic 14, 453–466, https://doi.org/10.1007/s10142-014-0372-5 (2014).
Wang, L., Qin, L., Liu, W., Zhang, D. & Wang, Y. A novel ethylene-responsive factor from Tamarix hispida, ThERF1, is a GCC-box- and DRE-motif binding protein that negatively modulates abiotic stress tolerance in Arabidopsis. Physiol Plant 152, 84–97, https://doi.org/10.1111/ppl.12159 (2014).
Liu, W. J., Wang, Y. C. & Gao, C. Q. The ethylene response factor (ERF) genes from Tamarix hispida respond to salt, drought and ABA treatment. Trees-Struct Funct 28, 317–327, https://doi.org/10.1007/s00468-013-0950-5 (2014).
Xie, X. L., Yin, X. R. & Chen, K. S. Roles of APETALA2/ethylene-response factors in regulation of fruit quality. Crit Rev Plant Sci 35, 120–130, https://doi.org/10.1080/07352689.2016.1213119 (2016).
Han, Y. C. et al. Banana transcription factor MaERF11 recruits histone deacetylase MaHDA1 and represses the expression of MaACO1 and Expansins during fruit ripening. Plant Physiol 171, 1070–1084, https://doi.org/10.1104/pp.16.00301 (2016).
Kuang, J. F. et al. The transcriptional regulatory network mediated by banana (Musa acuminata) dehydration-responsive element binding (MaDREB) transcription factors in fruit ripening. New Phytol 214, 762–781, https://doi.org/10.1111/nph.14389 (2017).
Xiao, Y. Y. et al. Banana ethylene response factors are involved in fruit ripening through their interactions with ethylene biosynthesis genes. J Exp Bot 64, 2499–2510, https://doi.org/10.1093/jxb/ert108 (2013).
Huang, J. et al. The jujube genome provides insights into genome evolution and the domestication of sweetness/acidity taste in fruit trees. PLoS Genet 12, https://doi.org/10.1371/journal.pgen.1006433 (2016).
Zhuang, J. et al. Genome-wide analysis of the putative AP2/ERF family genes in Vitis vinifera. Sci Hortic-Amsterdam 123, 73–81, https://doi.org/10.1016/j.scienta.2009.08.002 (2009).
Li, M. Y. et al. Genome-wide analysis of AP2/ERF transcription factors in carrot (Daucus carota L.) reveals evolution and expression profiles under abiotic stress. Mol Genet Genomics 290, 2049–2061, https://doi.org/10.1007/s00438-015-1061-3 (2015).
Zhuang, J. et al. Genome-wide analysis of the AP2/ERF gene family in Populus trichocarpa. Biochem Biophys Res Commun 371, 468–474, https://doi.org/10.1016/j.bbrc.2008.04.087 (2008).
Zhao, T., Xia, H., Liu, J. & Ma, F. The gene family of dehydration responsive element-binding transcription factors in grape (Vitis vinifera): genome-wide identification and analysis, expression profiles, and involvement in abiotic stress resistance. Mol Biol Rep 41, 1577–1590, https://doi.org/10.1007/s11033-013-3004-6 (2014).
Shu, Y., Liu, Y., Zhang, J., Song, L. & Guo, C. Genome-wide analysis of the AP2/ERF superfamily genes and their responses to abiotic stress in Medicago truncatula. Front Plant Sci 6, 1247, https://doi.org/10.3389/fpls.2015.01247 (2015).
Tang, Y. et al. Genome-wide analysis of the AP2/ERF gene family in physic nut and overexpression of the JcERF011 gene in rice increased its sensitivity to salinity stress. PLoS One 11, e0150879, https://doi.org/10.1371/journal.pone.0150879 (2016).
Zhang, C. H. et al. Genome-wide analysis of the AP2/ERF superfamily in peach (Prunus persica). Genet Mol Res 11, 4789–4809, https://doi.org/10.4238/2012.October.17.6 (2012).
Hu, L. & Liu, S. Genome-wide identification and phylogenetic analysis of the ERF gene family in cucumbers. Genet Mol Biol 34, 624–633, https://doi.org/10.1590/S1415-47572011005000054 (2011).
Ohta, M., Matsui, K., Hiratsu, K., Shinshi, H. & Ohme-Takagi, M. Repression domains of class II ERF transcriptional repressors share an essential motif for active repression. Plant Cell 13, 1959–1968 (2001).
Albrecht, V., Ritz, O., Linder, S., Harter, K. & Kudla, J. The NAF domain defines a novel protein-protein interaction module conserved in Ca2+-regulated kinases. EMBO J 20, 1051–1063, https://doi.org/10.1093/emboj/20.5.1051 (2001).
Xu, W., Li, F., Ling, L. & Liu, A. Genome-wide survey and expression profiles of the AP2/ERF family in castor bean (Ricinus communis L.). BMC Genomics 14, 785, https://doi.org/10.1186/1471-2164-14-785 (2013).
Reams, A. B. & Neidle, E. L. Selection for gene clustering by tandem duplication. Annu Rev Microbiol 58, 119–142, https://doi.org/10.1146/annurev.micro.58.030603.123806 (2004).
Yang, S., Luo, C., Song, Y. & Wang, J. Two groups of Thellungiella salsuginea RAVs exhibit distinct responses and sensitivity to salt and ABA in transgenic Arabidopsis. PLoS One 11, e0153517, https://doi.org/10.1371/journal.pone.0153517 (2016).
Aukerman, M. J. & Sakai, H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15, 2730–2741, https://doi.org/10.1105/tpc.016238 (2003).
Moose, S. P. & Sisco, P. H. Glossy15, an APETALA2-like gene from maize that regulates leaf epidermal cell identity. Gene Dev 10, 3018–3027, https://doi.org/10.1101/gad.10.23.3018 (1996).
Chung, M. Y. et al. A tomato (Solanum lycopersicum) APETALA2/ERF gene, SlAP2a, is a negative regulator of fruit ripening. Plant J 64, 936–947, https://doi.org/10.1111/j.1365-313X.2010.04384.x (2010).
Cramer, G. R. et al. Transcriptomic analysis of the late stages of grapevine (Vitis vinifera cv. Cabernet Sauvignon) berry ripening reveals significant induction of ethylene signaling and flavor pathways in the skin. BMC Plant Biol 14, 370, https://doi.org/10.1186/s12870-014-0370-8 (2014).
Xie, X. L. et al. Isolation, classification and transcription profiles of the AP2/ERF transcription factor superfamily in citrus. Mol Biol Rep 41, 4261–4271, https://doi.org/10.1007/s11033-014-3297-0 (2014).
Ziliotto, F. et al. Grape berry ripening delay induced by a pre-veraison NAA treatment is paralleled by a shift in the expression pattern of auxin- and ethylene-related genes. BMC Plant Biol 12, 185, https://doi.org/10.1186/1471-2229-12-185 (2012).
Tournier, B. et al. New members of the tomato ERF family show specific expression pattern and diverse DNA-binding capacity to the GCC box element. Febs Letters 550, 149–154, https://doi.org/10.1016/S0014-5793(03)00757-9 (2003).
Yang, Z., Tian, L. N., Latoszek-Green, M., Brown, D. & Wu, K. Q. Arabidopsis ERF4 is a transcriptional repressor capable of modulating ethylene and abscisic acid responses. Plant Mol Biol 58, 585–596, https://doi.org/10.1007/s11103-005-7294-5 (2005).
Wang, A., Tan, D., Takahashi, A., Li, T. Z. & Harada, T. MdERFs, two ethylene-response factors involved in apple fruit ripening. J Exp Bot 58, 3743–3748, https://doi.org/10.1093/jxb/erm224 (2007).
Yin, X. R., Allan, A. C., Chen, K. S. & Ferguson, I. B. Kiwifruit EIL and ERF genes involved in regulating fruit ripening. Plant Physiol 153, 1280–1292, https://doi.org/10.1104/pp.110.157081 (2010).
Lee, J. M. et al. Combined transcriptome, genetic diversity and metabolite profiling in tomato fruit reveals that the ethylene response factor SlERF6 plays an important role in ripening and carotenoid accumulation. Plant J 70, 191–204, https://doi.org/10.1111/j.1365-313X.2011.04863.x (2012).
Bolser, D., Staines, D. M., Pritchard, E. & Kersey, P. Ensembl plants: integrating tools for visualizing, mining, and analyzing plant genomics data. Methods Mol Biol 1374, 115–140, https://doi.org/10.1007/978-1-4939-3167-5_6 (2016).
Finn, R. D. et al. HMMER web server: 2015 update. Nucleic Acids Res 43, W30–38, https://doi.org/10.1093/nar/gkv397 (2015).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, https://doi.org/10.1038/msb.2011.75 (2011).
LetunicI, L. & Bork, P. Interactive tree of life (iTOL)v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44, W242–W245, https://doi.org/10.1093/nar/gkw290 (2007).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208, https://doi.org/10.1093/nar/gkp335 (2009).
Hu, B. et al. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297, https://doi.org/10.1093/bioinformatics/btu817 (2015).
Li, L., Stoeckert, C. J. Jr & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–2189, https://doi.org/10.1101/gr.1224503 (2003).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645, https://doi.org/10.1101/gr.092759.109 (2009).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111, https://doi.org/10.1093/bioinformatics/btp120 (2009).
Anders, S., Pyl, P. T. & Huber, W. HTSeq - a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, https://doi.org/10.1093/bioinformatics/btu638 (2015).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628, https://doi.org/10.1038/nmeth.1226 (2008).
Zhang, C. M., Huang, J. & Li, X. G. Identification of appropriate reference genes for RT-qPCR analysis in Ziziphus jujuba Mill. Sci Hortic-Amsterdam 197, 166–169, https://doi.org/10.1016/j.scienta.2015.09.026 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25, 402–408 (2001).
Metsalu, T. & Vilo, J. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 43, W566–W570, https://doi.org/10.1093/nar/gkv468 (2015).
Fujimoto, S. Y., Ohta, M., Usui, A., Shinshi, H. & Ohme-Takagi, M. Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box-mediated gene expression. Plant Cell 12, 393–404, https://doi.org/10.1105/tpc.12.3.393 (2000).
Acknowledgements
This work was financially supported by National Plan for Science & Technology Support Program (Grant No. 2013BAD14B03-03), and Shaanxi Science & Technology Co-ordination & Innovation Project (Grant No. 2013KTZB03-03).
Author information
Authors and Affiliations
Contributions
Z.Z. and X.L. designed the experiments. Z.Z. conducted data collection, experiments, and contributed to analysis and manuscript preparation. Both authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Z., Li, X. Genome-wide identification of AP2/ERF superfamily genes and their expression during fruit ripening of Chinese jujube. Sci Rep 8, 15612 (2018). https://doi.org/10.1038/s41598-018-33744-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33744-w
Keywords
This article is cited by
-
Genome-wide identification and expression profiling analysis of maize AP2/ERF superfamily genes reveal essential roles in abiotic stress tolerance
BMC Genomics (2022)
-
Identification and expression analysis of BURP domain-containing genes in jujube and their involvement in low temperature and drought response
BMC Genomics (2022)
-
Genome-wide identification and characterization of AP2/ERF gene superfamily during flower development in Actinidia eriantha
BMC Genomics (2022)
-
Physicochemical and antioxidant activity of fruit harvested from eight jujube (Ziziphus jujuba Mill.) cultivars at different development stages
Scientific Reports (2022)
-
Genome-wide survey and identification of AP2/ERF genes involved in shoot and leaf development in Liriodendron chinense
BMC Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
