
Abstract
Subduction zones impose an important control on the geochemical cycling between the surficial and internal reservoirs of the Earth. Sulphur and carbon are transferred into Earth’s mantle by subduction of pelagic sediments and altered oceanic lithosphere. Release of oxidizing sulphate- and carbonate-bearing fluids modifies the redox state of the mantle and the chemical budget of subduction zones. Yet, the mechanisms of sulphur and carbon cycling within subduction zones are still unclear, in part because data are typically derived from arc volcanoes where fluid compositions are modified during transport through the mantle wedge. We determined the bulk rock elemental, and sulphur and carbon isotope compositions of exhumed ultramafic and metabasic rocks from Syros, Greece. Comparison of isotopic data with major and trace element compositions indicates seawater alteration and chemical exchange with sediment-derived fluids within the subduction zone channel. We show that small bodies of detached slab material are subject to metasomatic processes during exhumation, in contrast to large sequences of obducted ophiolitic sections that retain their seafloor alteration signatures. In particular, fluids circulating along the plate interface can cause sulphur mobilization during several stages of exhumation within high-pressure rocks. This takes place more pervasively in serpentinites compared to mafic rocks.
Similar content being viewed by others

Introduction
Subduction of altered oceanic lithosphere returns water, C, S, B, and numerous other volatiles to Earth’s mantle (e.g.1,2,3). The recycling of these species modifies the redox state of the mantle and controls the chemistry of arc volcanoes through the ascent of fluids from the slab into the sub-arc mantle4,5,6, where they ultimately control the formation of arc-related porphyry deposits7. However, the sources, pathways, and speciation of sulphur and carbon species during subduction are still poorly constrained. Evidence for volatile transfer from slab to mantle wedge has been primarily inferred from arc-related tracers, such as 34S-enriched sulphur signatures in arc volcanoes that likely originate from subducted seawater sulphate8, the oxidized state of arc magmas, and oxidizing, sulphate-bearing veins within porphyry deposits (see references in9). However, these tracers retain an imprint from chemical interaction within the sub-arc mantle and do not provide direct evidence of sulphur and carbon cycling along the slab-mantle wedge interface. Better insight into the mechanisms of fluid transfer and element cycling during subduction processes can be gained from exhumed subduction-related rocks.
During subduction, volatiles are released by sediment compaction and subsequent metamorphic reactions, with associated metamorphic mineral growth indicative of some of these reactions (e.g.10,11,12). Fluids released during metamorphic dehydration can produce complex channel networks13,14 and are typically thought to be focused within a narrow mélange zone along the interface of the subducting slab and the mantle wedge15,16. This mélange is characterized by chemically hybridized rocks and provides a window into fluid circulation within the subduction zone channel15,17,18. Carbon cycling within subduction zones has been the focus of numerous recent studies to constrain the long-term carbon flux between the surface and deep-Earth reservoirs (e.g.19,20). However, sulphur isotope data from subduction-related rocks currently consists mostly of in situ sulphide mineral analyses21,22, with bulk rock sulphur compositions of high-pressure rocks known for just a few metamorphosed serpentinite samples23,24. Furthermore, sulphate microphases are typically absent within both seafloor altered serpentinites and high-pressure rocks, but oxidized sulphur species are incorporated into rock-forming phyllosilicates such as serpentine25. Due to the difficulty in detecting this sulphate, its impact during subduction metamorphism is often neglected and thus fails to capture the behaviour of the bulk sulphur species within both seafloor altered serpentinites and high-pressure equivalents.
To better understand both sulphur and carbon fluxes within subduction complexes, we investigated exhumed mafic and ultramafic samples from Syros, Greece. Bulk rock sulphur (sulphate and sulphide) and carbon (carbonate carbon and reduced/organic carbon) species have been chemically extracted and their isotopic compositions determined. The presence of sulphur in both oxidized (sulphate) and reduced (sulphide) states in altered oceanic lithosphere makes it an ideal tool for tracking fluid sources and redox conditions, and allows us to infer mechanisms of element redistribution in the subduction channel mélange.
Geological Setting
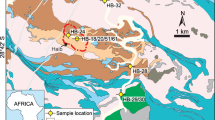
The island of Syros is dominated by interlayered schists and marbles, interpreted as metamorphosed flysch sediments, and local blueschist- to eclogite-facies meta-ophiolitic units of metabasic to ultramafic rocks (Fig. 1) (e.g.26,27). These mafic and ultramafic successions represent Cretaceous oceanic lithosphere formed within the Neo-Tethys between 76–80 Ma28,29 that was subducted to peak P-T conditions of >15 kbar and ~500 °C26 by ~50–53 Ma (e.g.10,30). The metabasic and ultramafic lithologies occur as metre to decametre blocks of eclogite, omphacite/jadeite fels, garnet glaucophane schist, meta-gabbro and serpentinite, representing the altered components of the former oceanic lithosphere. These lithologies are surrounded by a highly sheared matrix of chlorite (±amphibole) schist and serpentinite. The metabasic and ultramafic blocks and the chlorite (±amphibole) schist matrix are thought to represent the subduction channel mélange: the deformed and metasomatized interface between the subducting slab and the overlying mantle wedge18,31. For this study, we investigated serpentinites, metabasic rocks and chlorite (±amphibole) schists and felses from throughout the island of Syros (Fig. 1). These represent blocks of detached lithospheric slab and their surrounding mélange matrix, allowing us to evaluate the chemical exchanges between subducted oceanic lithosphere and surrounding matrix and provide new constraints on volatile fluxes within the subduction zone channel.

Simplified geologic map of Syros with the locations (red stars) of the studied samples. The box to the left locates the island of Syros (black box) and highlights in blue the Cycladic Blueschist Unit (CBU). Map modified after27.
Carbon and sulphur signatures compared to altered oceanic lithosphere
Carbon and sulphur concentrations and stable isotope compositions of the investigated samples fall within the ranges measured in unmetamorphosed (unsubducted), altered oceanic lithosphere (Figs 2, 3; Tables 1, 2). However, it is striking that i) sulphide concentrations are exceptionally low (mostly below detection limit) in all of the analysed samples (only one sample contained sulphide measureable for δ34Ssulphide; see Table 1) and ii) there is a positive and narrow range in δ34Ssulphate values measured in the serpentinites (15.7–20.8‰) and chlorite schists (18.5–19.7‰), whereas the metabasic samples have a larger range (δ34Ssulphate = 3.5 to 20.1‰; Fig. 2b, Table 1). These sulphur signatures are surprising since complex sulphur cycling processes during ocean floor alteration (e.g., microbial and thermochemical sulphate reduction) produce sulphur contents of up to 1 wt.% and sulphide and sulphate δ34S values of −45 to +27‰ (Figs 2b,c and 3)1,32. Previous studies suggest that subduction metamorphism does not considerably modify the sulphur isotope signatures gained during oceanic alteration, with a large range of sulphide and sulphate isotopic compositions preserved in seafloor-altered and subducted ophiolites (e.g., in high-P serpentinites in the Voltri massif, Italy; see Fig. 2)21,22,24. The main processes that can modify the bulk rock carbon and sulphur chemistry during subduction are metamorphic devolatilisation reactions21,23 or metasomatism through external fluid infiltration – with fluids produced by metamorphic dehydration reactions. In particular, dehydration of serpentinite to harzburgite and the transition from blueschist to eclogite facies have been shown to release 34S-enriched sulphur in the form of sulphate or SO2, facilitated by liberation of significant amounts of water20,21,23.

(a) Bulk rock carbon (total carbon, TC), (b) sulphate, and (c) sulphide isotope composition and contents of the studied samples compared to seafloor-altered serpentinites41,54,55,56,57,58,59, high-pressure serpentinites23,24, altered mafic oceanic crust32,38,60. Total carbon is comprised of carbonate carbon and reduced or organic carbon. Note, that sulphide contents are below detection limit in all serpentinites. Furthermore, seafloor-altered and high-pressure metamorphosed (up to eclogite facies) serpentinites from the Voltri Massif in Italy24 and the Cerro del Almires in Spain23 have ranges of carbon and sulphur signatures significantly larger than those in the Syros serpentinites and suggest preservation of the seafloor alteration signal.

(a) Schematic cross-section of a subduction zone (after15,31) showing the average range of carbon and sulphur composition in altered oceanic crust (AOC) and serpentinites, and the isotopic composition of the dominant carbon and sulphur species in pelagic sediments, Cretaceous seawater and the mélange matrix. The grey arrow labelled ‘mélange matrix’ indicates exhumation of the metabasic and meta-ultrabasic rocks along the subduction channel and modification of the mélange matrix chemistry by mechanical mixing. (b) Enlargement of the processes occurring in the subduction channel (square in (a)) where fluids are released by dehydration of sedimentary sequences (pelagic sediments), circulate and may interact to variable degrees with detached metabasic and meta-ultrabasic blocks under blueschist-facies conditions.
The mafic blocks from Syros are interpreted to be of oceanic origin based on bulk rock δ18O and δD values, retaining an imprint of seafloor alteration33,34. Carbon and sulphur in the metabasic blocks in this study overlap with altered oceanic crust (AOC) in terms of their isotopic composition, though sulphide contents are significantly lower (Fig. 2). Our results show positive correlations between Mg and δ34Ssulphate (Fig. 4a) and weak positive correlations between SO4-contents and MgO, K2O, Sr, and loss on ignition (bulk rock volatile content; not shown), suggesting increasing seawater alteration with increasing seawater sulphate incorporation35. However, some mafic blocks demonstrate interaction with secondary fluids. For example, blackwall reaction rinds at the contact between metabasic and ultramafic blocks and their surrounding mélange matrix are inferred to reflect fluid exchange during exhumation18,31. These rinds include tourmaline-bearing mafic rocks that formed during exhumation31. One tourmaline-bearing metagabbro studied here yields a δ34Ssulphate of 12.1‰, suggesting that fluids circulating during exhumation had an isotopic composition similar to this value, i.e., considerably lower than seawater sulphate compositions during the late Cretaceous (δ34Sseawater-sulphate ≈ 18.7–19.5‰36). Furthermore, the low sulphide contents provide strong evidence for metasomatism by an oxidizing fluid. Even though metamorphic devolatilisation reactions can cause H2S loss – primarily during blueschist-eclogite transition21, we consider it more likely that partial sulphide loss and local overprinting of the seafloor alteration signature was associated with metasomatism since these rocks did not reach eclogite-facies conditions. An even later stage of fluid circulation is recorded in two blueschist samples with distinctively higher total carbon (TC) contents of 0.3 to 0.9 wt.% (with δ13C of TIC [total inorganic carbon] of −0.7 and −7.4‰) – reflected by the presence of late carbonate veins – which is higher than average TC contents in AOC of ~0.21 wt.% (Fig. 2a)37,38. The origin and impact of these secondary fluids is discussed below.

(a–f) Selected element concentrations versus δ34Ssulphate showing a distinct negative correlation, and LaN/SmN against (g) δ34Ssulphate (h) Cs, and (i) Rb. All reflect mixing between a seawater-dominated fluid and a sediment-derived component with the grey arrows indicate increasing input of sedimentary-derived fluid. R2 (black line) is calculated for the serpentinites only. For As the R2-value (R2serp = 0.97*) excludes the outlier, as R2 for all data would not be meaningful.
Similarly, the serpentinites from Syros record a complex fluid history. Cooperdock et al.39 report δ18O and δD values of Syros serpentinites within the range of abyssal serpentinites. However, based on these data interaction with slab-derived fluids is possible. While our carbon data overlaps with abyssal serpentinites, the sulphur geochemistry of Syros serpentinites is distinctly different, with sulphide contents for all samples below detection limit (Fig. 2). Such low sulphide contents and δ34Ssulphate values close to seawater (modern δ34Sseawater-sulphate ≈ +21.15‰40) have previously only been observed in the active serpentinizing system of the Atlantis Massif along the Mid-Atlantic Ridge. There, carbonate and sulphate are actively incorporated into serpentinites by extensive seawater-peridotite interaction, with high water-rock ratios resulting in the oxidation of sulphide minerals41. In addition to abyssal metasomatism, carbonate and sulphate may be incorporated during fault-bend serpentinization by oxidizing fluids near the seawater-sediment interface along the trench prior to their subduction (Fig. 3). Recent studies have suggested that bending of the subducting plate and associated faulting facilitates fluid influx deep into the lithospheric mantle42. These areas may be subject to extensive serpentinization, sequestering significant quantities of water, which is eventually transferred into Earth’s interior42. Simultaneously, oxidizing fluids could have removed sulphide minerals formed during early phases of serpentinization near an oceanic spreading centre. Though sulphate and carbonate were possibly incorporated during mid-ocean ridge or bend fault serpentinization, considering the metasomatic rinds on the metabasic samples18,31 and comparing with other subducted serpentinites (e.g., Voltri Massif serpentinites; Fig. 2) it is more likely that the serpentinites were also affected by metamorphic or metasomatic reactions during subduction and/or exhumation to modify the sulphur and carbon compositions.
Sulphur and carbon cycling in the subduction zone channel
The serpentinites studied here lack mineralogical evidence for having experienced significant dehydration. Thus, the low sulphur contents cannot primarily be ascribed to devolatilization reactions, but likely reflect external fluid ingress. Based on δ18O and δD values and fluid-mobile trace element compositions of serpentinites from Syros (including those studied here), a chemical exchange between ophiolitic blocks and the surrounding matrix has been proposed39. In addition, the sulphur geochemistry of the analysed serpentinites indicates two endmember fluid compositions that interacted with these rocks: a seawater-sulphate-like fluid and a fluid characterized by lower δ34Ssulphate values (Fig. 4). In the case of the Syros serpentinites, seawater sulphate incorporation either during serpentinization near a mid-ocean ridge spreading centre41 or by bend-fault serpentinization (Fig. 3a) would have occurred in the Late Cretaceous with seawater δ34Ssulphate values close to ~18.7–19.5‰36. The second endmember composition (with lower δ34Ssulphate values) is associated with elevated concentrations in fluid-mobile elements (FME), particularly Rb, Li, and Cs (Fig. 4), which we interpret as reflecting secondary, external fluid ingress.
Even though FMEs are typically enriched in most serpentinites, independent of geotectonic origin, abnormal enrichments of certain elements document different fluid sources3. Overall, FMEs such as Rb, Sr, Ba, Li, As, Cs, Pb, and U are liberated during metamorphic dehydration reactions, whereas As, Cs, and U are characteristic of a sedimentary fluid input3,43. At the same time, the largest reservoir of 32S-enriched sulphur within subduction zones is pelagic sediments that contain significant amounts of biogenic sulphide (with average δ34Ssulphide of approximately −25 to −15‰44,45). In particular, carbonaceous shales are considered to produce the most H2S-rich (and 32S-enriched) fluids upon metamorphism46. Even though sulphur is also stored within AOC, dehydration of carbonaceous shales produces one to two orders of magnitude higher concentrations of H2S in metamorphic fluids compared to mafic rocks46. Furthermore, a sedimentary fluid input is suggested in our data by negative correlations between δ34Ssulphate and both Mg and, to a lesser extent, δ18O (Fig. 4a,f). Sediments are enriched in MgO and have δ18Oclay values of +10 to +30‰44. Thus, their dehydration results in Mg-rich and 18O-enriched fluids shifting the oceanic signature to higher MgO contents and δ18O values, respectively. Additionally, we observe a trend towards high (La/Sm)N at low (Ba/Th), as well as positive correlations between Cs and (La/Sm)N (Fig. 4h) and Rb and (La/Sm)N (Fig. 4i), which is attributable to the input of a sedimentary component within the mélange zone47. Thus, the studied serpentinites record numerous lines of evidence for a metasomatic overprint by a fluid produced by metamorphic dehydration of sedimentary sequences.
In contrast to the serpentinites and metabasic samples, the chlorite schists (representing the mélange matrix) have higher TC contents (up to 1.8 wt.%) with δ13CTIC values of −2.9 to −1.1‰ (Table 2), and an exceptionally narrow range in δ34Ssulphate (18.5–19.7‰; Table 1). The sulphur compositions match seawater sulphate compositions of Late Cretaceous age (δ34S ≈ 18.7–19.5‰36) and imply a seawater-dominated component. Their rare earth element (REE) concentrations are higher than in the serpentinites (see suppl. mat.), but in most samples their FME concentrations are lower. This indicates a different origin of these rocks to the serpentinites. Chlorite schists in subduction zones have previously been described as the result of metasomatic and mechanical mixing at the contact between metabasic rocks and serpentinites47 and at shallow depth associated with exhumation as proposed by geodynamic modelling48. Mechanical mixing explains the highly variable trace element patterns of the chlorite schists (see suppl. Fig. S4-1), reflecting heterogeneous incorporation of mafic and ultramafic material. The presence of carbonate (dolomite and calcite) as pockets, layers or veins (see Suppl. Mat.) indicates that sedimentary material was also incorporated. In addition, fluids were locally focused through the mélange matrix producing carbonate veins, which are locally also preserved in the blueschists. These veins have δ34Ssulphate values of +20.1 to +20.8‰ and δ13CTIC values of −7.4 to −1.0‰, and may have formed from fluids produced by compaction of younger sedimentary sequences that were isotopically heavier in their sulphur composition. This agrees well with exhumation ages from Syros of ~25–20 Ma27, at which time δ34Sseawater-sulphate was ~+21‰36. Thus, the late carbonate veins may be associated with late stage exhumation processes.
Geotectonic Evolution
The serpentinite samples studied here have previously been inferred to be of abyssal origin39. Using oxygen and hydrogen isotopes, and major and trace element geochemistry, Cooperdock et al.39 excluded a potential origin from the fore-arc mantle wedge, as well as the influence of meteoric water. The carbon and sulphur geochemistry of the studied mafic and ultramafic samples reflects several stages of water-rock interaction by fluids with different origins: Cretaceous seawater and metamorphic dehydration. Even though the samples studied here overlap with the carbon and sulphur signatures found in seafloor altered mafic rocks, the lack of sulphide in almost all studied samples – and thus orders of magnitude lower sulphide contents compared to abyssal serpentinites – suggests sulphide removal after alteration on the seafloor, most likely during exhumation (Fig. 3). On Syros, blackwall alteration rinds and tourmaline-bearing mafic rocks that formed during exhumation record retrograde metasomatic overprinting at depths of ~25 km and temperatures ~400 °C with a fluid that was slab-derived, acidic, and unusually Li-rich33. The isotopic value of the tourmaline-bearing metagabbro and the negative correlation between Li contents and δ34Ssulphate (Fig. 4b) suggests that interaction of the serpentinites with external fluids most likely took place during the same stages of exhumation. Most likely, water-rock ratios were sufficiently low to allow for the preservation of the seafloor δ18O and δD alteration signature. Furthermore, the abundance of hematite in the blueschists (see suppl. material) and the low sulphide contents implicate an oxidizing fluid. Oxidation of sulphide to sulphate is associated with minor isotope fractionation49. Thus, in the metabasic samples oxidation of sulphide with δ34S values as low as −2.4‰ – which is within the range of sulphide found within AOC (Figs 2c, 3) – possibly generated δ34Ssulphate values of 3.5 to 20.1‰ (Table 1). In the serpentinites, these fluids would have oxidized the sulphide – if present – entirely.
In contrast, the composition of the chlorite-amphibole schists documents carbon and sulphur incorporation at shallower depth, with an isotopic signature dominated by a seawater component. The origin of these rocks suggests mechanical incorporation of trace amounts of seawater sulphate and carbonate, either from AOC, sediments, or serpentinites47. Even though CO2-rich fluids could have been focused through these rocks, the low sulphur contents indicate that these lithologies did not serve as primary fluid pathways for sulphur transfer within the subduction zone channel unless fluids were in chemical and isotopic equilibrium with the rock and thus left little record of fluid-rock interaction. However, as seawater circulation results in significant addition of seawater sulphate into abyssal serpentinites41 and incorporation of the sulphate into the serpentine structure25, we infer that sulphur concentrations in the fluid within the subduction channel were most likely very low. This also agrees with the low solubility of anhydrite at these P and T conditions, which inhibits extensive sulphur mobility9.
We advocate that although large sequences of serpentinite can preserve sulphur and carbon signatures of the subducted slab (e.g., in the Voltri Massif serpentinites)24, small bodies of decoupled oceanic lithosphere may be subject to considerable fluid infiltration during exhumation. Most likely, sulphur geochemical modification and sulphur transfer to the sub-arc mantle occur near the transition from blueschist- to eclogite-facies conditions, driven by dehydration reactions20,21,23. If these conditions are not reached, oxidizing fluids circulating during exhumation can induce sulphide oxidation and removal of sulphide phases. In particular, sulphide dissolution is enhanced during deformation and the input of highly saline fluids, which also increases sulphate solubility9,46,50. Additional sulphur transfer takes place during late stages of exhumation upon interaction with large quantities of saline, oxidizing and seawater-sulphate-dominated fluids liberated during compaction of sediments and expulsion of pore fluid during initial stages of subduction within the accretionary prism51. These fluids are documented by late-stage carbonate veins with δ34Ssulphate around +20.8‰ and may have affected highly sheared serpentinites, providing an explanation for δ34S values higher than Cretaceous seawater sulphate (see Table 1) because release of seawater of <50 Ma age would be characterized by δ34Ssulphate values between 21.5–22.5‰36. Oxidation by such late, seawater-dominated saline fluids could have additionally caused sulphate addition and local sulphide loss. Interestingly, metamorphic dehydration fluids seem to have more extensively affected the serpentinites than the metabasic samples, where interaction with dehydration fluids was restricted to the metasomatic rinds (Fig. 3). In contrast with these sulphur data, carbon results do not readily imply a suite of fluid sources and cannot easily be used as a tracer for specific fluid interactions. Therefore, we conclude that carbon was largely immobile during metamorphism at these P-T conditions.
The lack of sulphide in the majority of our analysed samples, which contain orders of magnitude lower sulphide concentrations than typical seafloor altered oceanic lithosphere (Fig. 3b,c), documents that sulphur can be mobilized at various stages and by several processes during evolution within a subduction zone. Furthermore, we show that oxidizing fluids play a central role in the mobilization of sulphur, particularly in smaller blocks of detached slab material, and that numerous lithologies can provide a sulphur source during subduction zone processes. This study also highlights the importance of constraining bulk rock sulphur and carbon compositions, which can reveal important aspects of chemical cycles within subduction zones and allows the tracking of bulk element fluxes between the surface and Earth’s deep reservoirs.
Methods
Sample preparation
For all samples thin section blocks were cut for thin sections for optical microscopy and electron microprobe (EMP) analyses. Bulk rock powders were prepared for determination of the bulk rock carbon and sulphur geochemistry. Prior to crushing the samples, the outermost 1–2 cm of the rock samples were cut away to remove contamination from weathering. The samples were cleaned in an ultrasonic bath prior to powdering them with a shatter box using an alumina dish. Sample preparation was carried out at the Department of Geosciences at Virginia Tech.
Mineralogy
The mineralogy of the studied samples was determined by transmitted and reflected light microscopy. On selected samples EMP analyses were performed in order to identify the mineral chemistry, specifically determining the carbonate phases. EMP analyses were carried out on a JEOL superprobe electron microprobe at the Geological department at the Freie Universität Berlin using 15 kV acceleration potential, a 20 nA current and a 1 μm beam size using natural and synthetic mineral standards. Relative analytical error is better than 1% (1σ) except for element contents <1 wt.%, where the analytical error is better than 10% (1σ).
Major and trace element compositions
Major element concentrations of lithiumtetraborate fluxed fusion glasses were analysed with a PANalytical 2404×-ray fluorescence spectrometer at the Department of Earth and Environment, Franklin Marshall College (following the procedures of52). Volatile contents were determined by loss on ignition. Concentrations of some trace elements were determined from pressed whole rock powders. A subset of samples has been determined by Cooperdock et al.39 (marked with * in Table S4).
Determination of the carbon geochemistry
Carbon was detected either as oxidized/inorganic carbon (TIC), reduced/organic carbon (TOC), or as the total carbon (TC) composition of the bulk rock. For all samples TC and TIC contents, and δ13C of TC, δ13C of TIC and δ13C of TOC were analysed. All carbon and oxygen analyses were carried out at the Department of Geosciences at Virginia Tech. TC contents, δ13CTC and δ13CTOC were measured on a Vario ISOTOPE elemental analyser (EA) coupled to an Isoprime 100 isotope ratio mass spectrometer (IRMS). For analyses of δ13CTOC, bulk rock samples were reacted for three days with 3 N HCl to remove all acid soluble carbon (e.g., calcite, dolomite). They were then rinsed with H2O, dried at 40 °C in the oven and homogenized in the agate mortar. TC and TOC isotope values are reported in the standard δ-notation relative to the Vienna-Pee Dee Belemnite (V-PDB) standard and calibrated to this scale using international [IAEA-CH-6 (sucrose; δ13C = −10.449‰) and IAEA-CH-7 (polyethylene; δ13C = −32.151‰)] and commercial standards [elemental microanalysis wheat flour; δ13C = −27.21‰]. Reproducibility of δ13CTC and δ13CTOC is better than 0.1‰.
The δ13CTIC and δ18OTIC and TIC contents were analysed on a MultiFlowGeo headspace sampler attached to an Isoprime 100 IRMS. Samples were prepared in septum vials, flushed with helium and acidified with phosphoric acid. Samples were then reacted for at least 3 hours at room temperature. Carbon and oxygen isotope values are reported in the standard δ-notation relative to the V-PDB standard and calibrated to this scale using the international standards IAEA-CO-1 (marble; δ13C = +2.492‰, δ18O = −2.4‰), IAEA-CO-9 (BaCO3; δ13C = −47.321‰, δ18O = −15.6‰) and NBS18 (calcite, δ13C = −5.014‰, δ18O = −23.2‰). Reproducibility for the analysis of the samples was better than ±0.07‰ for δ13C and better than ±0.3‰ for δ18O.
Determination of the sulphur geochemistry
Sulphur extractions were carried out by reacting 22–28 g of bulk rock powder to determine the acid volatile sulphide (AVS, typically bound within monosulphides such as pyrrhotite), the chromium reducible sulphide (CRS, typically bound within disulphides such as pyrite) and the sulphate fraction (e.g., anhydrite). Extraction of the sulphur components was carried out following a modified version of the methods of Canfield et al.53. In a first step, AVS was extracted by reacting the bulk rock powder with 6 N HCl in an inert N2-atmosphere and in a second step the residual sample was reacted with an acidified CrCl2 solution to extract the CRS fraction. In both cases the liberated H2S was precipitated as ZnS in a zinc acetate solution and subsequently converted to Ag2S through reaction with a 0.1 M AgNO3 solution. The sulphate fraction was recovered by reacting the solution from the AVS extraction with BaCl2 to form BaSO4. Amounts of AVS, CRS and sulphate were determined gravimetrically and corrected based on the sulphur content of the precipitate as determined on the EA, since co-precipitation of other phases during the wet chemical extraction could not be completely prevented. Detection limit for sulphate and sulphide depend on the amount of sample powder processed; for 25 g of sample powder 1 ppm sulphate (corresponding to 0.0002 g of BaSO4 extract) and 3 ppm sulphide (corresponding to 0.0002 g of Ag2S extract) are still detectable.
The isotopic composition of the AVS, CRS and sulphate were determined on a Vario ISOTOPE EA attached to an Isoprime 100 IRMS. To ensure complete combustion during EA-analyses, vanadium pentoxide (V2O5) was added to the samples. Sulphur isotope values are reported in standard δ-notation relative to the Vienna-Canyon Diablo Troilite (V-CDT) standard. During measurements the international sulphide (Ag2S) standards IAEA-S-1 (δ34S = −0.3‰), IAEA-S-2 (δ34S = +22.7‰) and IAEA-S-3 (δ34S = −32.3‰) and the sulphate (BaSO4) standards IAEA-SO-5 (δ34S = +0.5‰), IAEA-SO-6 (δ34S = −34.1‰), and NBS127 (δ34S = +20.3‰) were used to place our samples on the V-CDT scale. Reproducibility is better than 0.2‰ for all sulphur analyses (samples and standards), and the relative precision of sulphur contents is within 3% determined by multiple extractions of the same sample.
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Alt, J. C. et al. The role of serpentinites in cycling of carbon and sulfur: Seafloor serpentinization and subduction metamorphism. Lithos 178, 40–54 (2013).
Früh-Green, G. L., Connolly, J. A., Plas, A., Kelley, D. S. & Grobéty, B. Serpentinization of oceanic peridotites: Implications for geochemical cycles and biological activity. In: the subseafloor biospere at Mid-Ocean Ridges, Vol. 144 (American Geophysical Union, 2004).
Deschamps, F., Godard, M., Guillot, S. & Hattori, K. Geochemistry of subduction zone serpentinites: A review. Lithos 178, 96–127 (2013).
Plank, T. & Langmuir, C. H. Tracing trace elements from sediment input to volcanic output at subduction zones. Nature 362, 739–743 (1993).
Elliott, T. In Inside the Subduction Factory Vol. 138 (ed. Eiler, J.) 23–45 (American Geophysical Union Gephysical Monograph, 2003).
Davidson, J. P. In Subduction Top to Bottom Vol. 99 (eds Bebout, G. E., Scholl, D. W., Kirby, S. H. & Platt, J. P.) 251–262 (American Geophysical Union Geophysical Monograph, 1996).
Richards, J. P. The oxidation state, and sulfur and Cu contents of arc magmas: implications for metallogeny. LIthos (2014).
Woodhead, J. D., Harmon, R. S. & Fraser, D. G. O, S, Sr, and Pb isotope variations in volcanic rocks from the Northern Mariana Islands: implications for crustal recycling in intra-oceanic arcs. Earth and Planetary Science Letters 83, 39–52 (1987).
Newton, R. C. & Manning, C. E. Solubility of Anhydrite, CaSO4, in NaCl–H2O Solutions at High Pressures and Temperatures: Applications to Fluid–Rock Interaction. Journal of Petrology 46, 701–716 (2005).
Dragovic, B., Baxter, E. F. & Caddick, M. J. Pulsed dehydration and garnet growth during subduction revealed by zoned garnet geochronology and thermodynamic modeling, Sifnos, Greece. Earth and Planetary Science Letters 413, 111–122 (2015).
Baxter, E. F. & Caddick, M. J. Garnet growth as a proxy for progressive subduction zone dehydration. Geology 41, 643–646 (2013).
Peacock, S. M. O. The importance of blueschist → eclogite dehydration reactions in subducting oceanic crust. Geological Society of America Bulletin 105, 684–694 (1993).
Plümper, O., John, T., Podladchikov, Y. Y., Vrijmoed, J. C. & Scambelluri, M. Fluid escape from subduction zones controlled by channel-forming reactive porosity. Nature Geosci 10, 150–156 (2017).
Li, J. L., Gao, J., John, T., Klemd, R. & Su, W. Fluid-mediated metal transport in subduction zones and its link to arc-related giant ore deposits: Constraints from a sulifde-bearing HP vein in lawsonite eclogite (Tianshan, China). Geochimica et Cosmochimica Acta 120, 326–362 (2013).
Bebout, G. E. & Penniston-Dorland, S. C. Fluid and mass transfer at subduction interfaces—The field metamorphic record. Lithos 240–243, 228–258 (2016).
Angiboust, S., Pettke, T., De Hoog, J. C. M., Caron, B. & Oncken, O. Channelized fluid flow and eclogite-facies metasomatism along the subduction shear zone. Journal of Petrology 55, 883–916 (2014).
van der Straaten, F., Schenk, V., John, T. & Gao, J. Blueschist-facies rehydration of eclogites (Tian Shan, NW-China): Implications for fluid–rock interaction in the subduction channel. Chemical Geology 255, 195–219 (2008).
Miller, D. P., Marschall, H. R. & Schumacher, J. C. Metasomatic formation and petrology of blueschist-facies hybrid rocks from Syros (Greece): Implications for reactions at the slab-mantle interface. Lithos 107, 53–67 (2009).
Collins, N. C. et al. Subduction zone metamorphic pathway for deep carbon cycling: II: Evidence from HP/UHP metabasaltic rocks and ophicarbonates. Chemical Geology 412, 132–150 (2015).
Scambelluri, M. et al. Carbonation of subduction-zone serpentinite (high-pressure ophicarbonate; Ligurian Western Alps) and implications for the deep carbon cycling. Earth and Planetary Science Letters 441, 155–166 (2016).
Evans, K. A., Tomkins, A. G., Cliff, J. & Fiorentini, M. L. Insights into subduction zone sulfur recycling from isotopic analysis of eclogite-hosted sulfides. Chemical Geology 365, 1–19 (2014).
Giacometti, F. et al. Sulfur isotope evolution in sulfide ores from Western Alps: Assessing the influence of subduction-related metamorphism. Geochemistry, Geophysics, Geosystems 15, 3808–3829 (2014).
Alt, J. C. et al. Recycling of water, carbon, and sulfur during subduction of serpentinites: A stable isotope study of Cerro del Almirez, Spain. Earth and Planetary Science Letters 327–328, 50–60 (2012).
Schwarzenbach, E. M. Serpentinization, fluids and life: Comparing carbon and sulfur cycles in modern and ancient environments Doctor of Science thesis, ETH Zurich (2011).
Debret, B. et al. Assessing sulfur redox state and distribution in abyssal serpentinites using XANES spectroscopy. Earth and Planetary Science Letters 466, 1–11 (2017).
Okrusch, M. & Bröcker, M. Eclogites associated with high-grade blueschists in the Cyclades archipelago, Greece: a review. European Journal of Mineralogy 451–478 (1990).
Keiter, M. C. & Tomaschek, F. A new geological map of the Island of Syros (Aegean Sea, Greece): Implications for lithostratigraphy and structural history of the Cycladic Blueschist Unit. Geological Society of America: Special Paper 481 (2011).
Tomaschek, F., Kennedy, A. K., Villa, I. M., Lagos, M. & Ballhaus, C. Zircons from Syros, Cyclades, Greece - Recrystallization and mobilization of zircon during high-pressure metamorphism. Journal of Petrology 44, 1977–2002 (2003).
Robertson, A. H. F., Clift, P. D., Degnan, P. J. & Jones, G. Palaeogeographic and palaeotectonic evolution of the Eastern Mediterranean Neotethys. Palaeogeography, Palaeoclimatology, Palaeoecology 87, 289–343 (1991).
Lagos, M. et al. High precision Lu–Hf geochronology of Eocene eclogite-facies rocks from Syros, Cyclades, Greece. Chemical Geology 243, 16–35 (2007).
Marschall, H. R., Ludwig, T., Altherr, R., Kalt, A. & Tonarini, S. Syros metasomatic tourmaline: Evidence for very high-d11B fluids in subduction zones. Journal of Petrology 47, 1915–1942 (2006).
Alt, J. C. & Shanks, W. C. Microbial sulfate reduction and the sulfur budget for a complete section of altered oceanic basalts, IODP Hole 1256D (eastern Pacific). Earth and Planetary Science Letters 310, 73–83 (2011).
Marschall, H. R., Altherr, R., Gméling, K. & Kasztovszky, Z. Lithium, boron and chlorine as tracers for metasomatism in high-pressure metamorphic rocks: a case study from Syros (Greece). Mineralogy and Petrology 95, 291 (2009).
Putlitz, B., Matthews, A. & Valley, J. W. Oxygen and hydrogen isotope study of high-pressure metagabbros and metabasalts (Cyclades, Greece): implications for the subduction of oceanic crust. Contributions to Mineralogy and Petrology 138, 114–126 (2000).
Staudigel, H. In Treatise on Geochemistry Vol. 3 (eds Holland, H. D. & Turekian, K. K.) Ch. 3.15, 511–535 (Elsevier Ltd., 2003).
Paytan, A., Kastner, M., Campbell, D. & Thiemens, M. H. Seawater sulfur isotope fluctuations in the Cretaceous. Science 304, 1663–1665 (2004).
Alt, J. C. & Teagle, D. A. H. The uptake of carbon during alteration of ocean crust. Geochimica et Cosmochimica Acta 63, 1527–1535 (1999).
Shilobreeva, S., Martinez, I., Busigny, V., Agrinier, P. & Laverne, C. Insights into C and H storage in the altered oceanic crust: Results from ODP/IODP Hole 1256D. Geochimica et Cosmochimica Acta 75, 2237–2255 (2011).
Cooperdock, E. H. G., Raia, N. H., Barnes, J. D., Stockli, D. F. & Schwarzenbach, E. M. Tectonic origin of serpentinites on Syros, Greece: Geochemical signatures of abyssal origin preserved in a HP/LT subduction complex. Lithos 352–364 (2018).
Johnston, D. T. et al. Placing an upper limit on cryptic marine sulphur cycling. Nature 513, 530 (2014).
Delacour, A., Früh-Green, G. L., Bernasconi, S. M. & Kelley, D. S. Sulfur in peridotites and gabbros at Lost City (30°N, MAR): Implications for hydrothermal alteration and microbial activity during serpentinization. Geochimica et Cosmochimica Acta 72, 5090–5110 (2008).
Ranero, C. R., Morgan, J. P., McIntosh, K. & Reichert, C. Bending-related faulting and mantle serpentinization at the Middle America trench. Nature 425, 367–373 (2003).
Hattori, K., Takahashi, Y., Guillot, S. & Johanson, B. Occurrence of arsenic (V) in forearc mantle serpentinites based on X-ray absorption spectroscopy study. Geochimica et Cosmochimica Acta 69, 5585–5596 (2005).
Sharp, Z. Principles of stable isotope geochemistry. (Pearson Education Inc., 2007).
Walker, J. C. G. Global geochemical cycles of carbon, sulfur and oxygen. Marine Geology 70, 159–174 (1986).
Tomkins, A. G. Windows of metamorphic sulfur liberation in the crust: Implications for gold deposit genesis. Geochimica et Cosmochimica Acta 74, 3246–3259 (2010).
Marschall, H. R. & Schumacher, J. C. Arc magmas sources from mélange diapirs in subduction zones. Nature Geoscience (2012).
Gerya, T. V., Stöckhert, B. & Perchuk, A. L. Exhumation of high-pressure metamorphic rocks in a subduction channel: A numerical simulation. Tectonics 21, 6-1-6-19 (2002).
Fry, B., Ruf, W., Gest, H. & Hayes, J. M. Sulfur isotope effects associated with oxidation of sulfide by O2 in aqueous solution. Chemical Geology 73, 205–210 (1988).
Reed, M. H. & Palandri, J. L. In Sulfide Mineralogy and Geochemistry Vol. 61 Reviews in Mineralogy and Geochemistry (ed. Vaughan, D. J.) Ch. 11, 609–632 (Geochemical Society Mineralogical Society of America, 2006).
Mottl, M. J., Wheat, C. G., Fryer, P., Gharib, J. & Martin, J. B. Chemistry of springs across the Mariana forearc shows progressive devolatiolization of the subducting plate. Geochimica et Cosmochimica Acta 68, 4915–4933 (2004).
Boyd, F. R. & Mertzman, S. A. In Magmatic Processes: Physicochemical Principles, Special Pub. No. (ed. Mysen, B. O.) 13–24 (The Geochemical Society, 1987).
Canfield, D. E., Raiswell, R., Westrich, J. T., Reaves, C. M. & Berner, R. A. The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales. Chemical Geology 54, 149–155 (1986).
Schwarzenbach, E. M. et al. Sulfur geochemistry of peridotite-hosted hydrothermal systems: Comparing the Ligurian ophiolites with oceanic serpentinites. Geochimica et Cosmochimica Acta 91, 283–305 (2012).
Alt, J. C. & Shanks, W. C. I. Sulfur in serpentinized oceanic peridotites: Serpentinization processes and microbial sulfate reduction. Journal of Geophysical Research 103, 9917–9929 (1998).
Alt, J. C. et al. Hydrothermal alteration and microbial sulfate reduction in peridotite and gabbro exposed by detachment faulting at the Mid-Atlantic Ridge, 15°20′ N (ODP Leg 209): A sulfur and oxygen isotope study. Geochemistry Geophysics Geosystems 8 n/a-n/a (2007).
Schwarzenbach, E. M., Früh-Green, G. L., Bernasconi, S. M., Alt, J. C. & Plas, A. Serpentinization and carbon sequestration: A study of two ancient peridotite-hosted hydrothermal systems. Chemical Geology 351, 115–133 (2013).
Delacour, A., Früh-Green, G. L. & Bernasconi, S. M. Sulfur mineralogy and geochemistry of serpentinites and gabbros of the Atlantis Massif (IODP Site U1309). Geochimica et Cosmochimica Acta 72, 5111–5127 (2008).
Schwarzenbach, E. M., Gill, B. C. & Johnston, D. Unraveling multiple phases of sulfur cycling during the alteration of ancient ultramafic oceanic lithosphere. Geochimica et Cosmochimica Acta 223, 279–299 (2018).
Alford, S. E., Alt, J. C. & Shanks Iii, W. C. Sulfur geochemistry and microbial sulfate reduction during low-temperature alteration of uplifted lower oceanic crust: Insights from ODP Hole 735B. Chemical Geology 286, 185–195 (2011).
Acknowledgements
This project was supported by NSF-EAR grant 1324566 to E.M.S. and B.C.G., and NSF EAR award 1250470 to M.J.C. We thank E.F. Baxter, J.C. Schumacher, J.S. Gorce, H.L. Brooks, E. Ramos, and J.A. Kendall for participation in field work and sample collection, and for subsequent discussions. We acknowledge support by the German Research Foundation and the Open Access Publication Fund of the Freie Universität Berlin.
Author information
Authors and Affiliations
Contributions
Samples were collected by M.J.C., E.H.G.C. and J.B. M.P. did all sample preparation and most analyses under the supervision of E.M.S., B.C.G. and M.J.C. B.C.G. and E.M.S. conducted some of the analyses. E.M.S. wrote the first draft of paper. All authors helped during data interpretation and revision of the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schwarzenbach, E.M., Caddick, M.J., Petroff, M. et al. Sulphur and carbon cycling in the subduction zone mélange. Sci Rep 8, 15517 (2018). https://doi.org/10.1038/s41598-018-33610-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33610-9
Keywords
This article is cited by
-
Lattice dynamics, sound velocities, and atomic environments of szomolnokite at high pressure
Physics and Chemistry of Minerals (2023)
-
Slab-derived devolatilization fluids oxidized by subducted metasedimentary rocks
Nature Geoscience (2022)
-
Uncovering and quantifying the subduction zone sulfur cycle from the slab perspective
Nature Communications (2020)
-
Subducting serpentinites release reduced, not oxidized, aqueous fluids
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
