
Abstract
Tissue engineering approaches offer alternative strategies for urinary diversion after radical cystectomy. Possible triggering of cancer recurrence remains, however, a significant concern in the application of stem-cell based therapies for oncological patients. Soluble mediators secreted by stem cells induce tissue remodelling effects, but may also promote cancer cells growth and metastasis. We observed a substantial increase in the concentration of IL-6 and IL-8 in the secretome of adipose-derived stem cells (ASCs) co-cultured with bladder cancer cells. Concentrations of GM-CSF, MCP-1 and RANTES were also elevated. Bioactive molecules produced by ASCs increased the viability of 5637 and HT-1376 cells by respectively 15.4% and 10.4% (p < 0.0001). A trend in reduction of adhesion to ECM components was also noted, even though no differences in β-catenin expression were detected. When HT-1376 cells were co-cultured with ASCs their migration and invasion increased by 24.5% (p < 0.0002) and 18.2% (p < 0.002). Expression of p-ERK1/2 increased in 5637 cells (2.2-fold; p < 0.001) and p-AKT in HB-CLS-1 cells (2.0-fold; p < 0.001). Our results confirm that ASCs crosstalk with bladder cancer cells in vitro what influences their proliferation and invasive properties. Since ASCs tropism to tumour microenvironment is well documented their application towards post-oncologic reconstruction should be approached with caution.
Similar content being viewed by others


Introduction
Bladder cancer (BC) is the fourth most common cancer worldwide. The highest incidence rates are observed in Southern and Western Europe, Northern America, and Western Asia1,2. Even though mortality rates have been decreasing in recent years, BC remains substantial health burden due to high recurrence rates3,4. Radical cystectomy (RC) is considered the gold standard for the treatment of muscle-invasive and high-risk non-muscle-invasive BC with minor or no significant differences in oncological outcomes when comparing open RC with laparoscopic and robot-assisted RC5,6,7. The procedure involves removal of the entire bladder, lymph nodes, part of the urethra, and nearby organs that may contain cancer cells. Still, patients after RC remain at risk of BC recurrence with remaining urethra as a common recurrence site8.
Both continent and incontinent diversions are available for bladder replacement after RC. Due to significant problems associated with the use of gastrointestinal segments for bladder augmentation, new methods for urinary tract reconstruction are being sought9,10. Most of these methods use cell-seeded matrices to build tissue-engineered tubular grafts11,12. New, biologically derived scaffolds seeded with autologous cells for bladder wall substitution are also investigated13,14,15. Several cell types, e.g. bladder epithelial cells, smooth muscle cells, adipose-derived stem cells, or urine-derived stem cells are used for seeding onto scaffolds to promote tissue regeneration. Still, most techniques for scaffolds production employ autologous adipose-derived stem cells (ASCs)16,17,18.
ASCs are considered as the most suitable source of cells for stem cell-based therapies mainly because they can be harvested in large quantities using minimally invasive procedures19,20,21. It has been shown that ASCs secrete a wide variety of soluble mediators that promote morphological regeneration and functional restoration of bladder defects22,23,24. Possible triggering of cancer recurrence during remission remains, however, a significant concern in the application of stem cell-based therapies for cancer patients. It is suggested that paracrine factors secreted by locally delivered ASCs may induce activation of persisting tumour-initiating cells25. Despite intensive investigation, the influence of ASCs on cancer progression remains mostly unclear.
Previously we showed that conditioned medium form ASCs culture (ASC-CM) reduces bladder cancer cells viability and increases their resistance to ciprofloxacin, an antibiotic used to treat many bacterial infections, including urinary tract infections26. To gain further insight into the nature of interactions between ASCs and bladder cancer cells we co-cultured both cell types in a transwell system that prevents passage of cells but allows bidirectional transport of soluble factors. Then we analysed the composition of ASC-CM, quantified changes in viability, proliferation, adhesion, and migration of cancer cells, and examined activation of critical pro-survival pathways that are known for promoting cell growth, regulating apoptosis, chemotherapeutic drug resistance, and cellular senescence.
Results
Multiplex protein analysis
Qualitative and quantitative analysis of ASC-CM composition is essential in order to identify key players influencing the biological activities of these cells. When ASCs were co-cultured with human primary bladder carcinoma cell lines (Table 1) a strong increase in protein concentration was observed for IL-6 (from 23-fold to 3.9-fold depending on the cell line) and for IL-8 (from 16.1-fold to 10.3-fold). A moderate increase in the concentration of GM-CSF (from 3.6-fold to 2.3-fold), MCP-1 (from 2.3-fold to 1.7-fold), and RANTES (from 4.5-fold to 1.5-fold) were also noted. No significant changes in the level of IL-1B, TNF-α, and TGF-β1 were observed in co-culture with cancer cells in comparison to monoculture. The presence of IL-1A, IL-4, IL-10, and IFN-γ in ASC-CM could not be detected.
Cell morphology
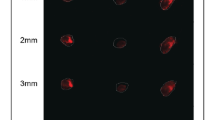
The culture of carcinoma cell lines in ASC-CM or co-culture with ASCs did not influence the morphology of cancer cells (Fig. 1). No signs of deterioration, e.g. granularity around the nucleus, detachment of the cells from the growth surface or cytoplasmic vacuolation were observed. Microscopic images were further used for quantitative analysis (cellSens Software, Olympus). No changes in shape, perimeter, as well as cell and nuclear volumes, were detected in comparison to monoculture (data not shown).

Morphology of bladder carcinoma cell lines cultured in ASC-CM or in co-culture with ASCs. Magnification ×100.
Proliferation
Co-culture with ASCs increased the number of HB-CLS-1 and HT-1376 cells by respectively 8.5% and 15.5% compared to control (Fig. 2A). These changes were not, however, statistically significant (p > 0.05). No differences in the number of 5637 cells were observed after co-culture with ASCs. Culture in ASC-CM did not influence the proliferation rate in any of the bladder carcinoma cell lines. No statistically significant differences were observed between the culture in ASC-CM and co-culture with cancer cells for any of the bladder carcinoma cell lines.

Proliferation and viability of carcinoma cells cultured in ASC-CM and in co-culture with ASCs. (A) Soluble mediators secreted by stem cells increased the number of HB-CLS-1 and HT-1376 cells by respectively 8.5% and 15.5% compared to control. Interestingly, this effect was only observed when cells were co-cultured with ASCs not when they were cultured in ASC-CM. Bars represent standard deviation. (B) Conditioned medium from ASCs increased the viability of 5637 and HT-1376 cells by respectively 15.4% and 10.4% compared to control. ASC-CM did not influence the metabolic activity of HB-CLS-1 cells. Similarly, co-culture with stem cells increased the viability of 5637 and HT-1376 cells (19,7% and 16.1%), but not of HB-CLS-1 cells. Bars represent standard deviation; p < 0.05.
Viability
The metabolic activity of 5637 and HT-1376 cells after culture in ASC-CM was increased by respectively 15.4% (p < 0.0001) and 10.4% (p < 0.0001) (Fig. 2B). Co-culture with ASCs increased the viability of both bladder carcinoma cell lines by respectively 19.7% (p < 0.001) and 16.1% (p < 0.0001). No changes were, however, observed for HB-CLS-1 cells. Interestingly, no statistically significant differences were noted between the viability of cancer cells cultured in conditioned medium and co-cultured with stem cells.
Cell cycle
The proportion of HB-CLS-1 and HT-1376 cells in G0/G1 phase was reduced by respectively 7.2% and 11.3% after incubation with ASC-CM (Fig. 3). At the same time, the proportion of cancer cells in S phase was increased by respectively 5.4% and 4.3%. These changes were not, however, statistically significant (p > 0.05). Co-culture with stem cells did not influence the cell cycle of both bladder cancer cell lines. Similarly, no changes in the distribution of 5637 cells in subsequent phases of the cell cycle were noted after incubation in ASC-CM and after co-culture with ASCs.

Cell cycle perturbations in carcinoma cells cultured in ASC-CM and in co-culture with ASCs. Culture in ASC-CM reduced the proportion of HB-CLS-1 and HT-1376 cells in G0/G1 phase by 7.2% and 11.3% and increased the proportion of cells in S phase by respective 5.4% and 4.3%. In contrast to incubation in conditioned medium, co-culture with ASCs did not influence the distribution of cells in subsequent phases of the cell cycle. No changes were observed for 5637 cell line. Bars represent standard deviation.
Apoptosis
Loss of plasma membrane asymmetry is one of the earliest features of apoptotic cells. Co-culture with ASCs did not induce translocation of phosphatidylserine from the inner to the outer leaflet of the plasma membrane of cancer cells (Fig. 4). A minor reduction in the number of live cells (6.1%) with a simultaneous increase in the number of late-apoptotic cells (4.0%) was observed for HT-1376 cell line after incubation in ASC-CM. These changes were not, however, statistically significant (p > 0.05).

Membrane phospholipid redistribution in carcinoma cells cultured in ASC-CM and in co-culture with ASCs. Annexin V positive/propidium iodide negative cells were classified as early-stage apoptotic cells, and double-positive cells were classified as late-stage apoptotic cells. Culture in ASC-CM did not induce changes in the membrane asymmetry. Co-culture with ASCs reduced the number of live HT-1376 cells by 6.1% and increased the number of late apoptotic cells by 4.0%. Bars represent standard deviation.
ECM adhesion
Cell adhesion plays a significant role in cellular communication and regulation, influencing migratory and invasive properties that promote cell motility. A trend in reduction of cell adhesion to all analysed ECM components (collagen I, collagen II, collagen IV, fibronectin, laminin, tenascin, vitronectin) after incubation with ASC-CM could be seen (Fig. 5). Adhesion of 5637, HB-CLS-1 and HT-1376 cells to collagen I was reduced by respectively 12.2%, 14.6% and 6.82% in comparison to control. Depending on the cell line, adhesion to collagen II was reduced by 9.0%, 5.9%, and 6.8% and to collagen IV by 6.7%, 1.1% and 10.8%. Adhesion of cancer cells to fibronectin was reduced by 2.0%, 13.0% and 11.5%, and to laminin by 11.9%, 2.6% and 3.8%. Similarly, adhesion to tenascin was reduced by 14.7%, 9.2%, and 5.5% and to vitronectin by 16.9%, 19.1%, and 7.7%. These changes were not, however, statistically significant (p > 0.05).

Adhesion of bladder carcinoma cells to ECM proteins after culture in ASC-CM. Even though observed changes did not reach statistical significance a trend in the reduction of cell adhesion to ECM components (collagen I, collagen II, collagen IV, fibronectin, laminin, tenascin, vitronectin) could be noted. Bars represent standard deviation.
Migration
The motility of cancer cells could be already detected in monoculture (Fig. 6A). When cultured in ASC-CM or co-cultured with ASCs the migration of HT-1376 cells was significantly increased respectively by 41.7% (p < 0.006) and 24.5% (p < 0.0002) in comparison to control. Motility of HT-1376 cells cultured in ASC-CM was significantly higher than cancer cells co-cultured with ASCs (p < 0.05). A decrease in migration of HB-CLS-1 cells was observed (10.7% and 9.2%). These changes were not, however, statistically significant (p > 0.05). No differences were noted for 5637 cells.

Bladder cancer cells migration and invasion. (A) When cultured in ASC-CM or co-cultured with ASCs HT-1376 cells showed significantly higher migration than in monoculture. Under the same conditions migration of HB-CLS-1 cells was reduced. No changes were observed for 5637 cells. (B) HB-CLS-1 and HT-1376 cells co-cultured with ASCs showed higher invasive properties than control cells. In turn, invasion of 5637 cells was significantly reduced. Bars represent standard deviation; p < 0.05. (C) Expression of B-catenin in bladder carcinoma cells did not change after culture in ASC-CM or co-culture with ASCs. Full-length blot is presented in Supplementary Figure 1.
Invasion
The invasion through the membrane pores could be observed in control cells (Fig. 6B). Invasive properties allowed them to digest the reconstituted basement membrane matrix of proteins derived from the Engelbreth Holm-Swarm mouse tumour, migrate through the ECM layer and cling to the bottom of the membrane. The invasion was significantly increased by 18.2% for HT-1376 cells (p < 0.002) and by 18.3% for HB-CLS-1 cells when cells were co-cultured with ASCs. Interestingly, a significant reduction of invasive properties (24.5%, p < 0.02) was noted for 5637 cells.
B-catenin expression
B-catenin plays an essential role in the regulation of cell adhesion. Expression of B-catenin in bladder carcinoma cells cultured in ASC-CM and co-cultured with ASCs was confirmed by Western blotting (Fig. 6C). Western blot analyses were performed on whole cell lysates (20 µg per lane). No differences in signal intensities quantified by the dedicated software were detected in comparison to control (data not shown).
AKT/ERK activation
The PI3K/AKT/mTOR and MAPK signalling pathways are known for promoting cell growth, regulating apoptosis, chemotherapeutic drug resistance and cellular senescence. The InstantOne ELISA Kit used for analyses recognise several phosphorylation sites, e.g. ERK1/2/3 (Thr202/Tyr204, Thr185/Tyr187), AKT1/2/3 (Ser473) and p70 S6K (Thr389). Expression of phosphorylated ERK1/2 was significantly (p < 0.001) increased in 5637 cells cultured in ASC-CM (2.2-fold) in comparison to control (Fig. 7). Elevated expression of ERK1/2 was also observed in HB-CLS-1 cells, however, these results were not statistically significant. In turn, expression of AKT, one of the principle kinases activated by phosphoinositide 3-kinase, was significantly (p < 0.001) increased in HB-CLS-1 cells (2.0-fold). Minor increase in AKT activity was also noted in 5637 cells. No differences in AKT/ERK activation were observed in HT-1376 cells. Soluble mediators synthesised by ASCs did not influence p70 S6K activation in any of the bladder carcinoma cell lines.

AKT/ERK/p70 S6K activation in bladder carcinoma cells cultured in ASC-CM. When cultured in ASCs secretome 5637 and HB-CLS-1 cells showed increased expression of phosphorylated ERK1/2 in comparison to control. Significant induction (2-fold) of AKT activation could also be noted in HB-CLS-1 cells. No differences in AKT/ERK activation was observed in HT-1376 cells. ASC-CM did not induce phosphorylation of p70 S6K, which activity is controlled by multiple signalling pathways, including the MAPK, PI3K, and mTOR pathways. Bars represent standard deviation; p < 0.05.
Discussion
Mesenchymal stem cells (MSCs) possess remarkable regenerative properties as they secrete a wide range of soluble mediators that induce tissue remodelling effects27,28,29. A growing body of evidence suggests that results of MSC therapy are caused by molecules, e.g. proteins, nucleic acids, lipids and extracellular vesicles, that are secreted to the extracellular environment, not to their abilities for local engrafting and differentiation. The composition of MSCs secretome is tissue-specific and changes in response to fluctuations in physiological states and pathological conditions27,30. Urological cancer patients, particularly those undergoing invasive surgery procedures, are considered as good candidates for regenerative therapies31. Possible triggering of persisting cancer cells remains, however, a significant concern in the application of autologous stem cell-based therapies during cancer remission25,32. Several studies reported that MSCs exhibit marked tropism for tumours33. Within tumour microenvironment they are thought to promote cancer cells stemness, proliferation, migration and invasion34,35,36,37. Through secretion of proinflammatory cytokines, chemokines and growth factors they may also contribute to epithelial to mesenchymal transition (ETM) that has been shown to play an essential role in the tumorigenic process38. Oh the other hand, several studies showed that ASCs induce a tumour suppressive effects in vitro and in vivo39,40. Oncologic follow-up at ten years after breast reconstruction with autologous fat (880 patients) showed no increased risk of local recurrence or development of new cancer41. Recently, this finding has been confirmed by other research groups42,43. However, as there has been a case of a late recurrence of osteosarcoma which occurred 13 years after the initial pathology and 18 months after a lipofiling procedure, some questions raised about the safety of MSCs usage in patients with cancer history44.
Giving the fact that bidirectional crosstalk between stem and cancer cells is still poorly understood, especially for urological cancers, we examined the indirect interaction between human ASCs and three human bladder carcinoma cell lines derived from primary tumours. Our results showed that co-culture of ASCs and bladder cancer cells leads to a significant change in the secreted protein levels, especially IL-6 and IL-8, proinflammatory cytokines involved in tumorigenesis (Table 1). IL-6 is one of the major cytokines present in the tumour microenvironment. Its overexpression has been reported in many types of cancers. IL-6 promotes tumour progression by regulating survival, apoptosis, proliferation, angiogenesis, and invasiveness of cancer cells. Even though emerging evidence links MSCs with metastasis, the mechanisms underlying this phenomenon are unclear45,46. Cortini et al. reported that high concentration of IL-6 in the tumour microenvironment is associated with metastatic potential of human osteosarcoma. Co-culture with bone marrow MSCs induced migratory properties through TGF-β1 and IL-6 secretion. They showed that MSCs in monoculture secreted IL-6, but when co-cultured with cancer stem cells (CSCs) derived from osteosarcoma cell lines (HOS and MG63) the levels of IL-6 significantly increased47. Increased secretion of IL-6 was also noted after a bone marrow, amniotic membrane, and decidua MSCs co-culture with ovarian cancer cell lines (SKOV-3 and IGROV-1) and endometrial cancer cell line (Ishikawa). IL-6 treatment decreased epithelial marker expression and increased mesenchymal markers expression, as well as MMP-2 and MMP-9 levels. A significant increase in migration and invasion of cancer cells was also noted what indicates that IL-6 facilitates metastasis and invasion by promoting EMT48. Similarly, Ma et al. reported that IL-8 and IL-6 secreted by umbilical cord MSCs activated the autocrine IL-8 and IL-6 signalling in breast cancer cell line (MCF-7) and induced CD44(+)/CD24(−) cells, which consequently promoted MCF-7 cell migration in vitro and metastasis in vivo49. ASCs also significantly stimulated the proliferation of MCF-7 cells in direct mixed co-cultures and transwell system, although their conditioned media displayed antiproliferative activity50.
Several studies reported that the synthesis of proinflammatory cytokines, notably IL-6 and IL-8, promotes activation of pro-survival pathways in cancer cells. CM from umbilical cord MSCs inhibited the expression of E-cadherin, increased the expression of N-cadherin and PCNA, as well as expression of ZEB1, a transcription factor involved in EMT through activation of the ERK pathway51. Chen et al. noted that umbilical cord MSCs activated the MEK/ERK signalling pathway in promyelocytic cells (NB4 cell line and primary acute promyelocytic leukaemia cells)52. Glioma-associated MSCs increased the proliferation and self-renewal of glioma stem cells in vitro and enhanced their tumorigenicity and mesenchymal features in vivo through the IL-6/gp130/STAT3 pathway activation53. It was also shown that bone marrow MSCs stimulated the proliferation invasion, survival, tumorigenicity and migration of colorectal cancer cells in a paracrine manner. CM downregulated the levels of phosphorylated AMPK, what indicates that MSCs promote the progression of colorectal cancer through AMPK/mTOR-mediated NF-κB activation54. In turn, Wnt signalling was reported to influence prostate cancer cell lines (LNCAaP, PC3 and DU145) migration toward bone marrow MSC-CM55. We showed that ASC-CM influences activation of critical components of two pro-survival pathways, PI3K/AKT/mTOR and MAPK, both of which are regulated by Ras (Fig. 7). When cultured in ASCs secretome two bladder carcinoma cell lines (5637 and HB-CLS-1) showed increased expression of phosphorylated ERK1/2. Additionally, the culture of HB-CLS-1 cells in ASC-CM resulted in 2-fold induction of AKT phosphorylation. Surprisingly, ASC-CM did not induce phosphorylation of p70 S6K, which activity is controlled by multiple signalling pathways, including the PI3K/AKT/mTOR and MAPK signalling pathways. Activation of ERK1/2 by MSC-CM was also reported by Scherzad et al. They showed that proliferation of head and neck squamous cell lines cultured in MSC-CM was induced by soluble mediators secreted by MSCs, mainly IL-656.
Paracrine activity is thought to play a significant role in the cell-cell communication during tumour development and progression. Wu et al. showed that microvesicles (MVs) derived from human umbilical cord Wharton’s jelly MSCs inhibited bladder tumour cells growth in vitro and in vivo. After 72 h incubation with MVs (200ug/ml protein), T24 cells exhibited significant growth reduction, by 48.4% in comparison to control. Moreover, dose-dependent increase in the accumulation of cells in the G0/G1 phase with simultaneous reduction of cells in the S phase was observed. They also showed an increase in AnnexinV-FITC positive cells and dramatic up-regulation of cleaved caspase 3, p-p53 and p2157. Other group reported that proliferation of EJ and T24 cells cultured in ASC-CM was significantly reduced in comparison to control. Yu et al. also showed that ASC-CM increased the number of cells in the S phase and the percentage of early and late apoptotic cells. Apoptosis induced by soluble mediators secreted by ASCs was mediated by Bcl-2 family modulation and caspase activation. Expression of p-Akt was also decreased58. Umbilical cord Wharton’s jelly MSCs were also shown to inhibit proliferation of PC-3 prostate cancer cells. Co-culture with MSCs induced the cleavage of caspase 9/3 and PARP, activated JNK, and attenuated phosphorylation of Akt and ERK59. These results are in contrary to ours. We showed that the proliferation of bladder cancer cells co-cultured with ASCs was increased in comparison to control (Fig. 2A). Similarly, the viability of cells cultured in ASC-CM and co-cultured with ASCs was significantly increased (Fig. 2B). We observed a reduction in the proportion of HB-CLS-1 and HT-1376 cells in G0/G1 phase and subsequent increase in the proportion of cells in the S phase (Fig. 3). These results were, however, not statistically significant. No significant differences in the number of AnnexinV-FITC positive cells were noted between cells cultured in ASC-CM or co-cultured with ASCs for all tested bladder carcinoma cell lines (Fig. 4). Increased proliferation of different cancer cell lines treated with MSC-CM was noted by many research groups60,61,62. Enhanced growth of DU145 prostate cancer cells, both co-cultured with bone-marrow MSCs and treated with MSC-CM, was reported by Zhang et al. They showed that co-cultures of MSCs with DU154 cells promoted tube formation ability of human umbilical vein endothelial cells. Moreover, MSCs exposed to DU145 environment increased expression of markers associated with neovascularisation63. Du and Ju et al. demonstrated that MVs derived from Wharton’s jelly MSCs promoted growth and aggressiveness of renal 786-0 cells both in vitro and in vivo. MVs facilitated the progression of the cell cycle from G0/G1 to S phase64. Those discrepancies may be caused by the fact that the secretome is cell‐specific. Moreover, MVs comprise only a part of molecules secreted to the extracellular environment, and both soluble components and MVs are capable of independent regulation of numerous biological processes27.
Multiple cell adhesion molecules take part in intercellular and extracellular matrix interactions with tumour-progressing cells. The cancer-associated ECM actively contributes to tumour histopathology and behaviour. We showed a trend in the reduction of cell adhesion to ECM components (Fig. 5). Subsequently, migration of HT-1376 and invasion of HB-CLS-1 and HT-1376 cells was significantly increased after culture in ASC-CM. On the other hand, the invasion of 5637 cells cultured in the same conditions was significantly decreased (Fig. 6A,B). Giving the fact that Yu et al. observed a decreased migration of EJ and T24 after incubation in ASC-CM its influence might be cell-line dependent58. McAndrews et al. showed that co-culture with MSCs increased directional migration of MCF7 and MDA-MB-231 breast cancer cell lines that was mediated by TGF-β and the migratory proteins Rho-associated kinase, focal adhesion kinase, and matrix metalloproteinases65. ASC-CM increased the migratory activity of primary breast cancer cells isolated from human donors (two out of four samples), even though it was not parallel with increased expression of cellular receptors involved in migration66. We did not observe any differences in the expression of β-catenin in cells cultured in ASC-CM or co-cultured with ASCs in comparison to the control (Fig. 6C). In turn, Devarajan et al. reported that ASC-CM induces breast cancer cells (4T1) to express mesenchymal markers, e.g. fibronectin, alpha-smooth muscle actin and vimentin. Other group showed that direct co-culture with bone-marrow MSCs increased expression of EMT-related genes, e.g. fibronectin, SPARC, and galectin 1 in colon cancer cells (KM12SM). Interestingly, their expression was not elevated in cancer cells indirectly co-cultured with MSCs67.
Giving the fact that ASCs isolated from adipose tissue of patients with urological cancers show growth kinetics, surface marker expression and differentiation potential similar to ASCs isolated from adipose tissue of non-oncogenic participants they may be considered as a promising material for cell-based therapies31. However, there is substantial evidence that multiple human cancer cell lines, including bladder cancer cells, are responsive to signals from MSCs. We showed that co-culture of ASCs with human bladder cancer cells strongly induces secretion of IL-6 and IL-8, major pro-inflammatory cytokines present in the tumour microenvironment. Soluble mediators secreted by ASCs increased the viability and invasion of cancer cells and induced phosphorylation of ERK1/2 and AKT. It confirms that crosstalk between ASCs and cancer cells is highly complex. Therefore, a better understanding of ASCs influence on biological characteristics of bladder cancer cells is of great importance before their implantation into clinical therapy.
Conclusion
Our results support the hypothesis that through pro-inflammatory phenotype ASCs may increase proliferation, viability, and invasiveness of bladder cancer cells. Activation of pro-survival pathways known for promoting cell growth, regulating apoptosis, chemotherapeutic drug resistance and cellular senescence indicates that through the secretion of paracrine factors stem cells may influence cancer progression. Since stem cell tropism to tumour microenvironment is well documented, increased oncological risk should be taken into account when considering the clinical use of ASCs in regenerative cell-based therapies for bladder reconstruction.
Materials and Methods
Cell lines
Human primary bladder cancer cell lines 5637 (cat. no. HTB-9, grade II carcinoma) and HT-1376 (cat. no. CRL1472, grade III carcinoma) were purchased from American Type Culture Collection (ATCC). HB-CLS-1 cells (cat. no. 300190, grade III carcinoma) were purchased from Cell Line Service (CLS). To exclude differences due to patient-to-patient variability ASC52telo hTERT immortalised adipose-derived mesenchymal stem cells (cat. no. SCRC-4000) obtained from ATCC were used in this study. These primary cells characterise with high viability and plating efficiency. They show high (>90%) expression of CD29, CD44, CD73, CD90, CD105, CD166 and low expression (<5%) of CD14, CD19, CD34, CD45. When maintained under optimal growth conditions ASC52telo cells have been shown to be multipotent, capable of differentiating down the adipogenic, osteogenic and chondrogenic lineages.
Cell culture
All media and supplements, if not noted separately, were purchased from Corning, USA. 5637 and HB-CLS-1 cells were routinely cultured in RPMI1640, and HT-1376 cells were cultured in EMEM. Both media were supplemented with 10% FBS. ASC52telo cells were cultured in DMEM/Ham’s F-12 supplemented with 10% FBS and 10 ng/ml bFGF (Thermo Fisher Scientific). Cells were maintained at 37 °C in a humidified atmosphere of 5% of CO2.
ASC-CM preparation
ASC52telo cells were seeded in cell culture flasks at a density of 10.000 cells/cm2. Conditioned medium containing soluble factors secreted by stem cells was collected after 72 h incubation, centrifuged, filtered through a vacuum-driven cup device (Millipore Express PLUS, Merck Millipore) and stored at −20 °C. Before use ASC-CM was thawed and mixed with growth medium used for cancer cells culture at ratio 1:1.
Co-culture and culture in ASC-CM
Cancer cells were seeded on multiwell plates at a density of 5.000 cells/cm2, and ASC52telo cells were seeded on cell culture inserts (1um PET membrane) at the same cell density. After 24 h preincubation, inserts were transferred to multiwell plates. To investigate the influence of ASC-CM on cancer cells growth medium from selected wells was exchanged for a previously prepared conditioned medium. All experiments were performed after 72 h incubation. Cancer cells cultured in standard growth medium served as a control.
ASC-CM composition
ASC52telo cells were seeded on 6-well plates at a density of 5.000 cells/cm2, and cancer cells were seeded on cell culture inserts (1 um PET membrane) at the same cell density. After 24 h preincubation inserts were transferred to 6-well plates. Cells were co-cultured up to 3 days without medium change. Inserts were then removed, and medium in 6-well plates was changed. Supernatants obtained after subsequent 24 h culture were analysed in the protein assay. ASC52telo cells from monoculture served as a control and were treated like the co-cultures. The level of cytokines/chemokines (IL-1A, IL-1B, IL-4, IL-6, IL-8, IL-10, IFN-γ, TNF-α, GM-CSF, MCP-1, TGF-β1, RANTES) was profiled with the Human Mix-N-Match Multi-Analyte ELISArray Kit (Qiagen) according to the manufacturer protocol. Briefly, supernatants and Antigen Standards were transferred to the ELISArray plate. After incubation and washing, Detection Antibody was added. Excess of Detection Antibody was removed with Washing Buffer. Then Avidin-HRP was pipetted to all wells. After incubation and washing to remove excess HRP conjugate, Development Solution was added. Finally, Stop Solution was added to each well and absorbance was read at 450 nm on a microplate reader (iMark, Bio-Rad).
Viability
Cell viability was measured with the use of the MTT Assay Kit (Abcam) according to the manufacturer protocol. Briefly, growth medium was removed, and cells were incubated with MTT Reagent for 3 h at 37 °C. After incubation, formazan crystals were dissolved in MTT Solvent. The absorbance was read at 570 nm on a microplate reader (iMark, Bio-Rad).
Cell cycle
Cell cycle progression was measured with the use of Tali Cell Cycle Kit (Thermo Fisher Scientific) followed by flow cytometry analysis. Cells detached from the wells were suspended in PBS and fixed in ice-cold ethanol. They were kept in ethanol overnight at −20 °C and then centrifuged. The obtained cell pellet was suspended in PBS. After subsequent centrifugation, cells were suspended in the Staining Solution composed of propidium iodide, RNase A, and Triton X-100 and analysed with the use of FACSCanto II flow cytometer (Becton Dickinson). Percentage of cells in G0/G1, S and G2 phases was calculated using the BD FACS Diva software (Becton Dickinson).
Apoptosis
Redistribution of phosphatidylserine was measured with the use of FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) followed by flow cytometry analysis. Cells detached from the wells were washed in PBS and suspended in Binding Buffer. Then FITC Annexin V and PI were added. After subsequent incubation, phosphatidylserine translocation to the outer leaflet of the plasma membrane was analysed with the use of FACSCanto II flow cytometer (Becton Dickinson).
ECM adhesion
Cell adhesion to selected components of extracellular matrix (collagen I, collagen II, collagen IV, fibronectin, laminin, tenascin, vitronectin) was measured with the use of ECM Cell Adhesion Array Kit (Merck) according to the manufacturer protocol. Cells were detached from the wells and suspended in Assay Buffer. Then they were seeded on ECM protein-coated wells. After 2 h incubation, cells were rinsed with Assay Buffer, and Cell Stain Solution was added to each well. Wells were then washed, and Extraction Buffer was added to solubilise cell-bound stain. The absorbance was read at 570 nm on a microplate reader (iMark, Bio-Rad).
B-catenin expression
Cell lysates used for immunoblotting were prepared in RIPA buffer (Abcam). Protein concentrations were analysed using the BCA Protein Assay Kit (Thermos Fisher Scientific). 20 ug of proteins were loaded and separated by SDS-PAGE and transferred to nitrocellulose membrane (Bio-Rad). After blocking with StartingBlock TBS (Thermo Fisher Scientific), membranes were incubated with primary monoclonal antibody against β-catenin (1:250, Thermo Fisher Scientific) and GAPDH (1:700, Thermo Fisher Scientific) at 4 °C overnight, washed and incubated with HRP-conjugated secondary antibody (1:1000, Thermo Fisher) at RT for 1 h. The membranes were then washed, stained using DAB Substrate Kit (Thermo Fisher Scientific) and visualised (Gel Doc XR + Gel Documentation System, Bio-Rad). The signal from each band was quantified by dedicated software for densitometric evaluation (Image Lab Software, Bio-Rad).
Migration
Cell migration was measured with the use of QCM Chemotaxis Cell Migration Assay (Merck) based on the Boyden chamber principle. Cells were detached from the wells and suspended in chemoattractant-free media. The cell suspension was then added to inserts (8um, PC membrane) and medium containing 10% FBS to the lower chamber. After incubation, inserts were transferred into clean wells containing Cell Stain. Cotton-tipped swabs were used to remove the non-migratory cells layer from the interior of the inserts. Then inserts were transferred to clean wells containing Extraction Buffer. After incubation, absorbance was read at 570 nm on a microplate reader (iMark, Bio-Rad).
Invasion
Cell invasion through a basement membrane model was measured with the use of QCM ECMatrix Cell Invasion Assay (Merck). Cells were detached from the wells and suspended in chemoattractant-free media. The cell suspension was then added to inserts (8um, PC membrane, ECMatrix) and medium containing 10% FBS to the lower chamber. After incubation, ECMatrix gel and non-invading cells were removed from the interior of the inserts with cotton-tipped swabs. Inserts were then transferred into clean wells containing staining solution. Next, inserts were washed with water, air-dried and transferred to clean wells containing EXTRACTION BUFFER. After incubation, absorbance was read at 570 nm on a microplate reader (iMark, Bio-Rad).
AKT/ERK activation
Activation of signalling pathways was measured with the use of ERK/AKT/p70 S6K Activation InstantOne ELISA Kit (Thermo Fisher Scientific) according to the manufacturer protocol. Cells were detached from the wells and suspended in HBSS containing 5% FBS. They were transferred to the InstantOne ELISA assay microplate and lysed with Cell Lysis Mix. Antibody Cocktail was then added to selected wells. After incubation, wells were washed with Washing Buffer and Detection Reagent was pipetted to all wells. Stop Solution was added to stop the reaction. The absorbance was read at 570 nm on a microplate reader (iMark, Bio-Rad).
Statistical analysis
Results from at least three independent experiments are presented as means ± standard deviation (SD). Statistical analyses were performed using STATISTICA 13.1 (StatSoft, Poland). Comparisons between two groups were analysed by Student’s t-test, and one-way ANOVA was used for multiple comparisons. A value of p < 0.05 was considered to be statistically significant.
References
Antoni, S. et al. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur. Urol. 71, 96–108 (2017).
Wong, M. C. S. et al. The global epidemiology of bladder cancer: a joinpoint regression analysis of its incidence and mortality trends and projection. Sci. Rep. 8, 1129 (2018).
Chamie, K. et al. Recurrence of high-risk bladder cancer: a population-based analysis. Cancer 119, 3219–27 (2013).
Liedberg, F. et al. Local recurrence and progression of non-muscle-invasive bladder cancer in Sweden: a population-based follow-up study. Scand. J. Urol. 49, 290–295 (2015).
Lin, T. et al. A prospective randomised controlled trial of laparoscopic vs open radical cystectomy for bladder cancer: perioperative and oncologic outcomes with 5-year follow-upT Lin et al. Br. J. Cancer 110, 842–9 (2014).
Xia, L. et al. Robotic versus Open Radical Cystectomy: An Updated Systematic Review and Meta-Analysis. PLoS One 10, e0121032 (2015).
Kim, T. H. et al. Oncological Outcomes in Patients Treated with Radical Cystectomy for Bladder Cancer: Comparison Between Open, Laparoscopic, and Robot-Assisted Approaches. J. Endourol. 30, 783–791 (2016).
Li, X., Wang, W., Zhu, G., He, W. & Gou, X. Risk factors, follow-up, and treatment of urethral recurrence following radical cystectomy and urinary diversion for bladder cancer: a meta-analysis of 9498 patients. Oncotarget 9, 2782–2796 (2018).
Pokrywczynska, M., Adamowicz, J., Sharma, A. K. & Drewa, T. Human urinary bladder regeneration through tissue engineering – An analysis of 131 clinical cases. Exp. Biol. Med. 239, 264–271 (2014).
Kates, M. et al. Tissue-Engineered Urinary Conduits. Curr. Urol. Rep. 16, 8 (2015).
Meng, L. et al. Tissue-engineered tubular substitutions for urinary diversion in a rabbit model. Exp. Biol. Med. 241, 147–156 (2016).
Liao, W. et al. Tissue-engineered tubular graft for urinary diversion after radical cystectomy in rabbits. J. Surg. Res. 182, 185–191 (2013).
Leonhäuser, D. et al. Two differentially structured collagen scaffolds for potential urinary bladder augmentation: proof of concept study in a Göttingen minipig model. J. Transl. Med. 15, 3 (2017).
Lee, J. N. et al. Human Urine-derived Stem Cells Seeded Surface Modified Composite Scaffold Grafts for Bladder Reconstruction in a Rat Model. J. Korean Med. Sci. 30, 1754 (2015).
Pokrywczynska, M. et al. Is the Poly (L- Lactide- Co– Caprolactone) Nanofibrous Membrane Suitable for Urinary Bladder Regeneration? PLoS One 9, e105295 (2014).
Kajbafzadeh, A.-M. et al. Bladder muscular wall regeneration with autologous adipose mesenchymal stem cells on three-dimensional collagen-based tissue-engineered prepuce and biocompatible nanofibrillar scaffold. J. Pediatr. Urol. 10, 1051–1058 (2014).
Pokrywczynska, M. et al. Does the Mesenchymal Stem Cell Source Influence Smooth Muscle Regeneration in Tissue-Engineered Urinary Bladders? Cell Transplant. 26, 1780–1791 (2017).
Shakhssalim, N. et al. Bladder smooth muscle cells on electrospun poly(ε-caprolactone)/poly(l -lactic acid) scaffold promote bladder regeneration in a canine model. Mater. Sci. Eng. C 75, 877–884 (2017).
De Francesco, F., Ricci, G., D’Andrea, F., Nicoletti, G. F. & Ferraro, G. A. Human AdiposeStem Cells: From Bench to Bedside. Tissue Eng. Part B. Rev. 21, 572–84 (2015).
Bajek, A. et al. Adipose-Derived Stem Cells as a Tool in Cell-Based Therapies. Arch. Immunol. Ther. Exp. (Warsz). 64, 443–454 (2016).
Uzbas, F. et al. Molecular Physiognomies and Applications of Adipose-Derived Stem Cells. Stem Cell Rev. Reports 11, 298–308 (2015).
Wang, Q. et al. The morphological regeneration and functional restoration of bladder defects by a novel scaffold and adipose-derived stem cells in a rat augmentation model. Stem Cell Res. Ther. 8, 149 (2017).
Zhe, Z. et al. Bladder Acellular Matrix Grafts Seeded with Adipose-Derived Stem Cells and Incubated Intraperitoneally Promote the Regeneration of Bladder Smooth Muscle and Nerve in a Rat Model of Bladder Augmentation. Stem Cells Dev. 25, 405–414 (2016).
Xiao, D. et al. Adipose-derived stem cells-seeded bladder acellular matrix graft-silk fibroin enhances bladder reconstruction in a rat model. Oncotarget 8, 86471–86487 (2017).
Zimmerlin, L., Park, T. S., Zambidis, E. T., Donnenberg, V. S. & Donnenberg, A. D. Mesenchymal stem cell secretome and regenerative therapy after cancer. Biochimie 95, 2235–45 (2013).
Maj, M. et al. Influence of Mesenchymal Stem Cells Conditioned Media on Proliferation of Urinary Tract Cancer Cell Lines and Their Sensitivity to Ciprofloxacin. J. Cell. Biochem. 118, 1361–1368 (2017).
Vizoso, F. J., Eiro, N., Cid, S., Schneider, J. & Perez-Fernandez, R. Mesenchymal Stem Cell Secretome: Toward Cell-Free Therapeutic Strategies in Regenerative Medicine. Int. J. Mol. Sci. 18, (2017).
Marote, A., Teixeira, F. G., Mendes-Pinheiro, B. & Salgado, A. J. MSCs-Derived Exosomes: Cell-Secreted Nanovesicles with Regenerative Potential. Front. Pharmacol. 7, 231 (2016).
Tran, C. & Damaser, M. S. Stem cells as drug delivery methods: Application of stem cell secretome for regeneration. Adv. Drug Deliv. Rev. 82–83, 1–11 (2015).
Kusuma, G. D., Carthew, J., Lim, R. & Frith, J. E. Effect of the Microenvironment on Mesenchymal Stem Cell Paracrine Signaling: Opportunities to Engineer the Therapeutic Effect. Stem Cells Dev. 26, 617–631 (2017).
García-Contreras, M., Vera-Donoso, C. D., Hernández-Andreu, J. M., García-Verdugo, J. M. & Oltra, E. Therapeutic Potential of Human Adipose-Derived Stem Cells (ADSCs) from Cancer Patients: A Pilot Study. PLoS One 9, e113288 (2014).
Zhang, L., Xiang, J. & Li, G. The uncertain role of unmodified mesenchymal stem cells in tumor progression: what master switch? Stem Cell Res. Ther. 4, 22 (2013).
Lam, P. Y. P. & Ho, I. A. W. In Stem Cell Therapeutics for Cancer 21–38 (John Wiley & Sons, Inc), https://doi.org/10.1002/9781118660423.ch3 (2013).
Hill, B. S., Pelagalli, A., Passaro, N. & Zannetti, A. Tumor-educated mesenchymal stem cells promote pro-metastatic phenotype. Oncotarget 8, 73296–73311 (2017).
Lazennec, G. & Lam, P. Y. Recent discoveries concerning the tumor - mesenchymal stem cell interactions. Biochim. Biophys. Acta - Rev. Cancer 1866, 290–299 (2016).
Chang, A. I., Schwertschkow, A. H., Nolta, J. A. & Wu, J. Involvement of mesenchymal stem cells in cancer progression and metastases. Curr. Cancer Drug Targets 15, 88–98 (2015).
Ridge, S. M., Sullivan, F. J. & Glynn, S. A. Mesenchymal stem cells: key players in cancer progression. Mol. Cancer 16, 31 (2017).
Martin, F. T. et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res. Treat. 124, 317–326 (2010).
Rhee, K.-J., Lee, J. & Eom, Y. Mesenchymal Stem Cell-Mediated Effects of Tumor Support or Suppression. Int. J. Mol. Sci. 16, 30015–30033 (2015).
Norozi, F., Ahmadzadeh, A., Shahrabi, S., Vosoughi, T. & Saki, N. Mesenchymal stem cells as a double-edged sword in suppression or progression of solid tumor cells. Tumor Biol. 37, 11679–11689 (2016).
Delay, E., Garson, S., Tousson, G. & Sinna, R. Fat Injection to theBreast: Technique, Results, and Indications Based on 880 Procedures Over 10 Years. Aesthetic Surg. J. 29, 360–376 (2009).
Silva-Vergara, C. et al. Breast Cancer Recurrence Is not Increased With Lipofilling Reconstruction. Ann. Plast. Surg. 79, 243–248 (2017).
Kronowitz, S. J. et al. Lipofilling of the Breast Does Not Increase the Risk of Recurrence of Breast Cancer. Plast. Reconstr. Surg. 137, 385–393 (2016).
Papaccio, F. et al. Concise Review: Cancer Cells, Cancer Stem Cells, and Mesenchymal Stem Cells: Influence in Cancer Development. Stem Cells Transl. Med. 6, 2115–2125 (2017).
Kumari, N., Dwarakanath, B. S., Das, A. & Bhatt, A. N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 37, 11553–11572 (2016).
Fisher, D. T., Appenheimer, M. M. & Evans, S. S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 26, 38–47 (2014).
Cortini, M., Massa, A., Avnet, S., Bonuccelli, G. & Baldini, N. Tumor-Activated Mesenchymal Stromal Cells Promote Osteosarcoma Stemness and Migratory Potential via IL-6 Secretion. PLoS One 11, e0166500 (2016).
So, K. A., Min, K. J., Hong, J. H. & Lee, J.-K. Interleukin-6 expression by interactions between gynecologic cancer cells and human mesenchymal stem cells promotes epithelial-mesenchymal transition. Int. J. Oncol. 47, 1451–1459 (2015).
Ma, F. et al. Human Umbilical Cord Mesenchymal Stem Cells Promote Breast Cancer Metastasis by Interleukin-8- and Interleukin-6-Dependent Induction of CD44+/CD24− Cells. Cell Transplant. 24, 2585–2599 (2015).
Trivanović, D. et al. Characteristics of human adipose mesenchymal stem cells isolated from healthy and cancer affected people and their interactions with human breast cancer cell line MCF-7 in vitro. Cell Biol. Int. 38, 254–265 (2014).
Li, T. et al. Umbilical cord-derived mesenchymal stem cells promote proliferation and migration in MCF-7 and MDA-MB-231 breast cancer cells through activation of the ERK pathway. Oncol. Rep. 34, 1469–1477 (2015).
Chen, F. et al. Mesenchymal Stem Cells Induce Granulocytic Differentiation of Acute Promyelocytic Leukemic Cells via IL-6 and MEK/ERK Pathways. Stem Cells Dev. 22, 1955–1967 (2013).
Hossain, A. et al. Mesenchymal Stem Cells Isolated From Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells 33, 2400–2415 (2015).
Wu, X.-B. et al. Mesenchymal stem cells promote colorectal cancer progression through AMPK/mTOR-mediated NF-κB activation. Sci. Rep. 6, 21420 (2016).
Jin, F. et al. Regulation of prostate cancer cell migration toward bone marrow stromal cell-conditioned medium by Wnt5a signaling. Mol. Med. Rep. 8, 1486–1492 (2013).
Scherzad, A. et al. Human mesenchymal stem cells enhance cancer cell proliferation via IL-6 secretion and activation of ERK1/2. Int. J. Oncol. 47, 391–397 (2015).
Wu, S., Ju, G.-Q., Du, T., Zhu, Y.-J. & Liu, G.-H. Microvesicles derived from human umbilical cord Wharton’s jelly mesenchymal stem cells attenuate bladder tumor cell growth in vitro and in vivo. PLoS One 8, e61366 (2013).
Yu, X. et al. Human Adipose Derived Stem Cells Induced Cell Apoptosis and S Phase Arrest in Bladder Tumor. Stem Cells Int. 2015, 1–12 (2015).
Han, I. et al. Umbilical cord tissue-derived mesenchymal stem cells induce apoptosis in PC-3 prostate cancer cells through activation of JNK and downregulation of PI3K/AKT signaling. Stem Cell Res. Ther. 5, 54 (2014).
Melzer, C., von der Ohe, J. & Hass, R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun. Signal. 16, 2 (2018).
Chu, Y. et al. Adipose-derived mesenchymal stem cells promote cell proliferation and invasion of epithelial ovarian cancer. Exp. Cell Res. 337, 16–27 (2015).
Koellensperger, E. et al. Alterations of gene expression and protein synthesis in co-cultured adipose tissue-derived stem cells and squamous cell-carcinoma cells: consequences for clinical applications. Stem Cell Res. Ther. 5, 65 (2014).
Zhang, T. et al. Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res. Ther. 4, 70 (2013).
Du, T. et al. Microvesicles Derived from Human Wharton’s Jelly Mesenchymal Stem Cells Promote Human Renal Cancer Cell Growth and Aggressiveness through Induction of Hepatocyte Growth Factor. PLoS One 9, e96836 (2014).
McAndrews, K. M., McGrail, D. J., Ravikumar, N. & Dawson, M. R. Mesenchymal Stem Cells Induce Directional Migration of Invasive Breast Cancer Cells through TGF-β. Sci. Rep. 5, 16941 (2015).
Eterno, V. et al. Adipose-derived mesenchymal stem cells (ASCs) may favour breast cancer recurrence via HGF/c-Met signaling. Oncotarget 5, 613–33 (2014).
Takigawa, H. et al. Mesenchymal Stem Cells Induce Epithelial to Mesenchymal Transition in Colon Cancer Cells through Direct Cell-to-Cell Contact. Neoplasia 19, 429–438 (2017).
Author information
Authors and Affiliations
Contributions
M.M. and A.B. designed experiments, M.M. and A.K. conducted experiments, M.M. analyzed data and wrote the paper, T.D. supervised works.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maj, M., Kokocha, A., Bajek, A. et al. The interplay between adipose-derived stem cells and bladder cancer cells. Sci Rep 8, 15118 (2018). https://doi.org/10.1038/s41598-018-33397-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33397-9
Keywords
This article is cited by
-
The effect of obesity on adipose-derived stromal cells and adipose tissue and their impact on cancer
Cancer and Metastasis Reviews (2022)
-
Epidural adipose tissue-derived mesenchymal stem cell activation induced by lung cancer cells promotes malignancy and EMT of lung cancer
Stem Cell Research & Therapy (2019)
-
Association of body mass index with bladder cancer risk in men depends on abdominal obesity
World Journal of Urology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
