
Abstract
Protein copy numbers can be measured by biochemical methods ranging from quantitative Western Blotting to several mass spectrometry approaches. Such methods only provide average copy numbers, obtained over large cell numbers. However, copy number estimates for single cells or single organelles could be obtained by combining biochemical characterizations with an imaging approach. We performed this here for synaptic proteins, in a protocol that we termed comparative synaptosome imaging for semi-quantitative copy numbers (CosiQuant). In brief, in CosiQuant we immunostain in parallel biochemically-characterized synaptosomes, for which we have already determined the average protein copy numbers, and the samples of interest (such as neuronal cultures). We then derive the copy numbers in the samples of interest by comparing the immunofluorescence intensities. We measured the intensities not only in arbitrary fluorescence units, but also as numbers of antibodies per synaptosome, for a large number of targets. This implies that other groups can immediately apply CosiQuant for these targets, by simply estimating the number of antibodies per structure of interest. CosiQuant should therefore be a useful addition to the growing set of imaging techniques for synaptic neuroscience.
Similar content being viewed by others

Introduction
The quantitative organization of neurons has been the subject of countless scientific studies during the past decades, with synapse physiology being one of the most studied areas of neuroscience1,2,3. The research focus has typically been on explaining the mechanisms of neuronal communication, and finally on understanding the causes for neurological disorders.
Such disorders generally produce slight but significant changes in the basic molecular anatomy of the neuron, meaning the spatial organization and copy numbers of proteins within the neuron. The information on protein copy numbers in compartments such as the synapse is especially important, as it enables us to identify potential bottlenecks of cellular and molecular mechanisms4,5,6. As an example, mapping the protein composition of the synaptic vesicle7 has shown that the vesicular proton pump is only present in 1–2 copies per vesicle, and is thus one of the most likely molecules to generate a bottleneck (a rate-limiting factor) in synaptic vesicle recycling mechanisms. As a second example, endocytosis cofactors have also been shown to be limiting in the vesicle recycling pathway, while the exocytosis-related proteins are far more abundant, and are unlikely to be limiting8,9. In addition, knowledge on absolute copy numbers is also desirable from a technical point of view, as it renders comparisons between independent studies far more precise.
Numerous methods have been therefore developed to determine protein copy numbers, mainly by biochemical means. These techniques comprise Quantitative Western Blotting10 and mass spectrometry (MS) approaches like iBAQ (intensity-based absolute quantification11) and AQUA (absolute quantification approach12). Quantitative Western Blotting relies on the comparison of SDS-PAGE band intensities between a sample of interest and purified recombinant variants of the protein of interest, run on the gels in known amounts9. In many mass spectrometry approaches the estimation of the abundance of particular proteins is accomplished by comparing the spectra of the analyzed proteins and/or peptides to a standard. In AQUA this standard comprises an isotopically labeled peptide that reproduces a region of the protein of interest12, and which is spiked into the analyzed sample. iBAQ is a label-free method, in which one spikes accurately quantified proteins in the samples of interest, followed by in-solution digestion and mass spectrometry analysis. The intensities of the different peptides identified are summed to generate the overall protein intensities (for each protein of interest), and these are correlated to the known spiked amounts. This enables the use of a relatively simple linear regression to identify the amounts of the native proteins of interest11,13,14.
In all of these approaches the samples are usually cell culture lysates or brain homogenates, which precludes the analysis of variations between single cells or cell regions. To account for this, methods combining mass spectrometry and an imaging technique have been developed, like MALDI-TOF-TOF (matrix-assisted laser desorption ionization coupled to a tandem time-of-flight analysis) imaging, which makes it possible to map the intensities of MS spectra to the corresponding position in a sample15. However, accurate protein quantification is difficult to achieve, and the spatial resolution is still too low (usually about 20–50 µm) to obtain sub-cellular information. Alternatively, approaches for protein quantification based purely on imaging techniques could, in principle, provide the resolution needed for comparisons of protein copy numbers between cells or sub-cellular regions. For example, one can express GFP-tagged proteins of interest in the sample, followed by immunostaining for the proteins of interest16. One can then obtain estimates for the copy numbers of native (wild-type) proteins present in each cellular location by comparing the immunostaining signals to an estimation of the GFP copy numbers. However, these techniques are often difficult, as both the GFP signals and the immunostainings need to be carefully calibrated, and can only be applied to samples where the expression of GFP-tagged proteins is efficient.
Here we take advantage of synaptosomes that we have previously characterized by biochemical methods9 to establish a relatively easy imaging-based method for the estimation of protein copy numbers in neurons: comparative synaptosome imaging for semi-quantitative copy numbers (CosiQuant). Our approach relies on immunostaining the samples of interest and these synaptosomes in parallel. We then compare the resulting intensities in a semi-automated fashion. Protein copy numbers in the sample of interest can then be inferred from the known values in the synaptosome preparations, for which we derived estimates for more than 1000 proteins9. We tested this technique on cultured hippocampal neurons, and found that it works well for the estimation of neuronal proteins in this system.
Furthermore, we generalized this method, by removing the need for other laboratories to compare directly fluorescence intensities in our biochemically-characterized synaptosomes and in their preparations of interest. We expressed the fluorescence intensities we measured in synaptosomes in the form of “average numbers of antibodies per synaptosome”. This implies that other laboratories can estimate the numbers of antibodies in their structures of interest, and can then compare them with the numbers we provide, which would enable the estimation of protein copy numbers. We were able to validate this approach by turning to older datasets from the laboratory, not collected for the purpose of this work, which thus served as an independent control.
Results
For an initial proof-of-principle, we tested the comparative imaging approach on a very well established system, the primary hippocampal neuron culture. The protocol works as follows (Fig. 1): frozen synaptosome preparations, which have been biochemically characterized in a previous study9, are thawed and are immobilized on coverslips. The synaptosomes and cultured hippocampal neurons are then immunostained in parallel for two synapse markers (synaptophysin for synaptic vesicles, and bassoon for the active zone) and for the protein of interest (POI). The fluorescent signals are acquired in the three separate channels, and the intensities of these signals are measured. The data are further processed, and afterwards signal intensities of the POI’s derived from both preparations are compared. Based on the knowledge about protein copy numbers in the synaptosome preparations, copy numbers of the POI in hippocampal neuron synapses can be interpolated.

Principle of the comparative imaging approach (CosiQuant). Biochemically characterized synaptosome preparations are immobilized on coverslips. Cultured hippocampal neurons are immunostained in parallel with the synaptosomes for the synaptic vesicle marker synaptophysin (green), the active zone marker bassoon (blue) and the protein of interest (orange). The insets show example images of the fluorescently labeled structures (synapses/synaptosomes). Fluorescent signals for all labeled proteins are acquired, and intensities are compared between the synaptosome and neuron samples. Finally, protein copy numbers in cultured neuronal synapses can be estimated by comparing the intensities. Scale bar in overview images = 25 µm; scale bar in insets = 2 µm.
We tested the comparative imaging approach for 10 synaptic markers: the synaptic vesicle marker synaptophysin, the active zone marker bassoon, as indicated above, followed by four additional synaptic vesicle proteins (the fusion SNARE VAMP2, the glutamate transporters vGlut1/2, the calcium sensor synaptotagmin 1, and the synaptophysin-related protein synaptogyrin), two plasma membrane SNAREs (SNAP25 and syntaxin 1), the soluble vesicle-binding protein synapsin 1/2, and the endosomal protein syntaxin 12/13 (see3 for details on the different proteins). We have previously derived copy numbers for all of these proteins in the synaptosome preparations9. At the same time, thoroughly characterized antibodies are available for immunostainings.
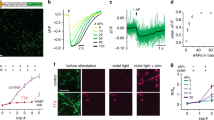
Example images for the immunostainings are shown in Fig. 2a, with synapsin 1/2 as the POI. The images, comprised of the three channels for synaptophysin, bassoon and the POI, were analyzed by a custom-written Matlab routine, as described in Methods. In a first step, potential synapses in each image were detected based on the local intensity maxima in the synaptophysin (synapse marker) signal (Fig. 2b). Application of an initial intensity threshold ensures that a huge fraction of the image noise is not taken into account. At the same time, the co-presence of the bassoon signal ensures that the detected objects are indeed synapses, and not only synaptophysin protein assemblies or vesicles transported along the neurites. For each synapse candidate the size, intensity and position are determined using a Gaussian fit (Fig. 2c). The R2 value for each fit is calculated, and is used in a filtering step later on. Subsequently, the intensities of all three channels for all detected synapses are plotted (Fig. 2d). This data set includes many synapses that are not fitted correctly, or might not represent real/single synapses. Thus, a final filtering step has been included in the data analysis, which excludes synapse candidates with a fit worse than a set R2 threshold. This results in a data set containing information about the size, position and intensities of synapses, carefully pruned to represent real synapses.

Image analysis for synapsin as protein of interest. (a) Example images of synaptosomes and cultured hippocampal neurons immunostained for synaptophysin, bassoon and synapsin. Scale bar = 10 µm (b) Automatic synapse selection by a custom-written Matlab routine in the areas indicated in the images in a. Synapse detection is based on the signal intensities of the synaptophysin staining (first channel). The selected synapses are numbered so fitted parameters (e.g. intensity) can later be assigned to the respective signals. Scale bar = 5 µm (c) Example of the Gaussian fits for the synapses indicated in panel b. The first image shows the original intensity data (scale in arbitrary units on the far right), the second image shows the model that is fitted to the data, and the third image indicates the residuals, i.e. the deviations of the data from the 2D Gaussian distribution. The R2 values of each fit are indicated on the right. Images are scaled in pixel. Scale bar = 800 nm (5 pixels) (d) Intensity histograms of all fitted synapses for the three channels (prior to final filtering).
For further analysis we collected the measured intensities for all POIs, derived from all images. Mean intensities were calculated and were compared between stainings of synaptosomes and cultured neurons (Fig. 3a). We first investigated synaptophysin, which is the most specific marker of synaptic vesicles7,10,17, being found almost exclusively in vesicles, unlike other vesicle proteins that are often also found on the plasma membrane (for example synaptotagmin 1 or VAMP218,19). The number of synaptic vesicles per synaptosome averages ~3809, measured in electron microscopy. In cultured hippocampal synapses only 250 ± 26 synaptic vesicles are present on average (mean ± SEM). This was calculated from 30 electron microscopy 3D reconstructions, with the following individual vesicle numbers: 36, 86, 89, 95, 119, 124, 128, 136, 137, 173, 185, 191, 197, 197, 212, 224, 233, 248, 249, 252, 257, 302, 315, 377, 403, 450, 509, 516, 526, 544. This analysis indicates that the synaptosomes contain on average 35% more synaptic vesicles. A similar value was found for the mean synaptophysin signal in our intensity comparison (29%), suggesting that these immunostaining signals are a reliable measure for the relative protein numbers in synaptosomes and hippocampal neurons.

Comparison of intensities derived from labeled proteins in synaptosomes and neuronal synapses. (a) Absolute intensities (in arbitrary units) of the 10 labeled proteins in synaptosome preparations and cultured hippocampal neurons, expressed as mean values derived from all selected synapses (after final filtering). The right panel shows example synapses, marked with the respective intensities for the synapsin 1/2 stainings (in arbitrary units). Scale bar = 5 µm. (b) Average synaptic protein copy numbers, obtained by dividing the synapse intensities by the average synaptosome intensities, and multiplying with the average copy numbers per synaptosome from9. Example synapses with the respective synapsin 1/2 copy numbers are shown in the right panel. Scale bar = 5 µm. In contrast to the graph in (a), the graph in (b) is shown in log scale. Bar graphs represent mean values with standard errors of the mean (SEM). N = 875 synaptosomes (for synaptophysin and bassoon), 24–184 synaptosomes (for all other POI’s), 1809 cultured synapses (for synaptophysin and bassoon), and 51–354 synapses (for all other POI’s).
Conclusions about the copy numbers of the other investigated proteins can now be drawn in a similar fashion, and absolute copy numbers can be estimated by comparisons with protein copy numbers from9, by dividing the fluorescence intensities of the neuronal synapses by the average synaptosome intensity, followed by multiplying the result with the (published) average copy numbers per synaptosome (Fig. 3b). Nevertheless, this is only useful if one can use the exact same synaptosome preparations analyzed by Wilhelm and colleagues9 for the comparative staining experiments. Since this is not the case for other laboratories, we also provide here the number of single antibodies (used for the immunostainings) per synaptosome for over 100 different targets (Tables 1 and 2). This will enable the comparison to immunostainings of any sample of choice performed with the same antibody, without the need for immunolabeling again the synaptosomes. This procedure only requires one to immunostain the protein of interest in the sample of choice, and to determine the number of single antibodies per structure of interest (Fig. 4). The number of antibodies can then be compared between synaptosomes (Tables 1 and 2) and the sample of choice, and thus protein copy numbers can be estimated.

A comparative approach based on using the “number of antibodies per structure” as a measure for immunostaining intensities. The intensity of the immunolabaled synaptosomes and structure of interest (here: cultured neuronal synapses) can be measured (upper panels). Fluorescent intensities from single antibodies absorbed to a glass coverslip and immunolabeled with the same secondary antibodies as used for the synaptosomes/synapses can be determined in parallel (lower panels). This makes it possible to express the intensity of labeled synaptosomes and structures of interest in terms of antibody copy numbers. This has been done by our laboratory for over 100 target proteins in synaptosomes, and can be done easily for any structure of interest by any other laboratory. Finally, intensities can be compared in terms of antibody copy numbers, and protein copy numbers can be calculated from this.
In order to validate this approach, we tested it on older data from our laboratory, which were not generated for this purpose, but were available from experiments performed previously by different investigators, using the same antibodies at different points in time, between 2012 and 2014. We analyzed super-resolution images (acquired using STED microscopy) of hippocampal neuron cultures immunostained for 4 proteins, which we had also investigated previously in the comparative imaging experiment showcased in Fig. 3 (synapsin, synaptogyrin, syntaxin12/13 and vGlut1/2). The intensities of single antibodies spotted on coverslips (derived from the same samples) were measured (Fig. 5a), and the number of single antibodies per neuronal synapse was calculated by dividing the total intensity in a synapse by that of the single antibodies. This provides a direct, measured number of antibodies per synapse in culture, from experiments performed between 2012 and 2014. This is the type of measurement any laboratory would be able to obtain directly, in immunostaining experiments.

Numbers of single antibodies as a measure for relative immunostaining intensity (a) Confocal and STED images of hippocampal neuron synapses and single antibodies bound to a glass coverslip. Synapses were stained for synaptophysin, bassoon and synapsin1/2 (confocal image). Super-resolution images of the signal derived from the anti-synapsin antibodies were taken in synapses (top right) or in a single-antibody configuration (bottom panels; left: scaled identically to synapses; right: autoscaled to visualize the signal of single antibodies). Scale bar = 500 nm (b) 2-dimensional scatter plot comparing numbers of antibodies per synapse derived from the measurement described in a), along the Y axis, or derived by calculation from synapse/synaptosome ratio (taken from results in Fig. 3), along the X axis. The spots represent mean values, and error bars represent the standard error of the mean (SEM). N = 632, 660, 1034 and 1009 synapses for synapsin, synaptogyrin, syntaxin 12/13 and vGlut 1/2 respectively, from two independent experiments, for the measured data. The estimated data are extracted from Fig. 3, and therefore have the same N values. The black line represents a linear regression with a slope of 1.34 and R2 of 0.955.
At the same time, we estimated the number of antibodies per synapse from our parallel immunostaining experiments, all performed in 2018. We simply multiplied the number of antibodies per synaptosome (from Tables 1 and 2) with the ratio between the synaptosome and the synapse intensities, obtained from the parallel experiments from Fig. 3. This estimate should be relatively precise. We then compared the overall results in a 2-dimensional scatter plot (Fig. 5b). The high correlation between the two methods indicates that the number of single antibodies per synapse is a reliable substitute for the relative intensity of an immunostaining. Therefore, numbers of antibodies per synaptosome provided by us in Tables 1 and 2 can be used for the comparative imaging approach. This makes CosiQuant an easily applicable method for the estimation of protein copy numbers, with sub-cellular resolution.
Discussion
We introduced here an imaging-based method for the estimation of protein copy numbers, which can be implemented relatively easy in every lab equipped for standard immunohistochemistry experiments. CosiQuant relies on the comparison of immunolabeling signals from the sample of interest and biochemically characterized synaptosome preparations. In brief, we derive the protein copy numbers for POIs in samples of interest by comparing the immunostaining signals to those derived from synaptosome preparations with known copy numbers. Furthermore, we provide a substitute for this comparison (Figs 4 and 5), so that other laboratories can apply the comparative imaging method to their sample of interest without having to use the exact same synaptosome preparations as used here.
In contrast to methods like Quantitative Western Blotting or mass spectrometry, no extensive purification steps are needed, and a much higher spatial resolution can be achieved. Furthermore, the use of synaptosomes instead of other standards, such as purified proteins or fluorescent beads, provides a more realistic comparison to neuronal samples. Effects such as the orientation of different proteins within membranes or molecular complexes, protein density, or steric hindrances, which all affect antibody binding, will apply in an identical fashion to both cultured synapses and synaptosomes, thus reducing the experimental differences between the standard sample and the sample to be measured.
Nevertheless, the comparative imaging approach also exhibits several limitations. A major one concerns the fact that CosiQuant assumes that the synaptosomes and the sample of interest are immunostained equally well. This should not be a major concern regarding the immunostaining of synaptosomes and of cultured neurons, since factors like penetration of antibodies should be comparable in both of these thin samples. For samples like whole brains or organs, however, this might differ substantially (although we found this not to be the case under optimal immunostaining conditions, for ~20 different antibodies9). For such samples discrepancies in antibody penetration could be tested by staining for a GFP-tagged variant, whose fluorescence is used as a standard, and the differences in staining efficiency could be corrected for. Another procedure to ensure relatively equal staining conditions while comparing synaptosomes and whole brain/organ samples would be to prepare and to immunostain thin sections of the samples. In this fashion the sample depth can be defined, and can be limited to ~10 µm. In order to achieve this, one can prepare cryosections of freshly dissected samples. Brains or organs can be either snap-frozen or chemically fixed first and then frozen in liquid nitrogen or on dry ice. Subsequently, the samples can be cut on a cryostat into 10 µm thin sections, which can be immunostained on coverslips or glass slides.
Another limitation concerns the usage of numbers of single antibodies as a substitute to staining intensity for the synaptosome preparations. We provide here the numbers of single antibodies per synaptosome (Tables 1 and 2) for over 100 different antibodies. But in order to use these numbers for the comparison of samples, one needs to stain the sample of interest under the exact same conditions (including fixation, antibody concentration, or blocking conditions) as the synaptosomes. These conditions might not be applicable to all samples. Furthermore, CosiQuant cannot be used for target proteins that have not been biochemically characterized yet in our synaptosomes. Nevertheless, the initial database of antibody numbers per synaptosome we provide here may already prove useful, since it covers pathways and networks ranging from the cytoskeleton to synaptic vesicle exo- and endocytosis, or to the postsynaptic density. Another limitation concerns the fact that antibody staining intensities may not sum linearly, and thus some estimates may not be correct. It has been our experience that this type of error is largely negligible, at least for structures that are not immensely bright. For example, immunostained single antibodies, which should be identified by ~3 secondary antibodies, are indeed ~3 fold brighter than individual secondary antibodies20,21, both in STED microscopy and in epifluorescence microscopy. This may not necessarily hold true in every imaging system, and should be tested carefully.
In summary, CosiQuant provides a comparably easy method for the estimation of protein numbers, and should in principle be applicable to a variety of different samples. This should be especially relevant in samples that are not purifiable (as cells from the sensory system or the peripheral nervous system22,23) and thus cannot be analyzed by methods as mass spectrometry, which require abundant, purified samples.
Methods
Synaptosome preparation
Synaptosomes were purified by simple differential centrifugation and ficoll gradient centrifugation, as described previously in9. The synaptosome preparations from that study were snap frozen and stored at −80 °C. The frozen synaptosomes were used by thawing them on ice and immobilization on glass coverslips (for details see immunocytochemistry section).
Primary hippocampal neuron culture
Rat primary hippocampal neuron cultures were prepared from newborn rats as described before24,25 and were cultured under standard conditions. Cultured neurons of 10 to 15 days in vitro (21 or 22 DIV for the calculation of single antibody numbers per synapse; Fig. 5 and Tables 1, 2) were used for immunocytochemistry experiments.
Immunocytochemistry of synaptosomes and primary hippocampal neurons
For synaptosome immobilization on glass coverslips, the coverslips were coated with 5% bovine serum albumin (BSA, AppliChem #A1391,0500) over night at 4 °C. Frozen synaptosome preparations were thawed on ice and dilutions (in PBS) were spun down in a centrifuge (VWR MegaStar3.0R) for 40 min at 4000 rpm, at 4°. Fixation of synaptosomes and cultured neurons was done with a 4% PFA solution for 15 min at 4 °C and another 45 min at room temperature. Subsequently, preparations were washed briefly in PBS and quenched for 30 min in 100 mM NH4Cl. Permeabilization and background epitope blocking were achieved by 30 min incubation in PBS containing 0.1% Triton X-100 and 5% BSA (blocking solution). Both samples were incubated with primary antibodies diluted in blocking solution (prepared as one master mix) for 60 min at room temperature. After washing for 30 min in PBS the preparations were incubated with secondary antibodies, diluted in blocking solution for 60 min at room temperature. Primary and secondary antibodies used for all stainings, and the respective dilutions from a 1 mg/ml stock can be found in Table 3. Final washing in high-salt PBS (containing 500 mM NaCl) and PBS was followed by embedding in Mowiol. The samples were imaged with an epifluorescent Nikon microscope.
Some immunostainings of cultured neurons and synaptosomes used for calculating the number of single antibodies per synapse/synaptosome (Fig. 5 and Tables 1, 2) were performed with slight variations to the protocol described above. Neurons cultured in the Banker arrangement (locally separated from the astrocyte feeder layer26) were fixed with a 3% glyoxal solution27 and the blocking and permeabilization was achieved with 2.5% BSA and 0.3% Tween in PBS. Secondary antibody incubation was done for 60 min with Cy3-labeled donkey anti-mouse or rabbit Fab fragments (Dianova, #715-166-150 and #711-166-152) and Atto647N-labeled goat anti-mouse or rabbit antibodies (Rockland, #610-156-121 and #610-156-122). All other steps of the staining protocol remained the same.
The immunolabeling protocol for cultured neurons and synaptosomes stained for super resolution imaging (data used in Fig. 5 and Tables 1, 2) can be found in9.
Information about all antibodies used for stainings, in addition to the ones described in Table 3, can be found in Tables 1 and 2.
Image acquisition
Comparative imaging of the immunolabled synaptosomes and cultured neurons was done with an inverted epifluorescence Nikon Eclipse Ti-E microscope. The microscope was equipped with an HBO 100 W lamp and images were acquired with an Andor IXON X3 897 camera or a Nikon DS-Qi2 camera (for images used to calculate synaptosome staining signals in terms of number of antibodies, Tables 1 and 2). The samples were imaged using a 100X PLAN APO oil immersion objective (NA 1.45). For multi-color imaging the following filter sets were used for Alexa488, Cy3 and Cy5 imaging: 470/40 nm (excitation, Alexa488), 525/50 nm (emission, Alexa488), 545/25 nm (excitation, Cy3), 605/70 nm (emission, Cy3), 620/60 nm (excitation, Cy5), 700/75 nm (emission, Cy5). Image acquisition software used was NiS-Elements AR (Nikon) and imaging parameters were kept the same for samples that were compared.
Super-resolution imaging (STED) and confocal imaging of neurons and synaptosomes (data used in Fig. 5 and Tables 1, 2) were performed with a Leica TCS SP5 STED microscope, exactly as described in9.
Image analysis
The image analysis was a two-step process carried out via custom-written Matlab (The Mathworks Inc.) routines. In the first step, the aim was to obtain initial guesses for the positions of the synapses. For this purpose, the script searches for the local intensity maxima in the images immunostained for synaptophysin (synapse marker). In order to avoid too many false positives caused by intensity noise, the images were filtered before searching for the local maxima using a Gaussian kernel with standard deviation σ = 480 nm. In addition, intensity thresholds were applied, i.e. spots with very small peak intensities, which are most likely due to noise, were not taken into account. The thresholds were carefully chosen by eye, and they were set low enough so that real synapses were not accidentally removed. Thus at this point, due to the low thresholds, the set of selected synapses included a fraction of false positive synapse candidates. The set of synapse candidates was further filtered at a later stage of the image analysis.
In the second step, the script goes back to the raw (unfiltered) images and fits the exact positions, sizes and intensities for all synapse candidates using the initial guesses as obtained in the first step. For the fit a square region of interest (2.2 μm × 2.2 µm) around each candidate is defined, and all candidates in all channels are fitted, i.e. those stained for synaptophysin, bassoon and the respective protein of interest (POI). The fit model is a 2D Gaussian function of variable size, position, orientation, amplitude and offset. For further analysis, mainly the total (integrated) intensity of each synapse candidate was used. The R2 between the model and the data was calculated as a measure for the goodness of the fit.
In a final step, all fitted synapse candidates were filtered according to the R2 value for each channel (synaptophysin, bassoon, POI). Candidates with a R2 value below 0.85 (synaptophysin), 0.7 (bassoon) and 0.6 (POI) for the fit were discarded. Thus, sets of well fitted synapses and synaptosomes were left, which could be compared in terms of intensity.
For display purposes only (Figs 2a and 5) images were adjusted in brightness and contrast using ImageJ (Wayne Rasband, US National Institutes of Health). If intensities were compared, image adjustments in brightness and contrast were equally applied to all conditions.
Data analysis
Intensities of the analyzed and filtered synapses and synaptosomes were collected and mean intensities were compared between neuron cultures and synaptosomes for each protein of interest. To analyze the signals in terms of antibody copy numbers, the intensities of the cellular structures (synaptosomes or cultured neurons) were divided by the average intensity of single antibodies immunostained on coverslips (thus equivalent to background antibody signals), as performed in the past20.
Protocol for imaging single antibodies
A simple procedure enables the analysis of the intensities of single immunostained primary antibodies. We typically rely on coverslips coated with poly-L-lysine, which are incubated for 10–30 minutes with primary antibody dilutions (final concentrations of 10 µg/ml), in PBS. The coverslips are then fixed, using the same fixation buffer as desired in the final application (for example 4% PFA, with or without 0.1% glutaraldehyde, or 3% glyoxal; see27, for further details on different fixation protocols). After fixation a quenching procedure is performed for 15–30 minutes, with 50–100 mM NH4Cl in PBS, or 50–100 mM glycine in PBS. This is followed by 2–3 rapid washes with PBS.
Alternatively, the fixation step can be avoided completely, as in most experiments the primary antibodies are not subjected to fixation during the immunostaining procedure. We only perform the fixation procedure when analyzing antibodies that are normally taken up by living cells, and therefore are fixed during the immunostaining process.
The coverslips are then incubated for 15–30 minutes with PBS containing 2–3% BSA (blocking buffer). The blocking buffer coats the poly-L-lysine surface with BSA, and prevents the extensive binding of secondary antibodies to this surface. This is followed by incubating the coverslips with secondary antibodies (diluted to 10 µg/ml) in PBS containing 2–3% BSA, for 30–60 minutes. The coverslips are then washed extensively: 3 × 5 minutes with PBS containing high salt (500 mM NaCl), and 3 × 5 minutes with normal PBS (150 mM NaCl). The coverslips can then be mounted in the desired mounting medium, and can be imaged.
For the image analysis, we recommend applying a bandpass filter on the images, to detect the antibody spots, followed by Gaussian fits on the spots, to obtain the total signal intensity associated to each spot. The population of antibody intensities obtained should be fitted well by a single Gaussian, whose peak position indicates the average single primary antibody intensity. Performing this experiment with super-resolution is very convenient, since then large spots (full width at half maximum, FWHM, larger than 50 nm) can be discounted. They do not represent single antibodies, but presumably are caused by dirt on the coverslips.
Importantly, a very simple and practical application for obtaining this type of value, without any additional experiments, has been to investigate the background spots obtained on the clean coverslip areas adjacent to cultured neurons, in the normal immunostaining experiments used for determining protein intensities in the cultured neurons. It has been our experience that the results obtained are indistinguishable from those obtained when immunostaining antibodies on coverslips in separate experiments, as described above, provided that large spots, indicating dirt or cell debris on the coverslips, are discounted. Nevertheless, a number of experiments in which antibodies on coverslips are measured as indicated above should be performed, to test that the background spots from the cultures can be indeed trusted, since inappropriate handling of the cultures (poor fixation or blocking, for example) may result in the formation of extensive antibody clusters, which would perturb the measurements.
Statistics
Bars and data points in Figs 3 and 5 show mean values. All error bars represent the standard error of the mean (SEM), calculated in Sigma Plot (Systat Software, Inc.), unless stated otherwise in the figure legend.
Column C in Table 1 shows the standard error of the mean (SEM) from typically several hundreds of analyzed synapses or the range of value (ROV) derived from several experiments, which were done by multiple investigators.
Animals
Wild type Wistar rats (Rattus norvegicus) for the preparation of primary hippocampal neuron cultures and synaptosomes were obtained from the University Medical Center Göttingen. All animals were handled according to the specifications of the University of Göttingen and of the local authority, the State of Lower Saxony (Landesamt für Verbraucherschutz, LAVES, Braunschweig, Germany). All animal experiments were approved by the local authority, the Lower Saxony State Office for Consumer Protection and Food Safety (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit).
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Sudhof, T. C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–47 (2004).
Haucke, V., Neher, E. & Sigrist, S. J. Protein scaffolds in the coupling of synaptic exocytosis and endocytosis. Nat. Rev. Neurosci. 12, 127–38 (2011).
Rizzoli, S. O. Synaptic vesicle recycling: steps and principles. 33, 788–822 (2014).
Haass, C. & Selkoe, D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–12 (2007).
Südhof, T. C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–11 (2008).
Kauer, J. A. & Malenka, R. C. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 8, 844–58 (2007).
Takamori, S. et al. Molecular Anatomy of a Trafficking Organelle. 831–846, https://doi.org/10.1016/j.cell.2006.10.030 (2006).
Denker, A., Kröhnert, K., Bückers, J., Neher, E. & Rizzoli, S. O. The reserve pool of synaptic vesicles acts as a buffer for proteins involved in synaptic vesicle recycling. 108, 17183–17188 (2011).
Wilhelm, B. G. et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344, 1023–8 (2014).
Jahn, R., Schiebler, W. & Greengard, P. A quantitative dot-immunobinding assay for proteins using nitrocellulose membrane filters. Proc. Natl. Acad. Sci. USA 81, 1684–7 (1984).
Schwanhäusser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Gerber, S. A., Rush, J., Stemman, O., Kirschner, M. W. & Gygi, S. P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. USA 100(12), 6940–5 (2003).
Lu, P., Vogel, C., Wang, R., Yao, X. & Marcotte, E. M. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25, 117–24 (2007).
Malmström, J. et al. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 460, 762–5 (2009).
Caprioli, R. M., Farmer, T. B. & Gile, J. Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. Anal. Chem. 69, 4751–60 (1997).
Sugiyama, Y., Kawabata, I., Sobue, K. & Okabe, S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. 2, 1–5 (2005).
Rizzoli, S. O. et al. Evidence for early endosome-like fusion of recently endocytosed synaptic vesicles. Traffic 7, 1163–76 (2006).
Wienisch, M. & Klingauf, J. Vesicular proteins exocytosed and subsequently retrieved by compensatory endocytosis are nonidentical. Nat. Neurosci. 9, 1019–27 (2006).
Truckenbrodt, S. et al. Newly produced synaptic vesicle proteins are preferentially used in synaptic transmission. EMBO J. 37(15), pii-e98044, https://doi.org/10.15252/embj.201798044 (2018).
Opazo, F. et al. Aptamers as potential tools for super-resolution microscopy. Nat. Methods 9, 938–9 (2012).
Truckenbrodt, S. et al. X10 expansion microscopy enables 25‐nm resolution on conventional microscopes. EMBO Rep. e45836, https://doi.org/10.15252/embr.201845836, (2018).
Scheffer, D. I., Shen, J., Corey, D. P. & Chen, Z.-Y. Gene Expression by Mouse Inner Ear Hair Cells during Development. J. Neurosci. 35, 6366–6380 (2015).
Matern, M. S. et al. Transcriptomic Profiling of Zebrafish Hair Cells Using RiboTag. Front. cell Dev. Biol. 6, 47 (2018).
Beaudoin, G. M. J. et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 7, 1741–54 (2012).
Opazo, F. et al. Limited intermixing of synaptic vesicle components upon vesicle recycling. Traffic 11, 800–12 (2010).
Kaech, S. & Banker, G. Culturing hippocampal neurons. Nat. Protoc. 1, 2406–15 (2006).
Richter, K. N. et al. Glyoxal as an alternative fixative to formaldehyde in immunostaining and super-resolution microscopy. EMBO J. 37, 139–159 (2018).
Acknowledgements
We thank Christina Schäfer for technical assistance, as well as Sven Truckenbrodt for experimental assistance. This work was funded by grants from the Deutsche Forschungsgemeinschaft through the Collaborative Research Center 889 (to S.O. Rizzoli), the Collaborative Research Center 1190 (to S.O. Rizzoli), and by a Consolidator Grant from the European Research Council (NeuroMolAnatomy, to S.O. Rizzoli).
Author information
Authors and Affiliations
Contributions
S.O.R. and K.N.R. designed the experiments. K.N.R. performed the experiments with exception of the immunostainings used for Tables 1 and 2 and Figure 5, which were performed by M.S.H., S.T. and C.S., H.W. wrote the Matlab routines. J.E.U. calculated the synaptic vesicles from 3D electron microscopy reconstructions of cultured neurons produced by T.S. S.O.R. and K.N.R. wrote the initial manuscript draft. All authors commented on and refined the manuscript. S.O.R. supervised the project.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Richter, K.N., Wildhagen, H., Helm, M.S. et al. Comparative synaptosome imaging: a semi-quantitative method to obtain copy numbers for synaptic and neuronal proteins. Sci Rep 8, 14838 (2018). https://doi.org/10.1038/s41598-018-33130-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33130-6
Keywords
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
