
Abstract
The aim of this study is to explore the effect of genetic variation on diabetic retinopathy (DR) risk in a Taiwanese population. The logistic regression model was used to evaluate the relationship between DR status and risk factors, including the conventional parameters and genetic risk score (GRS). Candidate single nucleotide polymorphisms (SNPs) in GRS were selected based on previous reports with a combined P < 10−4 (genome-wide association) and P < 0.05 (meta-analysis). In total, 58 SNPs in 44 susceptibility loci were selected, and four were used to calculate GRS. After adjustment for age, systolic blood pressure, diabetes duration, and HbA1c, the DR risk was 4.95 times higher for patients in the top GRS third tile than for those in the bottom third tile (95% CI = 2.99–8.18; P < 0.001). The addition of genetic information improved DR prediction, increasing the area under the curve (AUC) from 0.72 to 0.77 (P = 0.0024) and improving the sensitivity of the model such that 40 more subjects were reclassified into DR status. The developed multivariate logistic regression model combining conventional risk factors and the multilocus GRS can predict DR, thus enabling timely treatment to reduce blindness in T2D patients.
Similar content being viewed by others
Introduction
Diabetic retinopathy (DR) is a common microvascular complication of diabetes and a leading cause of blindness in adults1,2. The prevalence of DR among type 2 diabetes (T2D) patients is about 22%, and up to 80% of patients suffering from diabetes for over 10 years develop DR. The condition progresses from non-proliferative DR (NPDR) to proliferative DR (PDR) and early detection of NPDR may lead to a 60% reduction in PDR and 83% reduction in blindness3. However, many diabetic patients with mild NPDR are not aware of the condition because it does not usually affect their vision, and, since NPDR does not require treatment, it is difficult to prevent its progression to PDR. As DR is a consequence of diabetes, patients should take general precautions to improve their blood sugar control in order to stop or slow the disease progression, but there are no reliable biomarkers for predicting NPDR and/or its development into PDR. Therefore, it is important to identify risk factors for DR progression, which would enable implementation of timely and effective treatment to reduce blindness in T2D patients.
Previous studies showed that a number of traditional risk factors such as age, gender, diabetes duration, fasting plasma glucose, glycosylated hemoglobin (HbA1c), and systolic blood pressure (SBP) could predict the progression and severity of DR4,5,6,7. Multifocal electroretinogram (mfERG) with implicit time (IT) and Z-score, foveal thickness, and blood biomarkers, including lipid components (high- and low-density lipoprotein [HDL and LDL, respectively], cholesterol, and triglycerides [TGs]), apolipoprotein, advanced glycation end products, and cytokines could also be used to predict the DR status8,9,10,11. Furthermore, it is established that heredity plays a key role in the pathogenesis of diabetes and its complications12,13,14, and familial clustering of DR among T2D patients suggests strong contribution of genetic factors to the risk of developing the disease13.
A number of genome-wide association studies (GWASs) and candidate gene association studies have been performed to examine genetic susceptibility to DR in different ethnic populations15,16,17. However, a comprehensive composite model that can estimate the combined effect of conventional risk factors and genetic background to predict the occurrence of DR in diabetic patients is limited. Therefore, the aim of this study was to investigate the association between previously reported genetic variants and DR risk, and develop a multifactorial logistic regression model to predict DR in the Han Chinese population of Taiwan.
Results
Characteristics of the study participants
Among the 1,055 T2D patients enrolled in this replication study, 468 had DR (case) and 587 did not have DR (non-DR, control). The male to female ratios were 1.03 (238/230) for the DR group and 1.13 (311/276) for the control group, indicating no statistically significant difference in gender distribution (P = 0.492 by Pearson chi-square test). However, age, diabetes duration, age of onset, HbA1c, SBP, diastolic blood pressure (DBP) in the DR group were significantly higher compared to control (P < 0.001 for all parameters; Table 1). Additionally, DR groups had lower eGFR value and high ACR value in limited subjects (n = 564 for control and 162 for DR).
Association between individual single nucleotide polymorphisms and DR risk
Among the 58 susceptibility single nucleotide polymorphisms (SNPs), the majority (except rs4762 and rs5498) passed the Hardy-Weinberg equilibrium (HWE) test (P < 0.05). For most of the passed SNPs (except rs1801282, rs487083, rs7903146, rs10501943, rs3742872, and rs13306430), minor allele frequency (MAF) exceeded 5% and were similar to those in Han Chinese from Beijing (NCBI GRCh37.p13 assembly) according to the dbSNP website (based on 1000 Genomes project; https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) (Table S1). None of the 58 susceptibility SNPs showed statistical significance in the additive model after Bonferroni correction.
Then, we genotyped/imputed all SNPs located in 44 susceptibility genes; among them, 93 had P < 0.05 in the additive model (Table S2). After applying all selection criteria, including P ≥ 0.05 in the HWE test, MAF > 0.05, imputation info ≥0.4, and P < 5.38 × 10−4 after Bonferroni correction, four SNPs, rs4748644, rs11101385, rs61893374, and rs142644390, were proved significant in the additive models (P = 3.81E-04, 1.11E-05, 2.73E-06, and 1.63E-05, respectively). The identified SNPs are located in the PLXDC2 (Plexin domain-containing 2), ARHGAP22 (Rho GTPase-activating Protein 22), CNTN5 (Contactin 5), and FMN1 (Formin 1) genes, respectively (Table 2).
Cumulative effect of the four genetic loci on the DR risk
To build the model, we first calculated the multiplex genetic risk score (GRS) for each individual. The cumulative effect of the four significant SNPs was assessed by counting the number of risk genotypes in each individual, and the weighted GRS was calculated based on the logarithm odd ratio (OR) of the susceptibility SNPs. The mean number of risk alleles was 3.24 ± 1.07 (range 0–6), and the mean weighted GRS was 3.22 ± 0.99 (range 0–6.32) in the derivation sample. The distribution of risk alleles and weighted GRS is shown in Figure S1. All patients were divided into three groups based on the number of risk alleles. The data indicated that the DR risk increased with the number of risk genotypes (P = 5.98 × 10−12; Cochran-Armitage Trend test). Compared with individuals in the lowest range of weighted GRS, the ORs with 95% confidence intervals (CIs) for those in the middle and high range were 2.14 (1.22–3.74) and 4.95 (2.99–8.18), respectively (Table 3) in the derivation sample. These results suggest a cumulative effect of the four SNPs on the DR risk.
Compared models with and without genetic markers in the derivation sample
The best model retained four “conventional” variables (age, HbA1c, diabetes duration, and SBP) and the weighted GRS (computed based on the four SNPs) as significant independent variables (Table 3). The total risk score of the DR risk was calculated for each subject based on the regression coefficients of all variables (Table S3). The sensitivity was 74.9% (167/223) and specificity was 66.2% (178/269) when the optimal cutoff identified for risk score was ≥2.28 as screen positive and <2.28 as screen negative.
The probability of DR for each patient was calculated using the equation:
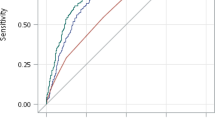
where α + β′X = −2.501 + 0.003 × Age + 0.497 × 1(HbA1c = 2) + 1.329 × 1(duration = 2) + 0.783 × 1(SBP = 2) + 0.759 × 1(weighted GRS = 2) + 1.599 × 1(weighted GRS = 3). The area under the curve (AUC) value was 0.77 (95% CI = 0.729–0.811), indicating that the model had a reasonably good discrimination ability. To determine the impact of the genetic factors on the model, a conventional model that included only age, diabetes duration, HbA1c, and SBP was built and the AUC value became 0.721 (95% CI = 0.677, 0.766). Comparison of the best and conventional models revealed a significant difference between the two models, indicating that the incorporation of the genetic data improved DR prediction compared to the conventional model (P = 2.4 × 10−3) (Fig. 1A). The total correct classification rate was 65.2% in the conventional model and 69.5% in the best model containing both conventional and genetic variables. The sensitivity of the model was increased such that 40 more DR subjects were reclassified into DR status after the inclusion of the genetic variables into the model (sensitivity = 47.1% vs. 65.0% in conventional and best models, respectively). However, the specificity of the model was decreased (19 non-DR subjects were miss-classified into DR groups) (specificity = 80.3% vs. 73.2% in conventional and best model, respectively).

Receiver Operating Characteristic (ROC) curve and area under the curve (AUC) in the Taiwanese population. ROC curves and AUC of the models built for derivation sample (A) and test sample (B) (AUC = 0.770 and 0.744, respectively). The diagonal line indicates zero predictive value of the model.
Assessment of the discriminatory ability of models in different datasets
To assess the discriminatory ability of the models, the model obtained from the derivation sample was applied to the test sample and validation sample and the AUC was determined for all datasets. The model showed a similar discrimination ability in the test sample (AUC = 0.744 [95% C.I. = 0.700, 0.787] (Fig. 1B)) compared to in the derivation dataset (AUC = 0.77 (95% CI = 0.729–0.811); P = 0.3926). However, the AUC value decreased when the best model was applied to the validation sample (AUC = 0.662 [95% CI = 0.601, 0.722]), and the difference between the two AUC values was significant (P = 0.0039) (Figure S2).
We also calculated positive and negative predictive values using the same risk score cutoff of 2.28 derived from derivation samples for test and validation samples. For the test set, the positive predictive value (PPV) was 72.0% (118 individuals with a risk score ≥2.28, of whom 85 were DR subjects) and the negative predictive value (NPV) was 62.7% (367 individuals with risk score <2.28, of whom 230 were non DR subjects). The corresponding values for the validation sample were PPV of 31.3% (32 individuals with a risk score ≥2.28, of whom 10 were DR subjects) and a NPV of 83.4% (494 individuals with risk score <2.28, of whom 412 were non DR subjects).
Discussion
In the present study, we investigated the DR risk among the Taiwanese population according to genetic variants identified by GWASs and meta-analysis, and built a prediction model. In the 44 replicated genes, four SNPs were identified to be associated with the DR risk in our population. Furthermore, a GRS based on the number of risk alleles from these four SNPs was calculated for each individual, and an independently cumulative genetic effect on the DR risk was observed in the multivariate models after adjusting for diabetes duration, HbA1c, and SBP. The addition of the genetic information to the conventional variables improved the discrimination ability for DR, increasing the AUC from 0.721 to 0.770. Furthermore, the sensitivity of model was increased after addition of GRS, which indicated that the model with genetic markers may be a clinical useful improvement, thus enabling timely treatment to reduce blindness in T2D patients.
Previous studies have identified genetic variants significantly associated with DR in various ethnic groups17,18,19,20,21. Here, we confirmed the association of PLXDC2, ARHGAP22, CNTN5, and FMN1 polymorphisms with DR in the Taiwanese population. The four identified SNPs were not linked to previously reported SNPs in the same loci and differed from them in allele frequencies. PLXDC2 and ARHGAP22 were earlier identified as DR risk genes by GWAS19 and meta-analysis20. SNPs rs1571942 and rs12219125 in the PLXDC2 locus were first reported as risk factors for T2D patients with retinopathy19 and showed nominally significant (P < 0.05) association with severe DR (≥severe NPDR or history of panretinal photocoagulation) for T1D patients. In the present study, rs4748644 was not linked to rs1571942 and rs12219125 (r2 = 0.045 for both; Table S4) and showed a different MAF (48.50% vs. 12.1% and 12.1%, respectively). SNPs rs483860517,19, rs1110135519, and rs1110135719 in the ARHGAP22 gene were identified as associated with DR in Han Chinese and Caucasian patients with both T1D and T2D. Rho GTPase-activating protein encoded by the ARHGAP22 gene is known to be involved in insulin response mechanisms regulating endothelial cell migration and cancer metastasis22. In the present study, rs11101385 was highly linked to the reported ARHGAP22 SNPs (rs4838605, rs11101355, and rs11101357; r2 = 0.712, 0.8, and 0.8, respectively; Table S4), with MAF ranging from 10.24% to 16.16% in the Taiwanese population. A previous GWAS identified CNTN5 and FMN1 as DR susceptibility genes among Mexican Americans: rs10501943 (P = 2.53 × 10−4) mapped to CNTN5 intron regions on chromosome 11q22 and rs10519765 (P = 6.21 × 10−5) mapped to FMN1 intron regions on chromosome 15q13 were found associated with severe DR18. In the present study, we identified other SNPs, rs61893374 in the CNTN5 gene and rs142644390 in the FMN1 gene as associated with DR in the Taiwanese population. These SNPs showed low linkage with those reported for Mexican Americans (r2 = 0.007 for rs61893374 vs. rs10501943 in CNTN5 and r2 = 0.003 for rs142644390 vs. rs10519765 in FMN1; Table S4) and had different MAFs (11.7% for rs61893374 vs. 4.9% for rs10501943 in CNTN5, and 11.2% for rs142644390 vs. 8.3% for rs10519765 in FMN1)18. These results indicate that the selected genes with critical biological functions play an important role in DR development in the Taiwanese population.
Our study has both strengths and limitations. In previous GWASs on DR, none of the regions reached genome-wide statistical significance18,19,23. The limitations of those studies include modest sample size by GWAS standards, heterogeneous phenotypes (PDR, NPDR, and diabetic macular edema), and poor DR standardization. Our study has several strengths. First, all participants, who were unrelated Han Chinese from Taiwan, underwent exhaustive ophthalmological examination following detailed protocols for both non-DR and DR patients, which limited chances of misclassification. Second, the genetic markers we selected were based on previous studies and all the genetic markers have been reported to be relevant to mechanisms of T2D or DR development. Therefore, these genetic markers represented robust and replicated variants for DR. Third, the difference in allelic frequencies for the four SNPs between the two groups ranged from 7.6% to 20.7% (sample size, 587 vs. 468 for T2D controls and DR cases, respectively) in the current study, thereby raising the statistical power for selecting the appropriate genetic markers of DR risk to over 96%.
We also recognized several limitations in the present study. First, not all of the suggested risk factors such as mfERG, foveal thickness, and certain blood biomarkers (cytokines) could be evaluated in the present study because the relevant information was not collected at the beginning of the study. Second, the cross sectional study design was used in current study. It is necessary to conduct a long-term follow-up evaluation of non-DR T2D patients who carry the risk genotypes to determine DR susceptibility depending on the identified SNPs and prediction model. Third, the AUC value of the model and PPV in the validation sample was decreased compared to that in the derivation sample. Additionally, inconsistency of the association for the genetic variants chosen for GRS was observed in the validation sample. This could be due to several reasons, including the small sample size, the self-reported DR status, and the fact that more participants had diabetic nephropathy (eGFR <90 mL/min/1.73 m2) in the validation sample.
In conclusion, we analyzed the association between a panel of genetic variants and the DR risk and developed a multivariate logistic regression model to predict DR in the Taiwanese population. Confirmatory studies in a cohort of a larger size should be performed in the near future to validate our model.
Methods
Study participants for derivation population
The study involved 1,055 T2D patients 20 years and older, who were recruited from the China Medical University Hospital (CMUH), Taichung, Taiwan. Diabetes was diagnosed based on medical records and fasting plasma glucose levels according to the American Diabetes Association Criteria24. Patients with type 1 diabetes (T1D), gestational diabetes, and maturity onset diabetes of the young (MODY) were excluded from this study. The participants were of Han Chinese ethnicity characteristic for 98% of the population in Taiwan. All T2D patients underwent complete ophthalmologic testing, including corrected visual acuity, fundoscopic examination, and fundus photography. An expert ophthalmologist graded DR according to the International Clinical DR Disease Severity Scale proposed by the American Academy of Ophthalmology25. The whole groups of subjects were randomly assigned to a derivation set (n = 528) and a test set (n = 527) at a 1:1 ratio. The two databases were found to be compatible (Table S5).
Study participants for validation
Another group of 542 T2D patients from three different hospitals, including CMUH, ChangHua Christian Hospital (CCH), and Taiwan Biobank, were selected for validation. The DR status was self-reported by the subjects. The characteristics of 95 DR and 447 non-DR patients are presented in Table S6.
The study was approved by the CMUH and CCH Institutional Review Board and informed consent was obtained from all participants. We confirm that all experiments of the study were performed in accordance with relevant guidelines and regulations.
Power calculation
With regard to the sample size, we estimated that for a type 1 error of 5% and a power of 80%, a total of 511 participants would be needed to detect an OR of 2.0 for a dependent and an independent variable both with a prevalence of 0.2. Thus, the sample size was sufficient to test small effect sizes given that the prevalence of the corresponding DR was not too low. G*Power Version 3.1.726,27 uses the algorithm described by Hsieh et al28. Table S7 was presented the sample size calculation based on different conditions.
Data collection
Data regarding age, gender, age at T2D diagnosis, and ocular history were collected from questionnaires. For each patient, SBP and DBP, waist and hip circumferences, and body mass index were determined, and blood samples were collected by venipuncture for genomic DNA isolation and serological tests, including fasting glucose and HbA1c, at the time of enrollment in the study.
Genetic marker selection and genotyping/imputation
All of the genetic markers were selected based on previous reports. The inclusion criteria of candidate SNPs were set as a combined P < 10−4 for GWAS18,19,23,29 and a P < 0.05 for meta-analysis20,21,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50. A total of 58 SNPs in 44 susceptibility loci were evaluated in our study (details are summarized in Table S1). Furthermore, we performed genotyping or imputation for all the SNPs in these 44 susceptibility loci using Illumina HumanHap550-Duo BeadChips or Affymetrix-TWB chips. Genotypic data were quality-controlled, and SNPs were excluded from further analysis if: (1) MAF was less than 5% in non-DR T2D controls, (2) the total call rate was less than 95% for both DR and control patients, or (3) an SNP significantly departed from HWE proportions for controls (P < 0.05). For the untyped SNPs, genotype imputation was performed according to the methodology of Howie et al.51 implemented in impute v2 (http://mathgen.stats.ox.ac.uk/impute/impute_v2.html). The panel from 1,000 Genomes Project was used as reference for imputation, and the software chose the best customized reference set for each individual. SNPs with low imputation quality (info < 0.4) and those in the same gene showing strong disequilibrium with each other (D′ >0.8) were excluded from further analysis. Each SNP was tested for association with DR in an additive model by multivariable logistic regression analysis after adjustment for T2D duration and HbA1c level. The genotypes were coded in the additive model as “0” for non-risk allele homozygotes, “1” for heterozygotes, and “2” for risk allele homozygotes. A total of 93 SNPs in 24 genetic loci were identified (Table S2); among them, four showed statistical significance after Bonferroni correction (cut-off P value for Bonferroni correction = 0.05/93 = 5.38 × 10−4). The flow chart for genetic marker selection is presented in Figure S3.
Statistical Analysis
Analyses were performed using the logistic regression model to evaluate the relationship between DR status and the following of two groups of factors: conventional parameters and genetic markers. These factors were selected based on literature review and information collected from databases. Conventional parameters included age, sex, DM duration, SBP, DBP, and serological markers (such as fasting plasma glucose, HbA1c, and lipid markers (HDL, LDL, cholesterol, and TGs). For the genetic markers, the four significant SNPs that passed the selection process described above were used to calculate the weighted GRS. The GRS for each individual was calculated based on the number of risk alleles weighted by the effect size (logarithm of ORs) according to the following equation52: weighted GRS = 4/3.462 × [(ARHGAP22_rs11101385_Risk × 0.932) + (FMN1_rs142644390_Risk × 1.077) + (PLXDC2_rs4748644_Risk × 0.451) + (CNTN5_rs61893374_Risk × 1.002)]. The weighted GRSs were divided into three equal groups to calculate the cumulative effect. The weighted GRS in the prediction model was calculated from the derivation sample and applied to test/validation samples.
The ORs with 95% CIs of predictor variables were estimated using logistic regression models to develop a best model of DR risk in the derivation sample and to assess the model’s discrimination ability in the test sample and validation sample. We used three steps to select independent variables that result in a “best” model53,54. First, we conducted a univariable analysis of each variable. Second, we selected variables P < 0.05 as a candidate in the multivariable model. Third, we constructed a multivariable model with candidate variables by the backward selection method. To determine the impact of genetic factors, two models were compared, including the model with “conventional” variables (age, diabetes duration, HbA1c, and SBP) and the model with conventional and GRS. The difference between AUC values from models was evaluated by Z statistics55.
Furthermore, the total risk score of the DR risk was calculated for each subject based on the regression coefficients of all conventional parameters and GRS in the best model56,57,58. Receiver operating characteristic (ROC) curves were generated to quantify the predictive accuracy of the models, and the AUC was used to assess the discriminatory ability of the models. To assess discrimination, the model obtained from the derivation sample was applied to the test sample and validation sample and the AUC was determined for all samples. All statistical analyses were performed using SPSS software, v. 12.0 for Windows (IBM, Armonk, NY, USA).
References
Caldwell, R. B. et al. Vascular endothelial growth factor and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev 19, 442–455, https://doi.org/10.1002/dmrr.415 (2003).
Taylor, H. R. & Keeffe, J. E. World blindness: a 21st century perspective. Br J Ophthalmol 85, 261–266 (2001).
Liu, W. J. et al. Assessing progression and efficacy of treatment for diabetic retinopathy following the proliferative pathway to blindness: implication for diabetic retinopathy screening in Taiwan. Diabetic medicine: a journal of the British Diabetic Association 20, 727–733 (2003).
Karoli, R., Fatima, J., Shukla, V., Garg, P. & Ali, A. Predictors of diabetic retinopathy in patients with type 2 diabetes who have normoalbuminuria. Annals of medical and health sciences research 3, 536–540, https://doi.org/10.4103/2141-9248.122087 (2013).
Maghbooli, Z., Pasalar, P., Keshtkar, A., Farzadfar, F. & Larijani, B. Predictive factors of diabetic complications: a possible link between family history of diabetes and diabetic retinopathy. Journal of diabetes and metabolic disorders 13, 55, https://doi.org/10.1186/2251-6581-13-55 (2014).
Cheng, Y. J. et al. Association of A1C and fasting plasma glucose levels with diabetic retinopathy prevalence in the U.S. population: Implications for diabetes diagnostic thresholds. Diabetes care 32, 2027–2032, https://doi.org/10.2337/dc09-0440 (2009).
Xu, J. et al. Prevalence and risk factors for diabetic retinopathy: the Beijing Communities Diabetes Study 6. Retina 32, 322–329, https://doi.org/10.1097/IAE.0b013e31821c4252 (2012).
Anitha, B., Sampathkumar, R., Balasubramanyam, M. & Rema, M. Advanced glycation index and its association with severity of diabetic retinopathy in type 2 diabetic subjects. Journal of diabetes and its complications 22, 261–266, https://doi.org/10.1016/j.jdiacomp.2007.05.005 (2008).
Saxena, R., Singh, D., Saklani, R. & Gupta, S. K. Clinical biomarkers and molecular basis for optimized treatment of diabetic retinopathy: current status and future prospects. Eye and brain 8, 1–13, https://doi.org/10.2147/EB.S69185 (2016).
Goldberg, R. B. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. The Journal of clinical endocrinology and metabolism 94, 3171–3182, https://doi.org/10.1210/jc.2008-2534 (2009).
Lim, L. S. & Wong, T. Y. Lipids and diabetic retinopathy. Expert opinion on biological therapy 12, 93–105, https://doi.org/10.1517/14712598.2012.641531 (2012).
Sobrin, L. et al. Candidate gene association study for diabetic retinopathy in persons with type 2 diabetes: the Candidate gene Association Resource (CARe). Investigative ophthalmology & visual science 52, 7593–7602, https://doi.org/10.1167/iovs.11-7510 (2011).
Rema, M., Saravanan, G., Deepa, R. & Mohan, V. Familial clustering of diabetic retinopathy in South Indian Type 2 diabetic patients. Diabetic medicine: a journal of the British Diabetic Association 19, 910–916 (2002).
Imperatore, G. et al. Sib-pair linkage analysis for susceptibility genes for microvascular complications among Pima Indians with type 2 diabetes. Pima Diabetes Genes Group. Diabetes 47, 821–830 (1998).
Hampton, B. M., Schwartz, S. G., Brantley, M. A. Jr. & Flynn, H. W. Jr. Update on genetics and diabetic retinopathy. Clinical ophthalmology 9, 2175–2193, https://doi.org/10.2147/OPTH.S94508 (2015).
Kuo, J. Z., Wong, T. Y. & Rotter, J. I. Challenges in elucidating the genetics of diabetic retinopathy. JAMA ophthalmology 132, 96–107, https://doi.org/10.1001/jamaophthalmol.2013.5024 (2014).
McAuley, A. K. et al. Replication of genetic loci implicated in diabetic retinopathy. Investigative ophthalmology & visual science 55, 1666–1671, https://doi.org/10.1167/iovs.13-13559 (2014).
Fu, Y. P. et al. Identification of Diabetic Retinopathy Genes through a Genome-Wide Association Study among Mexican-Americans from Starr County, Texas. Journal of ophthalmology 2010, https://doi.org/10.1155/2010/861291 (2010).
Huang, Y. C. et al. Genome-wide association study of diabetic retinopathy in a Taiwanese population. Ophthalmology 118, 642–648, https://doi.org/10.1016/j.ophtha.2010.07.020 (2011).
Hosseini, S. M. et al. The association of previously reported polymorphisms for microvascular complications in a meta-analysis of diabetic retinopathy. Human genetics 134, 247–257, https://doi.org/10.1007/s00439-014-1517-2 (2015).
Ding, Y., Hu, Z., Yuan, S., Xie, P. & Liu, Q. Association between transcription factor 7-like 2 rs7903146 polymorphism and diabetic retinopathy in type 2 diabetes mellitus: A meta-analysis. Diabetes & vascular disease research 12, 436–444, https://doi.org/10.1177/1479164115598274 (2015).
Larance, M. et al. Global phosphoproteomics identifies a major role for AKT and 14-3-3 in regulating EDC3. Molecular & cellular proteomics: MCP 9, 682–694, https://doi.org/10.1074/mcp.M900435-MCP200 (2010).
Sheu, W. H. et al. Genome-wide association study in a Chinese population with diabetic retinopathy. Human molecular genetics 22, 3165–3173, https://doi.org/10.1093/hmg/ddt161 (2013).
Diagnosis and classification of diabetes mellitus. Diabetes Care 36 Suppl 1, S67-74, https://doi.org/10.2337/dc13-S067 (2013).
Wilkinson, C. P. et al. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology 110, 1677–1682, https://doi.org/10.1016/S0161-6420(03)00475-5 (2003).
Faul, F., Erdfelder, E., Lang, A. G. & Buchner, A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior research methods 39, 175–191 (2007).
Faul, F., Erdfelder, E., Buchner, A. & Lang, A. G. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behavior research methods 41, 1149–1160, https://doi.org/10.3758/BRM.41.4.1149 (2009).
Hsieh, F. Y., Bloch, D. A. & Larsen, M. D. A simple method of sample size calculation for linear and logistic regression. Statistics in medicine 17, 1623–1634 (1998).
Hallman, D. M. et al. A genome-wide linkage scan for diabetic retinopathy susceptibility genes in Mexican Americans with type 2 diabetes from Starr County, Texas. Diabetes 56, 1167–1173, https://doi.org/10.2337/db06-1373 (2007).
Zhang, Z. et al. Association of chemokine ligand 5/chemokine receptor 5 gene promoter polymorphisms with diabetic microvascular complications: A meta-analysis. Journal of diabetes investigation 7, 212–218, https://doi.org/10.1111/jdi.12397 (2016).
Wang, W., He, M. & Huang, W. Association of monocyte chemoattractant protein-1 gene 2518A/G polymorphism with diabetic retinopathy in type 2 diabetes mellitus: A meta-analysis. Diabetes research and clinical practice 120, 40–46, https://doi.org/10.1016/j.diabres.2016.07.016 (2016).
Luo, S., Wang, F., Shi, C. & Wu, Z. A Meta-Analysis of Association between Methylenetetrahydrofolate Reductase Gene (MTHFR) 677C/T Polymorphism and Diabetic Retinopathy. International journal of environmental research and public health 13, https://doi.org/10.3390/ijerph13080806 (2016).
Zeng, Y. et al. Association between a vascular endothelial growth factor gene polymorphism (rs2146323) and diabetic retinopathy: a meta-analysis. BMC ophthalmology 15, 163, https://doi.org/10.1186/s12886-015-0155-3 (2015).
Fan, W. Y. & Liu, N. P. Meta-analysis of association between K469E polymorphism of the ICAM-1 gene and retinopathy in type 2 diabetes. International journal of ophthalmology 8, 603–607, https://doi.org/10.3980/j.issn.2222-3959.2015.03.30 (2015).
Meng, N., Zhang, Y., Li, H., Ma, J. & Qu, Y. Association of tumor necrosis factor alpha promoter polymorphism (TNF-alpha 238 G/A and TNF-alpha 308 G/A) with diabetic mellitus, diabetic retinopathy and diabetic nephropathy: a meta-analysis. Current eye research 39, 194–203, https://doi.org/10.3109/02713683.2013.834942 (2014).
Liu, L. et al. TGF-beta1 gene polymorphism in association with diabetic retinopathy susceptibility: a systematic review and meta-analysis. PloS one 9, e94160, https://doi.org/10.1371/journal.pone.0094160 (2014).
Han, L. et al. The associations between VEGF gene polymorphisms and diabetic retinopathy susceptibility: a meta-analysis of 11 case-control studies. Journal of diabetes research 2014, 805801, https://doi.org/10.1155/2014/805801 (2014).
Yuan, Y. et al. Meta-analysis of functional roles of DICCCOLs. Neuroinformatics 11, 47–63, https://doi.org/10.1007/s12021-012-9165-y (2013).
Qiu, M., Xiong, W., Liao, H. & Li, F. VEGF -634G > C polymorphism and diabetic retinopathy risk: a meta-analysis. Gene 518, 310–315, https://doi.org/10.1016/j.gene.2013.01.018 (2013).
Zhao, S., Li, T., Zheng, B. & Zheng, Z. Nitric oxide synthase 3 (NOS3) 4b/a, T-786C and G894T polymorphisms in association with diabetic retinopathy susceptibility: A meta-analysis. Ophthalmic genetics, https://doi.org/10.3109/13816810.2012.675398 (2012).
Yuan, D. & Liu, Q. Association of the receptor for advanced glycation end products gene polymorphisms with diabetic retinopathy in type 2 diabetes: a meta-analysis. Ophthalmologica. Journal international d’ophtalmologie. International journal of ophthalmology. Zeitschrift fur Augenheilkunde 227, 223–232, https://doi.org/10.1159/000335628 (2012).
Niu, W. & Qi, Y. An updated meta-analysis of methylenetetrahydrofolate reductase gene 677C/T polymorphism with diabetic nephropathy and diabetic retinopathy. Diabetes research and clinical practice 95, 110–118, https://doi.org/10.1016/j.diabres.2011.10.009 (2012).
Ma, J. et al. Meta-analysis of association between the Pro12Ala polymorphism of the peroxisome proliferator-activated receptor-gamma2 gene and diabetic retinopathy in Caucasians and Asians. Molecular vision 18, 2352–2360 (2012).
Tian, C., Fang, S., Du, X. & Jia, C. Association of the C47T polymorphism in SOD2 with diabetes mellitus and diabetic microvascular complications: a meta-analysis. Diabetologia 54, 803–811, https://doi.org/10.1007/s00125-010-2004-5 (2011).
Zhou, J. B. & Yang, J. K. Angiotensin-converting enzyme gene polymorphism is associated with proliferative diabetic retinopathy: a meta-analysis. Acta diabetologica 47, 187–193, https://doi.org/10.1007/s00592-009-0160-1 (2010).
Zhao, T. & Zhao, J. Association between the -634C/G polymorphisms of the vascular endothelial growth factor and retinopathy in type 2 diabetes: a meta-analysis. Diabetes research and clinical practice 90, 45–53, https://doi.org/10.1016/j.diabres.2010.05.029 (2010).
Abhary, S., Hewitt, A. W., Burdon, K. P. & Craig, J. E. A systematic meta-analysis of genetic association studies for diabetic retinopathy. Diabetes 58, 2137–2147, https://doi.org/10.2337/db09-0059 (2009).
Wiwanitkit, V. Angiotensin-converting enzyme gene polymorphism is correlated to diabetic retinopathy: a meta-analysis. Journal of diabetes and its complications 22, 144–146, https://doi.org/10.1016/j.jdiacomp.2006.09.004 (2008).
Zintzaras, E., Chatzoulis, D. Z., Karabatsas, C. H. & Stefanidis, I. The relationship between C677T methylenetetrahydrofolate reductase gene polymorphism and retinopathy in type 2 diabetes: a meta-analysis. Journal of human genetics 50, 267–275, https://doi.org/10.1007/s10038-005-0250-z (2005).
Fujisawa, T. et al. Meta-analysis of association of insertion/deletion polymorphism of angiotensin I-converting enzyme gene with diabetic nephropathy and retinopathy. Diabetologia 41, 47–53, https://doi.org/10.1007/s001250050865 (1998).
Howie, B., Marchini, J. & Stephens, M. Genotype imputation with thousands of genomes. G3(1), 457–470, https://doi.org/10.1534/g3.111.001198 (2011).
Ding, K., Bailey, K. R. & Kullo, I. J. Genotype-informed estimation of risk of coronary heart disease based on genome-wide association data linked to the electronic medical record. BMC cardiovascular disorders 11, 66, https://doi.org/10.1186/1471-2261-11-66 (2011).
Hosmer, D. W. L. S. Applied logistic regression. (Wiley-Interscience Publication, 2000).
Lin, C. C. et al. Development and validation of a risk prediction model for end-stage renal disease in patients with type 2 diabetes. Scientific reports 7, 10177, https://doi.org/10.1038/s41598-017-09243-9 (2017).
DeLong, E. R., DeLong, D. M. & Clarke-Pearson, D. L. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–845 (1988).
Seddon, J. M., Reynolds, R., Yu, Y. & Rosner, B. Validation of a prediction algorithm for progression to advanced macular degeneration subtypes. JAMA ophthalmology 131, 448–455, https://doi.org/10.1001/jamaophthalmol.2013.2578 (2013).
Seddon, J. M., Reynolds, R., Yu, Y., Daly, M. J. & Rosner, B. Risk models for progression to advanced age-related macular degeneration using demographic, environmental, genetic, and ocular factors. Ophthalmology 118, 2203–2211, https://doi.org/10.1016/j.ophtha.2011.04.029 (2011).
Seddon, J. M. et al. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Investigative ophthalmology & visual science 50, 2044–2053, https://doi.org/10.1167/iovs.08-3064 (2009).
Acknowledgements
This work was supported in part by research grants from the Biosignature project, Academia Sinica, Taiwan; from Ministry of Science and Technology of Taiwan (MOST 105-2314-B-039-014); and from China Medical University (DMR-106-067 and DMR-107-056). We thank the National Center for Genome Medicine of the National Core Facility Program for Biotechnology, Ministry of Science and Technology, for the technical/bioinformatics support. We also thank Dr. Li Tsai-Chung for her valuable advices in the statistical analysis.
Author information
Authors and Affiliations
Contributions
W.L.L. and F.J.T. conceived of and designed the research. J.M.L., W.L.C., M.C.H. and Y.C.H. conducted data collection. W.L.L., C.M.W. and Y.W.C. analyzed the data. W.L.L. wrote the manuscript. All the coauthors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liao, WL., Lin, JM., Chen, WL. et al. Multilocus genetic risk score for diabetic retinopathy in the Han Chinese population of Taiwan. Sci Rep 8, 14535 (2018). https://doi.org/10.1038/s41598-018-32916-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32916-y
Keywords
This article is cited by
-
A study of the genotyping and vascular endothelial growth factor polymorphism differences in diabetic and diabetic retinopathy patients
Egyptian Journal of Medical Human Genetics (2022)
-
Prognostic models of diabetic microvascular complications: a systematic review and meta-analysis
Systematic Reviews (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
