
Abstract
It is critical to understand how variations in chemical composition in surface seawater (SSW) affect the chemistry of marine atmospheric aerosols. We investigated the sea-to-air transfer of dissolved organic carbon (DOC) via cruise measurements of both ambient aerosols and SSW in the Oyashio and its coastal regions, the western subarctic Pacific during early spring. Sea spray aerosols (SSAs) were selected based on the stable carbon isotope ratio of water-soluble organic carbon (WSOC) (δ13CWSOC) and concentrations of glucose as a molecular tracer in marine aerosols together with local surface wind speed data. For both SSA and SSW samples, excitation-emission matrices were obtained to examine the transfer of fluorescent organic material. We found that the ratios of fluorescence intensity of humic-like and protein-like substances in the submicrometer SSAs were significantly larger than those in the bulk SSW (~63%). This ratio was also larger for the supermicrometer SSAs than for the SSW. The results suggest significant decomposition of protein-like DOC on a timescale of <12–24 h and/or preferential production of humic-like substances in the atmospheric aerosols regardless of the particle size. This study provides unique insights into the complex transfer of DOC from the ocean surface to the atmosphere.
Similar content being viewed by others

Introduction
Marine atmospheric aerosols play a key role in the climate system, as they act as cloud condensation nuclei (CCN) and ice nuclei (IN) and thus can control the atmospheric radiative budget through cloud formation1,2. The ocean surface is a major source of aerosols in both number and mass concentrations3. In particular, ocean-derived submicrometer particles contain a large fraction of water-soluble organic carbon (WSOC) as well as water-insoluble organic carbon (WIOC)4,5. This organic matter (OM) can significantly alter the hygroscopic property of aerosols6,7,8. -One of the largest global sources of directly emitted (primary) aerosols is wind-driven particle production at the ocean surface4,5,9. One important question that has remained unclear is how the production of sea spray aerosols (SSAs) with transfer of organic molecules influences the chemical properties of submicrometer SSAs at the ocean surface. In recent years, much effort has been devoted to examining linkages between the chemistry of SSAs and the biological and chemical conditions of surface seawater (SSW)10.
Characterization of SSAs requires sampling and analytical methods that allow isolation of these particles, preventing them from being modified by ambient gases and particles that preexist or are being transported. To assess the impact of marine biological activity on ambient aerosols, it is important to differentiate marine-derived aerosols from terrestrially (anthropogenic and plants) derived aerosols over the oceanic regions. For this purpose, some techniques need to be established to discriminate between ocean- and land-derived aerosols found in marine atmospheres. A method using the isotopic composition of aerosol carbon has been used successfully to determine the contributions of marine and terrestrial sources to aerosols found in the remote marine atmosphere11,12,13,14,15. In particular, WSOC-specific stable carbon isotope (13C) analysis in combination with molecular markers provides robust tools for the source apportionment of WSOC in marine aerosols16.
Three-dimensional excitation emission matrix (EEM) spectroscopy, which yields fluorescence intensities as a function of both excitation and emission wavelengths, has been widely applied to characterize chromophoric dissolved organic matter in terrestrial and oceanic systems17,18. However, the method has rarely been used for the analysis of complex organic matter in atmospheric aerosols19,20,21,22,23, particularly for marine aerosols. Classifications of organic matter with EEM spectra in SSA need to be employed in comparison with that in SSW, because their sources, chemical compositions, and transformation pathways are expected to be different from those in the marine aqueous environment. In this context, direct comparison of EEMs between SSAs and SSW in the field remains rarely carried out.
Whereas a large fraction of marine organic aerosols (OAs) remains chemically unresolved, a significant fraction of WSOC has been attributed to humic-like substances (HULIS), which are operationally defined as a complex mixture of heterogeneous compounds24. Moreover, there is growing evidence that primary producers in the surface ocean may contribute to the humic-like fluorescence in seawater25. The major pathways for HULIS production in aerosols include direct emissions (e.g., sea spray, soil resuspension) and secondary formation from both biogenic and anthropogenic precursors (e.g., oxidation, oligomerization, and polymerization). Their formation mechanism and alternative components should be considered, as they may explain the large missing source of marine OA in global models26.
Because bioorganic species can affect climate through cloud formation as well as atmospheric chemistry, it is critical to understand the factors that control the release of biomolecules and microbes from the ocean surface to the atmosphere and their roles in aerosol formation. Fundamental nascent chemical properties strongly depend on the geochemical state of source waters at the sea surface. In addition, it is important to understand how the production of SSAs influences the chemical composition of submicrometer and supermicrometer SSAs.
The Oyashio is a cold subarctic ocean current that flows southwestward as a western boundary current of the Western Subarctic Gyre in the North Pacific27. In the Oyashio and its coastal region, spring phytoplankton blooming occurs, namely from March to June28,29. This oceanic region in spring has the potential for enhanced concentration of OM at the sea surface, which enables us to study the chemical and biological linkages of OM at the ocean–atmosphere boundary.
The objective of this study is to elucidate the chemical transfer of major classes of organic compounds when they move from the bulk SSW to SSAs at the ocean–atmosphere boundary. Based on the shipboard measurements of both marine ambient aerosols and bulk surface seawater in the Oyashio and its coastal region in the western North Pacific (Supplementary Material, Fig. S1), we investigated the chemical transformation of organic compositions during SSW-to-SSA transfer using fluorescence EEM spectroscopy in combination with stable carbon isotopic ratios. Direct comparison of chemical information between SSAs and SSW obtained via EEM analysis enabled us to discuss possible processes of different chemical classes of OM. Our measurements will help to advance the understanding of the underlying processes that determine the distribution and composition of marine aerosols.
Results and Discussion
Identification and isolation of the data for sea spray aerosols (SSAs)
The median number concentrations of aerosol particles at dry diameters of 14–710 nm broadly ranged from 497 to 4,947 cm−3 for respective periods of the filter samplings with the number of aerosol samples of 22. While these values are similar to those of SSA produced from low temperature (~0–10 °C) seawater using a plunging jet SSA chamber30, the result suggests that SSAs could have co-existed with other types of aerosols, on the assumption that the number concentrations of SSAs in pristine marine air are typically <~1,000 cm−3 31. Other types of aerosols include particles formed during nucleation events in the open ocean, when their number concentrations can reach ~2,000 cm−3 32.
To specifically select the data for the organic component with the characteristics of sea spray, the elemental carbon (EC) concentrations were determined as a function of the stable carbon isotope ratio of WSOC (δ13CWSOC) in the observed aerosols (Fig. 1). EC is used here as a tracer of terrestrial origin. The δ13CWSOC values ranged from −18.1 to −22.9‰ with an average of −20.3 ± 1.7‰. Data with EC concentrations >0.02 µgC m−3 showed lower (isotopically lighter) δ13CWSOC values, suggesting that these aerosols were influenced by terrestrial sources, mostly anthropogenic ones. In contrast, the data with substantially low concentrations of EC (<0.02 µgC m−3) showed the δ13CWSOC values generally larger than −22‰. These δ13CWSOC values fall within the range of isotopic compositions enriched by marine-derived organic carbon (−22 to −18‰)16,33. The same is true for δ13C of total carbon (δ13CTC), whose values were also within the range of marine-derived organic carbon when EC concentrations were below 0.02 µgC m−3. Therefore, Fig. 1 clearly demonstrates that δ13C can successfully distinguish the contribution between terrestrial and marine origins of WSOC as well as TC in this study. Among the data set indicative of marine origin, six samples showed average local wind speeds of >5 m s−1 throughout the aerosol sampling, which were designated sea spray aerosol (SSA) samples in this study. It is noted that isotope equilibrium fractionation linked to photosynthesis of phytoplankton might affect the δ13C33,34. However, this relation was not distinctly observed between the δ13CWSOC in SSA samples and surface sea temperature (SST) during aerosol sampling (data not shown). This is partly attributed to the small variation in the average SST (~0.6–1.4 °C) during SSA sampling in this study.

Mass concentrations of elemental carbon (EC) as a function of δ13C for total carbon (TC) (δ13CTC) and water-soluble organic carbon (WSOC) (δ13CWSOC) in the submicrometer particles collected during the KH-15-1 cruise. A dashed box indicates the range of the data with characteristics of marine sources.
Table 1 summarizes the major parameters measured in the SSA samples and corresponding SSW samples. The average WSOC/sodium (Na+) mass ratio in the SSAs was 1.83 ± 1.30, which is close to the upper end of the OC/Na+ ratio range (0.1–2.0) previously reported for submicrometer marine primary OA35. If water-insoluble organic carbon (WIOC) is taken into account, the OC/Na+ ratio becomes larger than the typical range, probably because the ratio in the coastal region as observed in this study is generally larger than that in the open ocean36. The ratio obtained in the current study is consistent with our understanding that the submicrometer SSAs are enriched in OC relative to seawater4,37. The concentration of chlorophyll (Chl) a is used as a measure of phytoplankton biomass29 in the surface ocean. In general, the observed dissolved organic carbon (DOC) concentrations were larger with an increase in Chl a levels at the sea surface (Table 1).
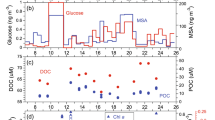
We also examined organic molecular markers to investigate the contribution of DOC to the submicrometer SSAs defined by the stable carbon isotope ratios. Previous studies in which the sea surface waters were analyzed for organics have revealed a significant carbohydrate concentration38. Figure 2a shows the relationship between WSOC and glucose in the SSA and all submicrometer aerosol samples. Here, glucose is used as a tracer for primary marine aerosols, as SSAs have been recognized to contain a substantial amount of monosaccharides, including glucose16,36. Indeed, Frossard et al.36 showed that primary marine aerosol particles generated from bubbled seawater had hydroxyl functional group compositions characteristic of monosaccharides and disaccharides, whereas organics in the seawater had those of polysaccharides, based on Fourier-transform infrared spectroscopy (FTIR) analysis. In our study, the concentrations of WSOC positively correlated with those of glucose, which was particularly evident for SSAs, supporting the nascent characteristic of the SSA samples identified here.

Concentrations of WSOC as a function of (a) glucose and (b) MSA during the cruise. The solid circles indicate the data of SSAs (i.e., the data with δ13CWSOC > −22‰, EC concentrations < 0.02 μgCm−3, and the average relative wind speeds >5 m s−1).
Figure 2b shows a scatter plot of the concentrations of WSOC versus methanesulfonic acid (MSA). An increase in the concentration of MSA did not necessarily accompany the increase in WSOC concentration, particularly for WSOC indicative of SSAs. The observed MSA is considered to be either produced by gas-phase MSA directly scavenged by aerosols or rapidly produced in aqueous phase from scavenged dimethylsulfoxide (DMSO) and methanesulfinic acid (MSIA)39, particularly under conditions with high relative humidity typical of the marine boundary layer (MBL). Furthermore, MSA is preferentially produced under low temperatures, such as the average ambient temperature of 2.5 ± 2.3 °C in the study area, due to the temperature-dependent kinetics of MSA formation40. Assuming a typical average OH radical concentration of 1 × 106 cm−3, a lifetime of DMS is roughly estimated to be <~1 day with respect to oxidation by OH radical in the MBL41. The secondary formation of MSA can explain the insignificant correlation with the SSA samples obtained in this study.
EEM spectra of submicrometer and supermicrometer aerosols vs. bulk seawater
Figure 3 presents the EEM spectra of WSOC in the submicrometer SSA samples in comparison with those of DOC in the SSW samples collected on March 9, 2015. The submicrometer SSA and SSW samples displayed a similar distinct peak in the EEM spectrum at wavelengths of excitation/emission (Ex/Em) = 300 nm/400 nm, which is typically defined as peak M42. This peak is generally considered of marine humic-like fluorophores42,43. It has been reported that HULIS and/or long-chain organics account for a significant fraction of marine aerosols20,44. In fact, the aerosol samples other than SSAs also showed distinct peaks of humic-like fluorophores. By using FTIR, Frossard et al.36 showed a larger fraction of alkane functional groups in SSAs generated from productive seawater than in SSAs generated from nonproductive seawater. Whereas our results corroborate the previous reports of the presence of organic materials with high concentrations observed in both the Pacific and Atlantic Oceans5,35,45, these data are significant because they show the consistent prevalence of HULIS and long-chain organics over a wide variety of sampling conditions in the Pacific Ocean.

Representative fluorescence excitation-emission matrices (EEMs) for (a) the submicrometer SSA samples and (b) the bulk surface seawater, obtained on March 9, 2015. Color codes indicate the fluorescence intensity in Raman Units (RU).
For the SSW samples, another obvious peak was observed at Ex/Em = 270–280 nm/320–340 nm (peak B and T), which generally corresponds to protein-like fluorophores42. These fluorophores are related to marine biological proteinaceous components, including cell or cell fragments, exopolymeric substances, and water-soluble amino acids, peptides, and protein46,47. The level of the protein-like component in the seawater was particularly high in the sea surface layers in the onshore during the same cruise, the spatial distribution of which was similar to that of the Chl a concentration48. For the SSA samples, on the other hand, the fluorescence intensity of the protein-like substances was not apparent in the EEM (Fig. 3a). These results clearly show that the submicrometer SSAs are enriched with humic-like compounds relative to the protein-like compounds, as compared to the SSW.
The biological index (BIX) has been used to estimate the contribution of autochthonous biological activity in surface water49. An increase in BIX is associated with an increase in the contribution of microbially derived organics. BIX values larger than 1 have been shown to correspond to DOM of aquatic microbial origin49, whereas lower values (e.g., <0.5) indicate contribution of soil origin50. The BIX values for the SSAs in the present study (0.74–1.15 with an average of 0.88 ± 0.18) (Table 2) correspond well to the typical ranges of microbial-origin DOM. Indeed, the BIX for the SSW ranged between 0.99 and 1.14, with an average of 1.09 ± 0.05. These results suggest that the majority of the DOM sampled was biologically derived, which significantly contributed to the SSAs observed in this study.
To investigate the dependence of the EEM spectra on the aerosol particle size, the spectra of WSOC in submicrometer and supermicrometer SSAs were compared with that of SSW collected on March 23, 2015 (Fig. 4). The EEM spectra of WSOC in the supermicrometer SSA were similar to that of the submicrometer SSA. The similarity in the EEM spectra supports the idea that the majority of protein-like compounds were not enriched in the supermicrometer size mode of SSAs, but rather preferentially remained in the SSW during the SSA production or were decomposed during the process of sea-to-air transfer. To summarize, these results suggest substantial transformation in composition of DOM during transfer from the SSW to SSAs, regardless of the aerosol particle size.

EEMs for the (a) the submicrometer SSAs, (b) supermicrometer SSAs, and (b) bulk surface seawater obtained on March 23, 2015. Note that the scales of color-coded EEM intensities in RU are different for each panel.
Implications for the sea-to-air transfer of dissolved organic matter
Figure 5 shows a scatterplot of the fluorescence intensity ratios of humic-like/protein-like compounds between the submicrometer SSAs and the SSW samples during the entire cruise. In spite of the limited number of the data, all the ratios for the SSAs were larger (by 63% in median) than those in the corresponding SSW samples. The higher ratios of the humic-like/protein-like fluorescence intensity in aerosols can be attributed to (i) the retention of protein-like DOM in the SSW, (ii) photochemical or biological depletion of protein-like compounds relative to humic-like compounds in the SSAs, and/or (iii) efficient formation of humic-like substances in the SSAs. These processes possibly occurred within the timescale of the aerosol sampling (~12–24 h).

A scatterplot of the fluorescence intensity ratios of humic-like (L)/protein-like (L) compounds between the submicrometer SSA and the SSW samples. A dashed line indicates a 1:1 line.
The retention of protein-like DOC relative to humic-like compounds in the SSW is likely the result of the protein-like DOC not being incorporated into the bubble films that produce sea spray particles. This preferential partitioning is attributable to proteins associated with exopolymer gels in SSW46, in which colloidal structures can be prevented from being incorporated into the bubble films to form SSAs51. In addition, if protein-like DOC formed a smaller amount of large particulate matter in SSW than did humic-like DOC, it would be less incorporated into SSAs relative to humic-like DOC.
Another possible explanation with regard to (ii) and (iii) is that protein-like compounds could be degraded via ultraviolet radiation and/or microbial activity at the sea surface, which reduced the abundance of protein-like DOC at the sea surface. Yamashita and Tanoue18 conducted a degradation experiment using natural phytoplankton to investigate temporal changes in marine humic- and tryptophan-like compound (one of the protein-like compounds) fluorescence intensities in artificial seawater samples. They demonstrated that marine humic-like fluorophores were rapidly produced on a timescale of a day, whereas tryptophan-like fluorophores from natural phytoplankton were rapidly degraded. That study supports that proteins can be converted to processed oligomers46 at the air–sea interface on timescale considered here, which is likely associated with microbial production of HULIS. The preferential formation of HULIS in the SSAs found in the current study can be explained by aggregation of the precursor materials of HULIS24,52, which include the degraded materials of protein-like compounds.
In this work, the observed fluorescent component in the SSAs with a spectral location at around Ex/Em = 300/400 nm was attributed to biologically derived HULIS. This indicates that selective formation and/or transition of HULIS during SSA production from DOC at the ocean surface can potentially play a significant role in determining the chemical and physical properties of aerosol particles in the MBL. Overall, this study demonstrated that the changes in chemical composition occur as freshly injected DOC into the atmosphere, which is chemically and/or biologically modified on a timescale of a half to one day. Future studies are required to conduct similar measurements of the linkage of chemical characteristics at the air–sea interface during phytoplankton blooms and post-blooms. Additional measurements are needed to elucidate the mechanism and relative contributions of photochemical and biological effects on the enrichment of HULIS rather than of protein-like substances compared to those in seawater. This study provides unique insights into the complex transfer of biologically derived organic species from the ocean surface to the atmosphere.
Methods
Ambient aerosol sample collection
Ambient submicrometer aerosol samples were collected from March 6–25, 2015, on board the R/V Hakuho Maru. The sampling was carried out during cruise KH-15–1 in the Oyashio and its coastal regions in the western North Pacific (Supplementary Material, Fig. S1)48. The aerosol samples were collected continuously using a high-volume air sampler (HVAS; Model 120SL, Kimoto Electric, Osaka, Japan) on the deck above the bridge of the ship. A cascade impactor (CI; Model TE-234, Tisch Environmental, Cleves, OH, USA) attached to the HVAS was used to collect size-segregated particles16 at a flow rate of 1130 L min−1 without temperature and humidity control. In this study, we used analytical results obtained from the bottom and upper stages of the impactor, which collected particles with aerodynamic diameter (Dp) < 0.95 μm and Dp > 0.95 μm, respectively. In the present study, aerosol particles collected at the bottom and upper stages were referred to as submicrometer and supermicrometer particles, respectively.
The aerosol sampling was conducted during local daytime and nighttime with the duration of each sampling approximately 12–24 hours. The samples were collected on quartz fiber filters (25 cm × 20 cm), which were pre-combusted at 410 °C for 6 hours to remove any contaminants. Collected filters were individually stored in glass jars with a Teflon-lined screwed cap at −20 °C to limit chemical reactions on the filter and losses of volatile compounds. Possible contamination from the ship exhaust was avoided by shutting off the sampling pump when air came from the beam and/or when the relative wind speed was low (<5m s−1). As a result of the data screening, we present 22 samples of the submicrometer particles for the data analysis out of the total 33 samples obtained in the present study.
Collection and analysis of bulk surface seawater (SSW) samples
SSW was collected using a bucket during each aerosol sampling period during the same cruise (Table S1). Average temperature of SSW at each time of the collection was 1.0 ± 0.6 °C. Further details of SSW properties during the cruise are given by Mizuno et al.48 The seawater samples were filtered with a 0.22-μm Durapore filter (Millipak, Millipore) under gentle vacuum. The filtrate was transferred into a pre-combusted glass vial with an acid-cleaned Teflon-lined cap48. The samples were then stored frozen at −20 °C in the dark until analysis.
For Chl. a measurement, duplicate seawater samples (each 0.5–1.2 L) were collected at 0 m of each sampling station using a clean plastic bucket, and filtered into 25 mm Whatman GF/F filters (nominal pore size of 0.7 µm) with gentle vacuum (<0.013 MPa). Each filter was blotted with a filter paper, frozen in liquid nitrogen, and then stored in an ultra-freezer (<−70 °C) until analysis on land. Following Suzuki et al.53, phytoplankton pigments including Chl. a were extracted with a DMF-bead-beating technique and analyzed with ultra-high performance liquid chromatography (UHPLC). In addition, DOC concentrations were measured according to Tanaka et al.54 Briefly, samples for DOC analysis were filtered with a 0.22-μm Durapore filter (Millipak, Millipore) under gentle vacuum and frozen (−20 °C) immediately after collection to store in precombusted borosilicate glass vials. DOC analysis was conducted by using a total organic carbon (TOC) analyzer (Model TOC-V, Shimadzu). Six SSW samples are presented for the data analysis out of the total 14 samples in this study.
Stable carbon isotopic characterization of water-soluble organic aerosols
For the determination of δ13CWSOC, a filter (14.14 cm2) for each sample was acidified to pH 2 with hydrochloric acid (HCl) to remove inorganic carbon prior to extraction. The decarbonated filter samples were then dried under a nitrogen stream for approximately 2 hours. WSOC was extracted from the filters in 20 mL of ultrapure water using the method described above for measuring the WSOC concentration. The extracted samples were concentrated via rotary evaporation, and 40 μL of each sample was transferred to be absorbed onto 10 mg of pre-combusted Chromosorb in a pre-cleaned tin cup. The 13CWSOC was then measured using an elemental analyzer (EA; NA 1500, Carlo Erba, Milan, Italy) interfaced with an isotope ratio mass spectrometer (IRMS; Finnigan MAT Delta Plus, Thermo Finnigan, San Jose, CA, USA). In addition, the δ13C of total carbon (δ13CTC) (i.e., without water extraction) was also measured with the EA–IRMS for the same aerosol filter samples. The 13C data were reported relative to an established reference of carbon Vienna Pee Dee Belemnite (VPDB). Further details of the analytical method used for isotopic analysis are given by Miyazaki et al.16.
Excitation fluorescence characterization
Fluorescence characteristics were obtained for both submicrometer/supermicrometer water-soluble aerosol and surface seawater samples. For the aerosol samples, a filter cut of 19.63 cm2 was extracted using 20 mL ultrapure water in an ultrasonic bath (5 min × 3). The extracts were filtered through a 0.22- µm pore syringe filter (Millex-GV, Millipore) to remove any insoluble particles with diameters larger than 0.22 µm.
Excitation-emission matrix (EEM) fluorescence was measured using a fluorometer (FluoroMax-4, Horiba)48,54. All fluorescence spectra were acquired in the S/R mode with instrumental bias correction. After this procedure, the EEM of Milli-Q water was subtracted from the sample EEMs. Finally, each EEM was calibrated to the water Raman signal, and the fluorescence is reported in Raman Units (RU; nm−1). Based on the ranges of Coble42, the fluorescence intensities of peak M (humic-like, Ex/Em = 300/400 nm) and T (protein-like, Ex/Em = 275/330 nm) were obtained from excitation/emission pairs. The biological index (BIX) is defined as the ratio of emission intensities at Em = 380 nm and 430 nm with excitation at Ex = 310 nm calculated from the EEM50.
It is noted that the EEM signal likely contains the effect of charge transfer processes55. This effect is likely insignificant in this study, because we discuss the relative intensities of the fluorescence of humic-like and protein-like compounds between SSAs and SSW samples. Furthermore, large responses of fluorescence to pH are known to be severe at extremes of pH56. However, within natural levels, typically observed in seawater as well as aerosol samples extracted with ultrapure water (almost neutral), the response to pH is considered to be insignificant57 in this study.
Chemical analysis of water-soluble aerosols
The term water-soluble aerosols in the present study is technically defined as particles sampled on the filter and extracted with ultrapure water followed by filtration through a syringe filter58. To determine the WSOC concentration of the submicrometer filter samples, another filter cut of 3.14 cm2 was extracted with 20 mL ultrapure water using an ultrasonic bath for 15 min. The extracts were filtered through the same type of 0.22-µm pore syringe filter as described above, before being injected into a TOC analyzer (Model TOC-LCHP, Shimadzu)59.
Additionally, another cut of the filter (3.14 cm2) was extracted with 10 mL of ultrapure water under ultrasonication to determine the concentration of major inorganic ions, including Na+. The same syringe filter type as described above was used, before the extract was injected into an ion chromatograph (Model 761 compact IC; Metrohm)59. Mass concentrations of elemental carbon (EC) were measured using a Sunset Laboratory OC-EC analyzer. A filter punch of 1.54 cm2 was used for this analysis.
Another portion of the filter (3.80 cm2) was extracted with dichloromethane/methanol to measure glucose as a biogenic molecular tracer. The –OH functional group in the extracts was reacted with N,O-bis-(trimethylsilyl) trifluoroacetamide (BSTFA) to form trimethylsilyl (TMS) ethers. The TMS derivatives were then analyzed using a capillary gas chromatograph (GC7890, Agilent) coupled to a mass spectrometer (MSD5975C, Agilent)59. The chemical measurement data are presented in Table 1.
Aerosol number concentrations
For measurement of aerosol number concentrations, ambient aerosol was aspirated at a flow rate of 17 L min−1 on the upper deck of the ship. The inlet was placed at the front edge of the upper deck facing the bow to avoid sampling of the ship exhausts. The aerosol was drawn to the laboratory beneath the deck, passed through the PM2.5 cyclone. It was then introduced to a scanning mobility particle sizer (SMPS), which is composed of a differential mobility analyzer (Models 3080 and 3081, TSI Inc.) and a condenstion particle counter (Model 3775, TSI Inc.). The data were corrected by taking account of diffusion loss of particles in the sampling line and the SMPS60. To remove data that were influenced by the ship exhaust, data obtained when the relative wind direction was within 60° from the bow and when relative wind speed was ≥5 m s−1 were used.
References
Intergovernmental Panel on Climate Change (IPCC). Climate Change 2013 - The Physical Science Basis. (Cambridge University Press, 2014), https://doi.org/10.1017/CBO9781107415324 (2014).
McFiggans, G. et al. The effect of physical and chemical aerosol properties on warm cloud droplet activation. Atmos. Chem. Phys. 6, 2593–2649 (2006).
Spracklen, D. V., Arnold, S. R., Sciare, J., Carslaw, K. S. & Pio, C. Globally significant oceanic source of organic carbon aerosol. Geophys. Res. Lett. 35, L12811, https://doi.org/10.1029/2008GL033359 (2008).
O’Dowd, C. D. et al. Biogenically driven organic contribution to marine aerosol. Nature. 431, 676–680 (2004).
Prather, K. A. et al. Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol. Proc. Natl. Acad. Sci. USA 110, 7550–7555, https://doi.org/10.1073/pnas.1300262110 (2013).
Ovadnevaite, J. et al. Primary marine organic aerosol: A dichotomy of low hygroscopicity and high CCN activity. Geophys. Res. Lett. 38, L21806, https://doi.org/10.1029/2011GL048869 (2011).
Vaishya, A. et al. Bistable effect of organic enrichment on sea spray radiative properties. Geophys. Res. Lett. 40, 6395–6398, https://doi.org/10.1002/2013GL058452 (2013).
Ovadnevaite, J. et al. Surface tension prevails over solute effect in organic-influenced cloud droplet activation. Nature. 546, 637–641 (2017).
Quinn, P. K., Collins, D. B., Grassian, V. H., Prather, K. A. & Bates, T. S. Chemistry and related properties of freshly emitted sea spray aerosol. Chem. Rev. 115, 4383–4399, https://doi.org/10.1021/cr500713g (2015).
O’Dowd, C. D. et al. Connecting marine productivity to sea-spray via nanoscale biological processes: Phytoplankton Dance or Death Disco? Sci. Rep. 5, 14883, https://doi.org/10.1038/srep14883 (2015).
Chesselet, R., Fontugne, M., Buatmenard, P., Ezat, U. & Lambert, C. E. The origin of particulate organic carbon in the marine atmosphere as indicated by its stable carbon isotopic composition. Geophys. Res. Lett. 8, 345–348 (1981).
Cachier, H., Buat-Ménard, M. P., Fontugne, M. & Chesselet, R. Long–range transport of continentally–derived particulate carbon in the marine atmosphere: Evidence from stable carbon isotope studies. Tellus, Ser. B. 38, 161–177 (1986).
Turekian, V. C., Macko, S. A. & Keene, W. C. Concentrations, isotopic compositions, and sources of size-resolved, particulate organic carbon and oxalate in near-surface marine air at Bermuda during spring. J. Geophys. Res.-Atmos. 108, 4157, https://doi.org/10.1029/2002jd002053 (2003).
Miyazaki, Y., Kawamura, K. & Sawano, M. Size distributions of organic nitrogen and carbon in remote marine aerosols: Evidence of marine biological origin based on their isotopic ratios. Geophys. Res. Lett. 37, L06803, https://doi.org/10.1029/2010GL042483 (2010).
Ceburnis, D. et al. Quantification of the carbonaceous matter origin in submicron marine aerosol by C-13 and C-14 isotope analysis. Atmos. Chem. Phys. 11, 8593–8606, https://doi.org/10.5194/acp-11-8593-2011 (2011).
Miyazaki, Y. et al. Contribution of dissolved organic matter to submicron water-soluble organic aerosols in the marine boundary layer over the eastern equatorial Pacific. Atmos. Chem. Phys. 16, 7695–7707, https://doi.org/10.5194/acp-16-7695-2016 (2016).
Murphy, K. R., Stedmon, C. A., Graeber, D. & Bro, R. Fluorescence spectroscopy and multi-way techniques. PARAFAC. Anal. Methods. 5, 6557–6566, https://doi.org/10.1039/C3AY41160E (2013).
Yamashita, Y. & Tanoue, E. In situ production of chromophoric dissolved organic matter in coastal environments. Geophys. Res. Lett. 31, L14302, https://doi.org/10.1029/2004GL019734 (2004).
Matos, J. T. V., Freire, S. M. S. C., Duarte, R. M. B. O. & Duarte, A. C. Natural organic matter in urban aerosols: Comparison between water and alkaline soluble components using excitation-emission matrix fluorescence spectroscopy and multiway data analysis. Atmos. Environ. 102, 1–10, https://doi.org/10.1016/j.atmosenv.2014.11.042 (2015).
Chen, Q. et al. Characterization of chromophoric water-soluble organic matter in urban, forest and marine aerosols by HR-ToF-AMS analysis and excitation–emission matrix spectroscopy. Environ. Sci. Tech. 50, 10351–10360, https://doi.org/10.1021/acs.est.6b01643 (2016).
Fu, P. et al. Fluorescent water-soluble organic aerosols in the High Arctic atmosphere. Sci. Rep. 5, 9845, https://doi.org/10.1038/srep09845 (2015).
Bones, D. L. et al. Appearance of strong absorbers and fluorophores in limonene-O3 secondary organic aerosol due to NH4 +-mediated chemical aging over long time scales. J. Geophys. Res. 115, D05203/1–D05203/14 (2010).
Aiona, P. K. et al. Effect of photolysis on absorption and fluorescence spectra of light-absorbing secondary organic aerosols. ACS Earth and Space Chem. 2(3), 235–245 (2018).
Graber, E. R. & Rudich, Y. Atmospheric HULIS: How humic-like are they? A comprehensive and critical review. Atmos. Chem. Phys. 6, 729–753, https://doi.org/10.5194/acp-6-729-2006 (2006).
Zhao, Z. et al. Picocyanobacteria and deep-ocean fluorescent dissolved organic matter share similar optical properties. Nature Communications. 8, 15284 (2017).
Fowler, D. et al. Atmospheric composition change: Ecosystems–Atmosphere interactions. Atmos. Environ. 43, 5193–5267, https://doi.org/10.1016/j.atmosenv.2009.07.068 (2009).
Qiu, B. Kuroshio and Oyashio Currents. In Encyclopedia of Ocean Sciences Academic Press, 1413–1425 (2001).
Kono, T. & Sato, M. A mixing analysis of surface water in the Oyashio region: Its implications and application to variations of the spring bloom. Deep-Sea Res. II. 57, 1595–1607, https://doi.org/10.1016/j.dsr2.2010.03.004 (2010).
Suzuki, K. et al. Population dyanamics of phytoplankton, heterotrophic bacteria, and viruses during the spring bloom in the western subarctic Pacific. Deep-Sea Res. I 58, 575–589, https://doi.org/10.1016/j.dsr.2011.03.003 (2011).
Salter, M. E. et al. An empirically derived inorganic sea spray source function incorporating sea surface temperature. Atmos. Chem. Phys. 15, 11047–11066, https://doi.org/10.5194/acp-15-11047-2015 (2015).
Kiponen, I. K., Virkkula, A., Hillamo, R. & Kerminen, V.-M. Number size distributions and concentrations of marine aerosols: Observations during a cruise between the English Channel and the coast of Antarctica. J. Geophys. Res. Atmos. 107, 4753, https://doi.org/10.1029/2002JD002533 (2002).
O’Dowd, C. D., Monahan, C. & Dall’Osto, M. On the occurrence of open ocean particle production and growth events. Geophys. Res. Lett. 37, L19805, https://doi.org/10.1029/2010GL044679 (2010).
Fontugne, M. R. & Duplessy, J. C. Organic carbon isotopic fractionation be marine plankton in the temperature range −1 to 30 °C. Oceanol. Acta. 4, 85–90 (1981).
Ceburnis, D. et al. Stable isotopes measurements reveal dual carbon pools contributing to organic matter enrichment in marine aerosol. Sci. Rep. 6, 36675, https://doi.org/10.1038/srep36675 (2016).
Russell, L. M., Hawkins, L. N., Frossard, A. A., Quinn, P. K. & Bates, T. S. Carbohydrate-like composition of submicron atmospheric particles and their production from ocean bubble bursting. Proc. Natl. Acad. Sci. USA 107, 6652–6657 (2010).
Frossard, A. A. et al. Sources and composition of submicron organic mass in marine aerosol particles. J. Geophys. Res. Atmos. 119(12), 977–13,003, https://doi.org/10.1002/2014JD021913 (2014).
Keene, W. C. et al. Chemical and physical characteristics of nascent aerosols produced by bursting bubbles at a model air-sea interface. J. Geophys. Res. 112, D21202, https://doi.org/10.1029/2007JD008464 (2007).
Aluwihare, L. I., Repeta, D. J. & Chen, R. F. A major biopolymeric component to dissolved organic carbon in surface sea water. Nature 387, 166–169 (1997).
Zhu, L., Nenes, A., Wine, P. H. & Nicovich, J. M. Effects of aqueous organosulfur chemistry on particulate methanesulfonate to nonsea salt sulfate ratios in the marine atmosphere. J. Geophys. Res. 111, D05316, https://doi.org/10.1029/2009GL038465 (2006).
Bates, T. S., Calhoun, J. A. & Quinn, P. K. Variations in the methanesulfonate to sulfate molar ratio in submicrometer marine aerosol particles over the South Pacific Ocean. J. Geophys. Res. 97, 9859–9865, https://doi.org/10.1029/92JD00411 (1992).
Davis, D. et al. Dimethyl sulfide oxidation in the equatorial Pacific: Comparison of model simulations with field observations for DMS, SO2, H2SO4(g), MSA(g), and MS, and NSS. J. Geophys. Res. 104, 5765–5784 (1999).
Coble, P. G. Marine optical biogeochemistry: the chemistry of ocean color. Chem. Rev. 107, 402–418 (2007).
Coble, P. G., Green, S. A., Blough, N. V. & Gagosian, R. B. Characterization of dissolved organic matter in the Black Sea by fluorescence spectroscopy. Nature. 348, 432–435 (1990).
Deng, C., Brooks, S. D., Vidaurre, G. & Thornton, D. C. O. Using Raman microspectroscopy to determine chemical composition and mixing state of airborne marine aerosols over the Pacific ocean. Aerosol Sci. Tech. 48, 193–206 (2014).
Cavalli, F. et al. Advances in characterization of size-resolved organic matter in marine aerosol over the North Atlantic. J. Geophys. Res. 109, D24215, https://doi.org/10.1029/2004JD005137 (2004).
Elliott, S. et al. Prospects for simulating macromolecular surfactant chemistry at the ocean–atmosphere boundary. Environ. Res. Lett. 9, 064012, https://doi.org/10.1088/1748-9326/9/6/064012 (2014).
Decho, A. W. & Gutierrez, T. Microbial extracellular polymeric substances (EPSs) in ocean systems. Front. Microbiol. 8, 922, https://doi.org/10.3389/fmicb.2017.00922 (2017).
Mizuno, Y. et al Determination of the freshwater origin of Coastal Oyashio Water using humic-like fluorescence in dissolved organic matter. Journal of Oceanography, 74, 509–521 (2018).
Huguet, A. et al. Properties of fluorescent dissolved organic matter in the Gironde Estuary. Org. Geochem. 40, 706–719 (2009).
Birdwell, J. E. & Valsaraj, K. T. Characterization of dissolved organic matter in fogwater by excitation-emission matrix fluorescence spectroscopy. Atmos. Environ. 44, 3246–3253 (2010).
Burrows, S. M. et al. A physically based framework for modeling the organic fractionation of sea spray aerosol from bubble film Langmuir equilibria. Atmos. Chem. Phys. 14, 13601–13629, https://doi.org/10.5194/acp-14-13601-2014 (2014).
Laskin, A., Laskin, J. & Nizkorodov, S. A. Chemistry of atmospheric brown carbon. Chemical reviews. 115, 4335–4382, https://doi.org/10.1021/cr5006167 (2015).
Suzuki, K., Kamimura, A. & Hooker, S. B. Rapid and highly sensitive analysis of chlorophylls and carotenoids from marine phytoplankton using ultra-high performance liquid chromatography (UHPLC) with the first derivative spectrum chromatogram (FDSC) technique. Mar. Chem. 176, 96–109, https://doi.org/10.1016/j.marchem.2015.07.010 (2015).
Tanaka, K., Kuma, K., Hamasaki, K. & Yamashita, Y. Accumulation of humic-like fluorescent dissolved organic matter in the Japan Sea. Sci Rep. 4, 5292, https://doi.org/10.1038/srep05292 (2014).
Del Vecchio, R. & Blough, N. V. On the origin of the optical properties of humic substances. Environ. Sci. Tech. 38, 3885–3891, https://doi.org/10.1021/es049912h (2004).
Patel-Sorrentino, N., Mounier, S. & Benaim, J. Y. Excitation–emission fluorescence matrix to study pH influence on organic matter fluorescence in the Amazon Basin rivers. Water Res. 36, 2571–2581 (2002).
Spencer, R. G. M., Bolton, L. & Baker, A. Freeze/thaw and pH effects on freshwater dissolved organic matter fluorescence and absorbance properties from a number of UK locations. Water Res. 41, 2941–2950 (2007).
Miyazaki, Y., Sawano, M. & Kawamura, K. Low-molecular-weight hydroxyacids in marine atmospheric aerosol: Evidence of a marine microbial origin. Biogeosciences. 11, 4407–4414, https://doi.org/10.5194/bg-11-4407-2014 (2014).
Müller, A., Miyazaki, Y., Tachibana, E., Kawamura, K. & Hiura, T. Evidence of a reduction in cloud condensation nuclei activity of water-soluble aerosols caused by biogenic emissions in a cool-temperate forest. Sci. Rep. 7, 8452, https://doi.org/10.1038/s41598-017-08112-9 (2017).
Mochida, M. et al. Size-segregated measurements of cloud condensation nucleus activity and hygroscopic growth for aerosols at Cape Hedo, Japan, in spring 2008. J. Geophys. Res. 115, D21207, https://doi.org/10.1029/2009JD013216 (2010).
Acknowledgements
This research was supported by Grants-in-Aid for Scientific Research (B) (25281002) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. We thank H. Kawakami for her help with the chemical analysis of the samples. This study was also supported by the Grant for Joint Research Program of the Institute of Low Temperature Science, Hokkaido University.
Author information
Authors and Affiliations
Contributions
Y.M., Y.Y., K.S. and J.N. designed the research. Y.M., E.T. and Y.Y. performed the laboratory experiments and Y.M. wrote the manuscript. K.K., S.K., Y.Y. and K.S. collected the samples. K.K. performed the online measurements of aerosol number concentrations with significant contributions from M.M. K.S. measured chlorophyll a concentrations. Y.M. and E.T. analyzed the data with significant contributions from Y.Y. J.N. coordinated the cruise measurement.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miyazaki, Y., Yamashita, Y., Kawana, K. et al. Chemical transfer of dissolved organic matter from surface seawater to sea spray water-soluble organic aerosol in the marine atmosphere. Sci Rep 8, 14861 (2018). https://doi.org/10.1038/s41598-018-32864-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32864-7
Keywords
This article is cited by
-
Virus infection of phytoplankton increases average molar mass and reduces hygroscopicity of aerosolized organic matter
Scientific Reports (2023)
-
Factors controlling the transfer of biogenic organic species from seawater to sea spray aerosol
Scientific Reports (2022)
-
New index of organic mass enrichment in sea spray aerosols linked with senescent status in marine phytoplankton
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
