
Abstract
Insects’ exoskeleton, gut, hemocoel, and cells are colonized by various microorganisms that often play important roles in their host life. Moreover, insects are frequently infected by vertically transmitted symbionts that can manipulate their reproduction. The aims of this study were the characterization of bacterial communities of four developmental stages of the fungivorous species Hoplothrips carpathicus (Thysanoptera: Phlaeothripidae), verification of the presence of Wolbachia, in silico prediction of metabolic potentials of the microorganisms, and sequencing its mitochondrial COI barcode. Taxonomy-based analysis indicated that the bacterial community of H. carpathicus contained 21 bacterial phyla. The most abundant phyla were Proteobacteria, Actinobacteria, Bacterioidetes and Firmicutes, and the most abundant classes were Alphaproteobacteria, Actinobacteria, Gammaproteobacteria and Betaproteobacteria, with different proportions in the total share. For pupa and imago (adult) the most abundant genus was Wolbachia, which comprised 69.95% and 56.11% of total bacterial population respectively. Moreover, similarity analysis of bacterial communities showed that changes in microbiome composition are congruent with the successive stages of H. carpathicus development. PICRUSt analysis predicted that each bacterial community should be rich in genes involved in membrane transport, amino acid metabolism, carbohydrate metabolism, replication and repair processes.
Similar content being viewed by others
Introduction
Insects are by far the most diverse and abundant animal group, in numbers of species globally, in ecological habits, and in biomass1. They are chronically colonized by various microorganisms that are not overtly pathogenic and are often beneficial or even required by the insect host. These microorganisms colonize on the insect exoskeleton, in the gut and hemocoel, and within insect cells2. Bacterial communities are known to play important roles in many crucial aspects of their hosts life, e.g. nutrition, development, pathogen defense, community interactions and survival in harsh environments by metabolizing toxins3,4,5,6,7,8,9,10,11,12,13. Moreover, insects are also frequently infected by vertically transmitted symbionts that manipulate their reproduction14. The lack of vertical transmission through male hosts has led to the evolution of five commonly recognized manipulation schemes: feminization, parthenogenesis induction, early and late male killing, and cytoplasmic incompatibility15. One of the microorganisms that induce these alterations is Wolbachia. It is estimated to infect more than 65% of all insect species16,17. However, Wolbachia is just one of the known reproductive manipulators, others are Cardinium, Rickettsia, Arsenophonus and Spiroplasma18. Cardinium has been shown to induce cytoplasmic incompatibility, feminization and parthenogenesis, while Rickettsia can cause parthenogenesis and male killing. Arsenophonus and Spiroplasma can also induce male killing. All of induced phenomena lead to female biased reproduction, which can be beneficial for infected matrilines, but simultaneously facilitate drastic evolutionary trajectories of their hosts15,19,20,21. The wide range of phenomena in which bacteria are involved during insects life-cycle makes research focused on defining the microbiome profile of insects species (especially those exposed to harsh environmental conditions) of particular interest.
Analyses of bacterial communities associated with insects is facilitated by Next Generation Sequencing (NGS) of the 16S rRNA gene (e.g.22,23,24). Excluding bacteria, insects are known to represent more than half of the world’s biodiversity. A growing number of studies concern the analysis of bacterial communities associated with insects in a particular context, e.g. transfer of gut bacteria in social insects25, endosymbiosis and intracellular symbionts transmission15,26 as well as development of new strategies to prevent transmission of human pathogens27. Thrips on the whole have received relatively little attention regarding their bacterial communities. Previous studies have largely focused on the most frequently encountered bacterium (identified as a near-Erwinia species) within a single thrips species Frankliniella occidentalis28,29,30,31,32 with a few studies on other species (i.e. Aptinothrips species33, Thrips tabaci34, Scirtothrips dorsalis35, Frankliniella fusca36 and F. tritici37). However, nothing is known about the structure of bacterial communities associated with fungivorous thrips species.
Hoplothrips carpathicus occurs in central and northern Europe38,39,40,41. It belongs to the Phlaeothripidae family and the Phlaeothripinae subfamily of the insects order Thysanoptera. Fungivorous species of the Phlaeothripinae subfamily have a narrow maxillary canal and feed on mycelium or spores of fungi covering decaying wood, sucking the contents of their cells. H. carpathicus, like most of fungivorous species of this genus, lives in small cavities under the bark of dead trees or on/in fruiting bodies of different species of fungi. Their hidden lifestyle and haphazard spatial dispersion are the reasons of difficulties in observing them in their natural habitat. Females with fully developed wings (macropterous form) can be reliably collected by using IBL-2 screen traps hanging on 1.5 m high in deciduous forests41 (Supplementary Fig. S1) while the immature stages together with adults with or without wings can be collected by extracting from Fomes fomentarius (L.) Fr. (Basidiomycota) growing on different trees: beech (Fagus sylvatica L.), birch (Betula pendula Roth.), oak (Quercus robur L.) and Norway spruce (Picea abies (L.) H. Karst). The fruiting body of F. fomentarius provides many cavities, where H. carpathicus may live and find food. Recent studies have shown that biological compounds produced by F. fomentarius have many activities, such as antioxidant, anti-inflammatory, apoptotic, and anti-diabetic42,43,44,45,46. These compounds (e.g. triterpenoids) may be considered as toxins, especially for bacteria associated with fungivorous species. Bacterial communities associated with invertebrates which are the would-be colonizers need to overcome the presence of these substances through resistance or tolerance mechanisms.
In the present study we used NGS of the 16S rRNA gene to define whether the bacterial composition varies among the different developmental stages of H. carpathicus. We tested the hypothesis that a known endosymbiont might be present in the microbiome profiles. We also predicted the metabolic activity of the microorganisms associated with different developmental stages of H. carpathicus. Lastly, we sequenced the mitochondrial COI barcode of H. carpathicus to facilitate future molecular identification.
Results
COI sequence analysis
The barcode COI region of two specimens representing different developmental stages of H. carpathicus (pupa and adult) was sequenced. With an consensus length of 653 nucleotides, the sequences obtained contained no insertions, deletions or stop codons. Both sequences represented the same haplotype. Comparison with GenBank records and homology search shown high similarity of obtained H. carpathicus COI sequences with those obtained for other representatives of order Thysanoptera (≥86% similarity among the top 100 Blast hits on 100 subject sequences). All top 100 Blast hits were to Thysanoptera.
General description of 16S rRNA gene sequencing results
For each developmental stage of H. carpathicus, we obtained >65 000 good quality 16S rRNA gene sequences, ranging between 66 241 for the first stage larvae (L1) and 295 697 for the imago stage (Im). More details for sequence data for each stage of H. carpathicus as well as the number of the observed OTUs and values of Chao1 index, are shown in Table 1. At least 352 OTUs, ranging from 352–1406, were observed in different developmental stages, which indicates that the microbial population is highly complex. Moreover, rarefaction analysis of the obtained data revealed trends indicating that sampling of microbial communities varied in their degree of completion by life stage (Supplementary Fig. S2). Diversity indices for each microbiome are reported in Supplementary Table S1.
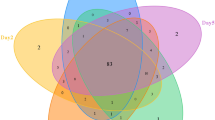
The number of shared OTUs among developmental stages of H. carpathicus is shown as a Venn diagram (Fig. 1). We were able to classify 99.80% of sequences to phylum. Detailed taxonomic analyses on different ranks are available in supplementary data as sunburst charts (Supplementary Fig. S3) and also in a table (Supplementary Table S2).

Analysis of OTUs at 97% similarity among different developmental stages of H. carpathicus. (A) Venn diagram showing overlaps of OTUs, in which 76 OTUs (core microbial community) were common for each tested stage. (B) Percentage distribution of core microbial community (76 OTU) at phylum level, identified in all developmental stages. Abbreviations: L1 – first-stage larva, L2 – second-stage larva, P – pupa, Im – adult.
Microbial community composition
For all stages, analysis of microbial communities showed that >99.78% of the total reads were affiliated with Bacteria (Supplementary Fig. S3 and Supplementary Table S2). The remaining percentage comprised Archaea and unassigned records. The four microbiomes contained 21 phyla (Fig. 2, Supplementary Fig. S3 and Supplementary Table S2).

Abundance of bacterial 16S rRNA gene sequences at the phylum level with UPGMA clustering of H. carpathicus samples at different developmental stages according to community composition and structure. Abbreviations: L1 – first-stage larva, L2 – second-stage larva, P – pupa, Im – adult.
The most abundant phyla across all stages tested were Proteobacteria, Actinobacteria, Bacterioidetes and Firmicutes with each comprising a different share of the microbiome depending on developmental stage. In each developmental stage, these four phyla comprised >80.00% of the total microbial sequences obtained. Separately, Proteobacteria comprised on average 57.49% (32.71% in L1 to 82.20% in the pupal stage – P), Actinobacteria on average 29.07% (7.77% in P to 56.94% in the second stage larvae – L2), Firmicutes on average 5.10% (0.63% in Im to 15.13% in L1), and Bacteroidetes on average 2.36% (0.20% in L1 to 4.58% in P) of the total reads (Fig. 2 and Supplementary Table S2). SIMPER analysis showed that the share of Proteobacteria, Actinobacteria and Firmicutes was primarily responsible for the difference between samples (Table 2). The bacterial structure of all-but-one developmental stage pair differed significantly (Table 2). Only in the pupa-imago pair did the microbial communities not differ significantly (χ2 = 7.20, df = 21, P > 0.05).
The most abundant classes among bacterial communities of all development stages were Alphaproteobacteria, Actinobacteria, Gammaproteobacteria, Betaproteobacteria and Bacilli accounting for >72.00% of the total reads (Supplementary Fig. S3 and Supplementary Table S2). For pupa and adult stages the most dominant class was Alphaproteobacteria, and the most dominant order among Alphaproteobacteria was Rickettsiales (69.95% and 56.11%, respectively). The most dominant family was Rickettsiaceae and among that family the most dominant genus was Wolbachia, which comprised 69.95% and 56.11% of sequences in pupa and adult respectively. In the L2 larva microbiome, the most dominant genus was Tsukamurella (phylum Actinobacteria), which accounted 45.26% of total microbial reads. Among first-stage larva Actinobacteria was the most dominant phylum, which accounted 34.34%, and order Actinomycetales consisted 28.56% of that phylum.
Similarity analysis of bacterial communities based on UPGMA clustering of samples at different developmental stages (Fig. 2) showed that changes in microbiome composition are congruent with the successive stages of H. carpathicus development. Microbiome profiles of pupa and adult are the most similar, whereas the bacterial community composition of first-stage larva was less similar to those associated with other stages tested.
PICRUSt analysis predicted functional potentials of the bacterial community associated with different developmental stages. In all of the tested stages bacterial communities seem to be rich in genes involved in membrane transport, amino acid metabolism, carbohydrate metabolism, and replication and repair processes (Fig. 3). Predicted genes involved in membrane transport are thought to be connected with ATP-binding cassette transporters (ABC transporters), the phosphoenolpyruvate (PEP)-dependent phosphotransferase system (PTS system) and bacterial secretion system (not shown).

Inferred functions of bacterial communities associated with different developmental stages of H. carpathicus. All of the predicted KEGG metabolic pathways are shown at the second hierarchical level and grouped by major functional categories.
In both larval stages, predicted genes thought to be involved in metabolism of terpenoids and polyketides, and xenobiotics biodegradation and metabolism were relatively abundant. Based on OTU abundance, predicted genes thought to be involved in xenobiotics biodegradation and metabolism would be increased in L2 larvae relative to L1. In the pupa-associated bacterial community several functional categories (i.e. carbohydrate and lipid metabolism, signal transduction, and membrane transport) are decreased compared to larval stages. However, relative abundance of predicted genes thought to be involved in energy and nucleotide metabolism, replication and repair, and translation is increased in the pupa. In the adult stage, several predicted pathways should be more abundant than in pupal stage based on differential OTU relative abundance (e.g. amino acids, carbohydrate and lipid metabolism, as well as xenobiotics biodegradation and metabolism) (Fig. 3).
Discussion
Identification of most thrips to a species level is difficult due to their small size, subtle morphological differences47,48, high intraspecific polymorphism49, sexual dimorphism50, and the presence of cryptic species and species complexes48,51,52. Molecular identification of Thysanoptera species is desirable as it overcomes these complexities. Therefore, in this study the barcode COI region was sequenced for the first time for H. carpathicus. This species can now be included in future phylogenetic analyses focused on resolving relationships among thrips species. Nevertheless, the main part of the present study was the characterization of developmental-stage-specific microbiomes associated with this fungivorous thrips species.
Microorganisms display a wide diversity of specialized interactions with their insect hosts. These associations are ubiquitous and often beneficial to the insect53. Nonetheless, most studies on insect microbiomes have focused on the gut, most frequently for termites54,55,56, ants57,58,59, fire bugs60,61,62, fruit flies63,64,65, beetles66,67,68, and bees69,70,71,72. Thrips, thus far, have received little attention regarding these important microbiome-host interactions. Previous work has focused largely on the bacterium identified as near-Erwinia species and frequently encountered in Frankliniella occidentalis, F. fusca and Thrips tabaci (e.g.34,36,73). Analyses of thrips microbiomes with NGS techniques is much more limited (but see35,37).
This study resolves the complex microbial population structure of different developmental stages of H. carpathicus. In several studies of insect’s microbiome, authors recommend surface sterilization prior to DNA extraction74,75. The individuals tested here were rinsed three times in sterile distilled water without soaking in ethanol. However, Hammer et al.76 found that surface sterilization did not change bacterial community structure as compared to unsterilized specimens, which may be due to most of the bacteria residing inside the insect body relative to its surface. In the present study, we investigated the structure and relationships of bacterial communities associated with four developmental stages of H. carpathicus, without division into endo- and ectomicrobiome.
The present study also allowed us to track changes in the microbiome profiles associated with the species development. Using the UPGMA clustering method, we found that microbial communities of pupa and adult are the most similar (~90.00%) whereas microbiome associated with first-stage larva is less similar (~40.00%). This observation is consistent with numbers of observed OTUs – 352 in L1 larvae and ~3X higher in subsequent developmental stages. This is indicative of an increasingly complex bacterial community as development progresses.
In all tested developmental stages of H. carpathicus the most dominant phyla were Proteobacteria, Actinobacteria, Bacteroidetes. and Firmicutes. This is not surprising and has been found in other insects77,78,79. Although studies focused on bacterial communities associated with Thysanoptera species are limited, recent studies confirmed that Proteobacteria and Actinobacteria are the most abundant phyla in bacterial community of other thrips species, i.e. Scirtothrips dorsalis35 and Thrips palmi79. Proteobacteria comprises ~60% of bacteria detected in S. dorsalis and similar abundance was observed in H. carpathicus (~57%). In T. palmi this phylum was slightly less abundant and comprises ~50% of detected bacteria. The average abundance of Actinobacteria was similar for S. dorsalis and H. carpathicus (~33% vs. ~29%) while it was lower in T. palmi (~20%). Firmicutes and Bacterioidetes, although less abundant than Proteobacteria and Actinobacteria, in all tested in here stages of H. carpathicus, are also listed among dominant phyla in T. palmi79. Firmicutes comprises ~15% of bacteria detected in this species. Similar abundance of this phylum was identified in L1 sample, while in the case of other samples tested this phylum was less abundant and comprised no more than 4%. Differences in abundance was also observed in the case of Bacteroidetes. This phylum comprised ~10% of bacteria detected in T. palmi, but comprised no more than 5% of bacteria in H. carpathicus. The proportion of most abundant phyla is not stable during ontogenesis. In first- and second stage larvae Actinobacteria and Proteobacteria were the most dominant, but in pupa and adult the percentage of these two phyla changed significantly. The number of Actinobacteria decreased >2-fold, while the amount of Proteobacteria increased >2-fold. Higher relative abundance of Proteobacteria in adults than in larvae has been noted other insects74,80. Moreover, in pupa and adult of H. carpathicus, the abundance of Bacteroidetes declined. Studies comparing bacterial communities associated with insect’s successive life stages remains limited (~10 published papers in the last 6 years)66,74,79,81,82,83,84,85,86,87. Therefore, extrapolating these results to other studies is difficult. However, it is possible that change in ratio of main bacterial phyla is connected with pupation, when the gut is remodeled. During the transition from the second instar to adult in the propupa and pupa, many imaginifugal characters are broken down and reformed. According to Parker et al.88, the length of the midgut decreases greatly during development and the gut epithelium becomes pycnotic and is replaced by new gut cells. In adults midgut forms a long tube divided into three different histological regions89. The larval pygidial gland, which is typical imaginifugal structure, degenerates in the pupal stage88. These changes may indirectly impact the microbial communities and thus cause changes in bacterial composition.
We identified five genera: Agrobacterium, Erwinia, Methylobacterium, Pseudomonas, Serratia, which have previously been associated with other thrips species. Pseudomonas, Serratia and Erwinia have been identified internally from F. occidentalis and T. tabaci30,32,34,35,73, while Agrobacterium, Methylobacterium and Pseudomonas have been identified from S. staphylinus90 and S. dorsalis35. Analogous to Dickey et al.35, in our study DNA was extracted from whole body of tested individuals and therefore, it is impossible to clearly distinguish internal symbionts and those associated with the surface of the insect. Based on the previous reports of Yamoah et al.90, we think that Agrobacterium, Methylobacterium and Pseudomonas genera may be associated with exoskeleton of H. carpathicus.
It is known that F. fomentarius has antimicrobial, antioxidant, anti-inflammatory, apoptotic, and anti-diabetic activities42,43,44,45,46,91. Specifically, Kolundžíć et al.92 tested antimicrobial activity of F. fomentarius extracts of different polarity using nine different laboratory strains of gram negative and positive bacteria, respectively (Staphylococcus aureus, Staphylococcus epidemidis, Micrococcus luteus, Bacillus subtilis, Enterococcus feacalis, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa and Salmonella abony). They reported higher antimicrobial activity from methanol and aqueous extracts than cyclohexane and dichloromethane extracts, but this activity was lower compared to semi-synthetic antibiotics (ampicillin and amikacin, respectively). In total, they reported significant antimicrobial activity of F. fomentarius extracts against nine bacterial strains. Their results and other studies suggest that the antimicrobial activity results from elevated polyphenols and β-glucan. Hence, based on these results and other studies93,94,95,96 polyphenols and β-glucan could be connected with significant antimicrobial activity. As we mentioned in Introduction, such compounds may be considered as toxins and bacteria associated with invertebrates which are the would-be colonizers of F. fomentarius fruiting bodies need to overcome the presence of these substances through resistance or tolerance mechanisms. While we did not distinguish internal and external bacteria on H. carpathicus, any in latter category should exhibit properties that make possible to survive under such conditions.
The present study concerns the activities of the most dominant microbial groups, as well as identification of predicted metabolic functions of the bacterial community associated with H. carpathicus. PICRUSt analysis showed, that in microbial communities associated with all of the tested stages genes involved in membrane transport, amino acid metabolism, carbohydrate metabolism, and replication and repair processes should be elevated (Fig. 3). Higher relative abundance of predicted genes involved in membrane transport (especially those connected with ABC transporters) might be related to antibiotic resistance, because ATP-binding cassette (ABC)-type multidrug transporters use a free energy of ATP hydrolysis to pump drugs out of the cell97. In second-stage larvae, which intensively feed on polypore tissue, the microbiome was more enriched for predicted genes thought to be involved in metabolism of terpenoids and polyketides, and xenobiotics biodegradation and metabolism. Analysis of bacterial community associated with pupae showed that several functional categories are predicted to be reduced, such as those thought to be involved in carbohydrate and lipid metabolism, signal transduction, and membrane transport. Genes thought to be involved in energy and nucleotide metabolism, replication and repair, as well as translation pathways should be elevated in the pupae – the stages which do not feed and move, suggesting a mechanism by which bacteria extract energy for surviving inside pupal case. At the adult stage, a higher abundance of several pathways was predicted, such as amino acid, carbohydrate and lipid metabolism. Nevertheless, the in silico predicted functions need to be validated in vitro in future studies.
Genes thought to be involved in metabolism of terpenoids and polyketides, as well as xenobiotics and biodegradation metabolism are predicted by our results to be elevated in adults. This suggests a mechanism by which bacteria associated with adult H. carpathicus could tolerate triterpenoids, the major class of secondary metabolites produced by F. fomentarius’ fruting body (~75.00%)98. Our OTU-to-gene-to-pathway prediction scheme identified also polysaccharide metabolism as important. It would be interesting to test whether specific members of microbiome are indeed metabolizing some of α- and β-glucans produced by F. fomentarius98,99. Among α-glucans presence in fungi are cellulose and chitin98. H. carpathicus feeding on F. fomentarius needs a way to process these α-glucans and to access to intercellular fungal nutrients. The present study predicts an association between Tsukamurella and cellulose degradation. This genus was identified as dominant in intensively feeding second-stage larvae and suggests a mechanism by which L2 larvae might process cellulose. Microbiome studies of other wood and fungus associated insects100,101 provide anecdotal confirmation of this suggestion. Other bacterial genera: Staphylococcus, Bacillus, Sphingomonas, Brevibacterium and Chryseobacterium, which we identified in this study, have also been linked to cellulose degradation102,103,104. Interestingly, the genus Methylobacterium was found in all developmental stages of H. carpathicus. Some species of Methylobacterium are facultative methylotrophic bacteria, and have been reported mostly from soil90, but also in plants105. They can use a variety of organic substrates with carbon-carbon bonds as sources of carbon and energy106. The role of Methylobacterium in H. carpathicus might be similar to that suggested by studies of leafhopper and weevil, which also feed on a complex carbon source90,105. Other predicted functions of the H. carpathicus bacterial community were determined according to comprehensive analysis of the functional microbiome of arthropods107 where Agrobacterium, Methylobacterium and Serratia were joined together within single group of microorganisms containing nitrogen fixing and anaerobic metabolic traits.
Beside general insight into the bacterial community associated with H. carpathicus, this study identified the presence of known endosymbionts. In previous studies of Thysanoptera species it has been argued that the bacteria are facultative and acquired from the environment through feeding28,34,73. Nevertheless, detailed analyses focused on bacterial endosymbionts (especially Cardinium and Wolbachia) showed that vertically transmitted symbionts are present among thrips species108,109,110,111. Our identification of Wolbachia, often described as a ‘master manipulator’112, is particularly interesting. This bacterium can eliminate males, turn them into females, sterilize uninfected females or behave as a mutualistic symbiont113. Wolbachia has been detected both in arrhenotokous and thelytokous thrips species108,109,111 although the frequency of this reproductive manipulators in Thysanoptera is unknown. The absence of Wolbachia, identified by conventional PCR, has been shown for three thrips: Frankliniella occidentalis108, Thrips tabaci108,114 and Heliothrips haemorrhoidalis115. Nevertheless, recent studies have demonstrated that conventional PCR can fail to detect low-level infections116. In the present study, Wolbachia was shown to be the most dominant genus in pupa and adult stages of H. carpathicus (69.95% and 56.11%, respectively), while in first and second larval instars it occurred at a lower frequency (1.25% in L1 and 10.67% in L2, respectively). The reason of observed disproportion is unknown. It could be that Wolbachia infection increased with development or is biased based on sex. In our study both males and females were seen but only adult females were used in microbiome characterization and the sex of non-adults characterized is impossible to determine. We also could not calculate the sex ratio because the specimens of all developmental stages were selected successively and the breeding was finished when only adults, mainly macropterous females, left the fruiting body of F. fomentarius. The presence of both sexes indicates arrhenotokous reproduction, where females develop from fertilized eggs and males from unfertilized ones. In this study, the presence of Wolbachia in a fungivorous thrips species H. carpathicus has been identified for the first time. Nevertheless, its impact on the host reproduction system remains unclear and follow-up analyses are planned.
In conclusion, this paper presents data of bacterial community analysis of H. carpathicus with the use of NGS 16S rRNA sequence data, which allowed us to nearly fully characterize its microbiome. Moreover, it is the first report of Wolbachia endosymbiosis in this thrips species. Additionally, the sequenced COI fragment of H. carpathicus may be useful to identify all developmental stages of this species using barcoding approach and can be applied in further phylogenetic studies. Finally, the results obtained raise new and important questions regarding the role of Wolbachia and Tsukamurella in H. carpathicus biology.
Methods
Sample collection
Fruiting bodies of F. fomentarius were collected in Roztocze National Park, Białowieża NP and Polesie NP in summer and autumn 2016. A single fruiting body of F. fomentarius, which was colonized by H. carpathicus, was collected on 19th of November 2016 in Białowieża Primeval Forest (north-eastern Poland, geographical coordinates: 52°43′36.7″N and 23°47′22.7″E). Branches of dead oak were the host for this fungus. Singular specimens of H. carpathicus were extracted from the fruiting body using Tullgren funnel. It was subsequently placed into a plastic box with a damp paper towel at the bottom and stored in 24/10 °C, 16/8 h (L/D) conditions. The breeding was conducted in Department of Zoology Maria Curie-Sklodowska University in Lublin until January 23, 2017 when all adults left the fungus. During the breeding period, four developmental stages of H. carpathicus were successively selected: intensively feeding first and second larval instars (L1 and L2, respectively) and adults (Im), and pupae (P) that do not feed. Among adults, females had fully functional wings (macropterous form) and were more numerous, while males were wingless (apterous form) and less numerous. Therefore, for further analyses only adult females were used. Every stage was separately placed into tubes and stored in −30 °C. Afterwards the tubes with insects were sent for further analyses to the Department of Genetics and Biosystematics, University of Gdansk. Some specimens of all developmental stages (first and second larval instars, propupae, pupae, females with fully functional wings and wingless males) were mounted in Canada Balm. The examined materials are deposited in the collection of Department of Zoology, Maria Curie-Sklodowska University in Lublin.
DNA extraction
Due to the small sizes of individuals (<2 mm), DNA was extracted from the whole insects at different developmental stages by the Sherlock AX Purification Kit (A&A Biotechnology). Insects were rinsed three times in sterile distilled water prior to DNA extraction without soaking in ethanol. To avoid cross contamination of the samples, the process was performed with sterile equipment. The quantity and quality of the extracted DNA were evaluated by using a Nano Drop ND-1000 spectrophotometer (NanoDrop Technologies). After extraction, the DNA was stored at −20 °C until further use. Twelve samples consisting of genetic material isolated from first and second larval instars, pupae and adults (one individual per DNA isolate and three isolates per developmental stage) were used for microbiome analyses.
COI mitochondrial marker amplification and sequencing
The 5′ COI gene fragment was amplified for two individuals of H. carpathicus (pupa and adult), using universal primer set: HCO-2198 – GGTCAACAAATCATAAAGATATTGG and LCO-1490 – TAAACTTCAGGGTGACCAAAAAATCA117. PCR reactions were performed in 20 µL volume containing 0.8x JumpStart Taq ReadyMix (1 U JumpStart Taq DNA polymerase, 4 mM Tris-HCl, 20 mM KCl, 0.6 mM MgCl2, 0.08 mM dNTP; Sigma-Aldrich, Germany), 0.4 µM of forward and reverse primers and ~100 ng of DNA. The COI gene fragment was amplified under conditions as follows: initial denaturation at 94 °C for 5 min followed by 35 cycles of 94 °C for 1 min, 51.2 °C for 1 min and 72 °C for 1 min and ending with 72 °C for 5 min. All PCR products were purified by alkaline phosphatase and exonuclease I (Thermo Scientific, USA) treatment according to manufacturer’s protocols and sequenced with a BigDyeTM terminator cycle sequencing kit (Applied Biosystems, USA) at Macrogen (Amsterdam, Netherlands).
16S rRNA gene amplification and sequencing
The V3-V4 hypervariable regions of bacterial 16S rRNA gene region were amplified using the following primer set: 341F-CCTACGGGNGGCWGCAG and 785R-GACTACHVGGGTATCTAATCC. The targeted gene region has been shown to be suitable for Illumina sequencing118. Each library was prepared with a two-step PCR protocol based on Illumina’s “16S metagenomic library prep guide” (15044223 Rev. B), NEBNext® Q5 Hotstart High-Fidelity DNA polymerase (New England BioLabs Inc.) according to the manufacturer’s protocol using Q5® Hot Start High-Fidelity 2X Master Mix (NEBNext - New England BioLabs, PCR under the following conditions: 98 °C for 30 sec for initial denaturation of the DNA, followed by 25 cycles of 98 °C for 10 sec, 55 °C for 30 sec, 72 °C for 20 sec and additionally 72 °C for 2 min), and the Nextera Index kit (2 × 250 bp). Paired-end (PE, 2 × 250 nt) sequencing with a 5% PhiX spike-in was performed with an Illumina MiSeq (MiSeq Reagent kit v2) at Genomed, Warsaw, Poland; following the manufacturer’s run protocols (Illumina, Inc., San Diego, CA, USA). The automatic primary analysis and the de-multiplexing of the raw reads were performed on the MiSeq machine, with the use of MiSeq Reporter (MSR) v2.6 (16S Metagenomics Protocol).
Sequencing data analysis and statistical analysis
The COI sequences were edited and corrected (primer removal and alignment trimming) using BioEdit software119. Comparison with GenBank records and homology search was carried out on 9th of November 2017 using megablast algorithm120,121 and the Nucleotide database.
The data obtained for the 3 independent DNA extractions for each developmental stage of H. carpathicus were merged and considered as one sample in taxonomic analyses. The intension of this approach was to obtain a more reliable view into the “average” bacterial communities’ structure. The exact age of insects was not determined, because particular developmental stages were successively collected from fruiting body to avoid disturbing the colony excessively during breeding. Adults age have been estimated to be ~1 month.
The samples were processed and analyzed using the Quantitative Insights Into Microbial Ecology (QIIME, version 1.9.1) pipeline122. Paired-end reads from MiSeq sequencing were quality trimmed and joined with PANDAseq version 2.8123 with a quality threshold of 0.9. The sequences that did not meet the quality criteria were removed from further analysis (mean quality >20). Chimeric reads detection was performed with VSEARCH, version 1.7.0124, an open-source replacement of USEARCH software. Clustering of operational taxonomic units (OTUs) at 97% similarity was performed by using the uclust method, version 1.2.22q125. OTUs were assigned to taxa using the GreenGenes release 13.5 database as the reference126, with the taxonomy assignment tool PyNAST127. The Biological Observation Matrix (BIOM) table was used as the core data for downstream analyses128. Any sequences that were classified as Mitochondria or Chloroplast, as well as singletons, were filtered out of the dataset. Trends in rarefaction analysis of the obtained data (plateau curves) have been used for estimation of completeness of microbial communities sampling. Based on clusters, the diversity indices were estimated, including the Chao1, PD (a quantitative measure of phylogenetic diversity), Shannon, and Simpson indices and also the number of observed OTUs. The Chao1 index is a non-parametric richness estimator that calculates the minimal number of OTUs present in a sample. Shannon index measures the average degree of uncertainty in predicting the identity of an individual sequence chosen at random. Its value increases as the number of species increases and as the distribution of individuals among the species becomes more even. The Simpson index is the probability of sampling successively at random two individuals of the same species. It varies from 0 to 1 and increases as the number of species increases and as the distribution of individuals among the species becomes less even129. Comparison of the bacterial community structure was performed with the use of UniFrac130 and Emperor131. A membership Venn diagram was computed using the MetaCoMET132 web platform to determine the specific and shared OTUs across the developmental stages of H. carpathicus. Similarity percentage (SIMPER) analysis133 was performed to calculate the average dissimilarities in bacterial community structures between different developmental stages of H. carpathicus and to assess which phylum was responsible for the observed differences. The differences in bacterial community structures were tested by the χ2 test. Statistical analyses were performed using PAST 3.16134 and PopTools 3.2135 software.
All obtained sequential data were deposited in open-source databases. Both COI sequences have been deposited in GenBank under accession numbers MG491887 and MG491888 (pupa and adult, respectively). NGS raw reads were deposited under study accession number PRJEB22873 in ENA – the European Nucleotide Archive.
The software PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States136) was used to infer metabolic capacity of the microbiome from the 16S sequences. PICRUSt functional inference is implemented in two steps. First, a reference phylogenetic tree is constructed from the Greengenes database126 and gene contents are assigned to nodes in the tree if sequenced genomes are available, or otherwise predicted using ancestral state reconstruction algorithms136. Representative sequences from OTUs derived from experimental data and associated with Greengenes identifiers are normalized by 16S rRNA gene copy number and then mapped to the corresponding Greengenes identifiers in the reference tree. The final result is an annotated table of predicted gene counts per sample. Predicted annotations can be linked with PICRUSt software to the Kyoto encyclopedia of genes and genomes (KEGG) orthology (KO) accession numbers137.
Data Availability
The NGS data sets were deposited at the European Nucleotide Archive (ENA) under the Accession number PRJEB22873. COI barcode sequences were deposited in GenBank database under the Accession numbers MG491887 and MG491888. Sunburst charts of the relative abundance of microbial 16S rDNA sequences at different taxonomic levels are available for download from the Dropbox repository (https://www.dropbox.com/sh/2pseqdq09cgi9kg/AAAH_tcwOAF0aL1xhGStgJ_ja?dl=0).
References
Basset, Y. et al. Arthropod Diversity in a Tropical Forest. Science 338, 1481–1484 (2012).
Douglas, A. E. Multiorganismal Insects: Diversity and Function of Resident Microorganisms. Annu. Rev. Entomol. 60, 17–34 (2015).
Pinto-Tomas, A. A. et al. Symbiotic Nitrogen Fixation in the Fungus Gardens of Leaf-Cutter Ants. Science (80-). 326, 1120–1123 (2009).
Koch, H. & Schmid-Hempel, P. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc. Natl. Acad. Sci. USA 108, 19288–92 (2011).
Shin, S. C. et al. Drosophila Microbiome Modulates Host Developmental and Metabolic Homeostasis via Insulin Signaling. Science (80-). 334, 670–674 (2011).
Zheng, L. et al. Bacteria mediate oviposition by the black soldier fly, Hermetia illucens (L.), (Diptera: Stratiomyidae). Sci. Rep. 3, 2563 (2013).
Sun, Z. et al. Effects of BmCPV Infection on Silkworm Bombyx mori Intestinal Bacteria. Plos One 11, e0146313 (2016).
Douglas, A. E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 23, 38–47 (2009).
Douglas, A. E. Nutritional Interactions in Insect-Microbial Symbioses: Aphids and Their Symbiotic Bacteria. Buchnera. Annu. Rev. Entomol. 43, 17–37 (1998).
Adams, A. S., Adams, S. M., Currie, C. R., Gillette, N. E. & Raffa, K. F. Geographic Variation in Bacterial Communities Associated With the Red Turpentine Beetle (Coleoptera: Curculionidae). Environ. Entomol. 39, 406–414 (2010).
Michalkova, V., Benoit, J. B., Weiss, B. L., Attardo, G. M. & Aksoy, S. Vitamin B6 generated by obligate symbionts is critical for maintaining proline homeostasis and fecundity in tsetse flies. Appl. Environ. Microbiol. 80, 5844–53 (2014).
Benemann, J. R. Nitrogen Fixation in Termites. Science (80-). 181, 164–165 (1973).
Dillon, R. J. & Dillon, V. M. The Gut Bacteria of Insects: Nonpathogenic Interactions. Annu. Rev. Entomol. 49, 71–92 (2004).
Ferrari, J. & Vavre, F. Bacterial symbionts in insects or the story of communities affecting communities. Philos. Trans. R. Soc. B 366, 1389–1400 (2011).
Engelstädter, J. & Hurst, G. D. D. The Ecology and Evolution of Microbes that Manipulate Host Reproduction. Annu. Rev. Ecol. Evol. Syst 40, 127–49 (2009).
Hilgenboecker, K., Hammerstein, P., Schlattmann, P., Telschow, A. & Werren, J. H. How many species are infected with Wolbachia?–A statistical analysis of current data. FEMS Microbiol. Lett. 281, 215–20 (2008).
Lewis, Z. & Lizé, A. Insect behaviour and the microbiome. Curr. Opin. Insect Sci. 9, 86–90 (2015).
Rey, O. et al. Distribution of endosymbiotic reproductive manipulators reflects invasion process and not reproductive system polymorphism in the little fire ant Wasmannia auropunctata. Plos One 8, e58467 (2013).
Cordaux, R., Bouchon, D. & Grève, P. The impact of endosymbionts on the evolution of host sex-determination mechanisms. Trends Genet. 27, 332–341 (2011).
Moran, N. A., McCutcheon, J. P. & Nakabachi, A. Genomics and Evolution of Heritable Bacterial Symbionts. Annu. Rev. Genet. 42, 165–190 (2008).
Duron, O. et al. The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol. 6, 27 (2008).
Deutscher, A. T. et al. Near full-length 16S rRNA gene next-generation sequencing revealed Asaia as a common midgut bacterium of wild and domesticated Queensland fruit fly larvae. Microbiome 6, 85 (2018).
Ramalho, M. O., Bueno, O. C. & Moreau, C. S. Microbial composition of spiny ants (Hymenoptera: Formicidae: Polyrhachis) across their geographic range. BMC Evol. Biol. 17, 96 (2017).
Mereghetti, V., Chouaia, B. & Montagna, M. New Insights into the Microbiota of Moth Pests. Int. J. Mol. Sci. 18, 2450 (2017).
Engel, P. & Moran, N. A. The gut microbiota of insects – diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735 (2013).
Chrostek, E., Pelz-Stelinski, K., Hurst, G. D. D. & Hughes, G. L. Horizontal Transmission of Intracellular Insect Symbionts via Plants. Front. Microbiol. 8, 2237 (2017).
Dennison, N. J., Jupatanakul, N. & Dimopoulos, G. The mosquito microbiota influences vector competence for human pathogens. Curr. Opin. Insect Sci. 3, 6–13 (2014).
de Vries, E. J., Jacobs, G. & Breeuwer, J. A. Growth and Transmission of Gut Bacteria in the Western Flower Thrips, Frankliniella occidentalis. J. Invertebr. Pathol. 77, 129–137 (2001).
de Vries, E. J., Vos, R. A., Jakobs, G. & Breeuwer, H. A. J. Western flower thrips (Thysanoptera: Thripidae) preference for thrips-damaged leaves over fresh leaves enables uptake of symbiotic gut bacteria. Eur. J. Entomol. 103, 779–786 (2006).
Chanbusarakum, L. & Ullman, D. Characterization of bacterial symbionts in Frankliniella occidentalis (Pergande), Western flower thrips. J. Invertebr. Pathol. 99, 318–325 (2008).
Chanbusarakum, L. J. & Ullman, D. E. Distribution and Ecology of Frankliniella occidentalis (Thysanoptera: Thripidae) Bacterial Symbionts. Environ. Entomol. 38, 1069–1077 (2009).
de Vries, E. J., van de Wetering, F., van der Hoek, M. M., Jacobs, G. & Breeuwer, J. A. J. Symbiotic bacteria (Erwinia sp.) in the gut of Frankliniella occidentalis (Thysanoptera: Thripidae) do not affect its ability to transmit tospovirus. Eur. J. Entomol. 109, 261–266 (2012).
van der Kooi, C. J. & Schwander, T. Evolution of asexuality via different mechanisms in grass thrips (Thysanoptera: Aptinothrips). Evolution (N. Y). 68, 1883–1893 (2014).
de Vries, E. J., van der Wurff, A. W. G., Jacobs, G. & Breeuwer, J. A. J. Onion thrips, Thrips tabaci, have gut bacteria that are closely related to the symbionts of the western flower thrips, Frankliniella occidentalis. J. Insect Sci. 8, 1–11 (2008).
Dickey, A. M. et al. Estimating bacterial diversity in Scirtothrips dorsalis (Thysanoptera: Thripidae) via Next Generation Sequencing. Florida Entomol. 97, 362–366 (2014).
Wells, M., Gitaitis, R. & Sanders, F. H. Association of Tobacco Thrips, Frankliniella fusca (Thysanoptera: Thripidae) with Two Species of Bacteria of the Genus Pantoea. Ann. Entomol. Soc. Am. 95, 719–723 (2009).
Powell, C. M., Montiel, A. L., Beddingfield, B., Hanson, J. D. & Bextine, B. R. Comparison of Bacterial Communities of Flower Thrips (Frankliniella tritici) and Potato Psyllid (Bactericera cockerelli). Southwest. Entomol. 40, 765–773 (2015).
Kucharczyk, H. & Wyrozumski, Ł. Hoplothrips carpathicus PELIKÁN, 1961 (Thysanoptera: Phlaeothripidae) – a new thrips species in the Polish fauna. Polish J. Entomol. 84, 73–83 (2015).
Kobro, S. & Solheim, H. Hoplothrips carpathicus Pelikán in Norway. In Thrips and Tospoviruses: Proceedings of the 7th International Symposium on Thysanoptera 293–294 (2001).
Kobro, S. & Rafoss, T. Identification of adult males and females of Hoplothrips species (Thysanoptera: Tubulifera) known from Norway, and some deductions on their life history. Entomol. Fenn. 17, 184–192 (2006).
Kucharczyk, H., Kucharczyk, M. & Wyrozumski, Ł. Screen traps as an efficient method in faunal research on fungus-feeding thrips (Tubulifera: Phlaeothripidae). Polish J. Entomol. 84, 201–210 (2015).
Lee, J.-S. Effects of Fomes fomentarius supplementation on antioxidant enzyme activities, blood glucose, and lipid profile in streptozotocin-induced diabetic rats. Nutr. Res. 25, 187–195 (2005).
Park, Y.-M. et al. Anti-inflammatory and Anti-nociceptive Effects of the Methanol Extract of Fomes fomentarius. Biol. Pharm. Bull. 27, 1588–1593 (2004).
Ito, H., Sugiura, M. & Miyazaki, T. Antitumor polysaccharide fraction from the culture filtrate of Fomes fomentarius. Chem. Pharm. Bull. (Tokyo). 24, 2575 (1976).
Chen, W., Zhao, Z., Chen, S.-F. & Li, Y.-Q. Optimization for the production of exopolysaccharide from Fomes fomentarius in submerged culture and its antitumor effect in vitro. Bioresour. Technol. 99, 3187–3194 (2008).
Kim, S. H., Jakhar, R. & Kang, S. C. Apoptotic properties of polysaccharide isolated from fruiting bodies of medicinal mushroom Fomes fomentarius in human lung carcinoma cell line. Saudi J. Biol. Sci. 22, 484–90 (2015).
Brunner, P. C., Fleming, C. & Frey, J. E. A molecular identification key for economically important thrips species (Thysanoptera: Thripidae) using direct sequencing and a PCR-RFLP-based approach. Agric. For. Entomol. 4, 127–136 (2002).
Timm, A. E., Stiller, M. & Frey, J. E. A molecular identification key for economically important thrips species (Thysanoptera: Thripidae) in southern Africa Article. African Entomol. 16, 68–75 (2008).
Murai, T. & Toda, S. Variation on Thrips tabaci in colour and size. in Thrips and Tospoviruses: in Proceedings of the 7th International Symposium on Thysanoptera 377–378 (2001).
Tong, X., Wang, Z. & Zhao, C. Remarkable sexually dimorphic Aroidothrips longistylus newly recorded from China (Thysanoptera: Thripidae). Zootaxa 4028, 148–150 (2015).
Tyagi, K. et al. DNA Barcoding studies on Thrips in India: Cryptic species and Species complexes. Sci. Rep. 7, 4898 (2017).
Iftikhar, R., Ashfaq, M., Rasool, A. & Hebert, P. D. N. DNA Barcode Analysis of Thrips (Thysanoptera) Diversity in Pakistan Reveals Cryptic Species Complexes. Plos One 11, e0146014 (2016).
Duron, O. & Hurst, G. D. Arthropods and inherited bacteria: from counting the symbionts to understanding how symbionts count. BMC Biol. 11, 45 (2013).
Köhler, T., Dietrich, C., Scheffrahn, R. H. & Brune, A. High-Resolution Analysis of Gut Environment and Bacterial Microbiota Reveals Functional Compartmentation of the Gut in Wood-Feeding Higher Termites (Nasutitermes spp.). Appl. Environ. Microbiol. 78, 4691–4701 (2012).
Su, L. et al. Comparative Gut Microbiomes of Four Species Representing the Higher and the Lower Termites. J. Insect Sci. 16, 1–9 (2016).
Tholen, A., Schink, B. & Brune, A. The gut microflora of Reticulitermes flavipes, its relation to oxygen, and evidence for oxygen-dependent acetogenesis by the most abundant Enterococcus sp. FEMS Microbiol. Ecol. 24, 137–149 (2006).
Poulsen, M. & Sapountzis, P. Behind every great ant, there is a great gut. Mol. Ecol. 21, 2054–2057 (2012).
Funaro, C. F. et al. Army ants harbor a host-specific clade of Entomoplasmatales bacteria. Appl. Environ. Microbiol. 77, 346–50 (2011).
Russell, J. A. et al. Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. PNAS 106, 21236–21241 (2009).
Sudakaran, S., Salem, H., Kost, C. & Kaltenpoth, M. Geographical and ecological stability of the symbiotic mid-gut microbiota in European firebugs, Pyrrhocoris apterus (Hemiptera, Pyrrhocoridae). Mol. Ecol. 21, 6134–6151 (2012).
Salem, H., Kreutzer, E., Sudakaran, S. & Kaltenpoth, M. Actinobacteria as essential symbionts in firebugs and cotton stainers (Hemiptera, Pyrrhocoridae). Environ. Microbiol. 15, 1956–1968 (2013).
Kaltenpoth, M., Winter, S. A. & Kleinhammer, A. Localization and transmission route of Coriobacterium glomerans, the endosymbiont of pyrrhocorid bugs. FEMS Microbiol. Ecol. 69, 373–383 (2009).
Chandler, J. A., Morgan Lang, J., Bhatnagar, S., Eisen, J. A. & Kopp, A. Bacterial Communities of Diverse Drosophila Species: Ecological Context of a Host–Microbe Model System. Plos Genet. 7, e1002272 (2011).
Wong, C. N. A., Ng, P. & Douglas, A. E. Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster. Environ. Microbiol. 13, 1889–1900 (2011).
Roh, S. W. et al. Phylogenetic characterization of two novel commensal bacteria involved with innate immune homeostasis in Drosophila melanogaster. Appl. Environ. Microbiol. 74, 6171–7 (2008).
Arias-Cordero, E. et al. Comparative Evaluation of the Gut Microbiota Associated with the Below- and Above-Ground Life Stages (Larvae and Beetles) of the Forest Cockchafer, Melolontha hippocastani. Plos One 7, e51557 (2012).
Reid, N. M., Addison, S. L., Macdonald, L. J. & Lloyd-Jones, G. Biodiversity of active and inactive bacteria in the gut flora of wood-feeding huhu beetle larvae (Prionoplus reticularis). Appl. Environ. Microbiol. 77, 7000–6 (2011).
Hulcr, J. et al. Mycangia of Ambrosia Beetles Host Communities of Bacteria. Microb. Ecol. 64, 784–793 (2012).
Engel, P., Martinson, V. G., Moran, N. A. & Robinson, G. E. Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. 109, 11002–11007 (2012).
Mohr, K. I. & Tebbe, C. C. Diversity and phylotype consistency of bacteria in the guts of three bee species (Apoidea) at an oilseed rape field. Environ. Microbiol. 8, 258–272 (2006).
Martinson, V. G. et al. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol. Ecol. 20, 619–628 (2011).
Cox-Foster, D. L. et al. A Metagenomic Survey of Microbes in Honey Bee Colony Collapse Disorder. Science (80-). 318, 283–287 (2007).
Gitaitis, R. D., Diaz Perez, J. C. & Sanders, F. H. Transmission of Pantoea ananatis, Causal Agent of Center Rot of Onion, by Tobacco Thrips, Frankliniella fusca. Plant Desease 87, 675–678 (2003).
Chen, B. et al. Biodiversity and Activity of the Gut Microbiota across the Life History of the Insect Herbivore Spodoptera littoralis. Sci. Rep., https://doi.org/10.1038/srep29505 (2016).
Wei, G. et al. Insect pathogenic fungus interacts with the gut microbiota to accelerate mosquito mortality. Proc. Natl. Acad. Sci. USA 114, 5994–5999 (2017).
Hammer, T. J., Dickerson, J. C. & Fierer, N. Evidence-based recommendations on storing and handling specimens for analyses of insect microbiota. Peer J 3, e1190 (2015).
Kim, J. M. et al. Effects of diet type, developmental stage, and gut compartment in the gut bacterial communities of two Cerambycidae species (Coleoptera). J. Microbiol. 55, 21–30 (2017).
Colman, D. R., Toolson, E. C. & Takacs-Vesbach, C. D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 21, 5124–5137 (2012).
Yun, J.-H. et al. Insect Gut Bacterial Diversity Determined by Environmental Habitat, Diet, Developmental Stage, and Phylogeny of Host. Appl. Environ. Microbiol. 80, 5254–5264 (2014).
Wang, Y., Gilbreath, T. M., Kukutla, P., Yan, G. & Xu, J. Dynamic Gut Microbiome across Life History of the Malaria Mosquito Anopheles gambiae in Kenya. Plos One 6, e24767 (2011).
Hu, X., Wang, C., Chen, H. & Ma, J. Differences in the structure of the gut bacteria communities in development stages of the Chinese white pine beetle (Dendroctonus armandi). Int. J. Mol. Sci. 14, 21006–21020 (2013).
Kim, C.-H., Lampman, R. L. & Muturi, E. J. Bacterial Communities and Midgut Microbiota Associated with Mosquito Populations from Waste Tires in East-Central Illinois. J. Med. Entomol. 52, 63–75 (2015).
Mereghetti, V., Chouaia, B., Limonta, L., Locatelli, D. P. & Montagna, M. Evidence for a conserved microbiota across the different developmental stages of Plodia interpunctella. Insect Sci. 00, 1–13 (2017).
Mason, C. J. & Raffa, K. F. Acquisition and Structuring of Midgut Bacterial Communities in Gypsy Moth (Lepidoptera: Erebidae) Larvae. Environ. Entomol. 43, 595–604 (2014).
Staudacher, H. et al. Variability of Bacterial Communities in the Moth Heliothis virescens Indicates Transient Association with the Host. Plos One 11, e0154514 (2016).
Montagna, M. et al. Evidence of a bacterial core in the stored products pest Plodia interpunctella: the influence of different diets. Environ. Microbiol. 18, 4961–4973 (2016).
Hammer, T. J., McMillan, W. O. & Fierer, N. Metamorphosis of a Butterfly-Associated Bacterial Community. Plos One 9, e86995 (2014).
Parker, B. L., Skinner, M. & Lewis, T. Thrips Biology and Management. (Springer Verlag, 2013).
Moritz, G. Structure, Growth and Development. In Thrips as Crop Pests (ed. Lewis, T.) 15–64 (CAB International, 1997).
Yamoah, E. et al. Microbial population and diversity on the exoskeletons of four insect species associated with gorse (Ulex europaeus L.). Aust. J. Entomol. 47, 370–379 (2008).
Dharmaraj, K., Kuberan, T. & Sivasankari, R. Studies on antimicrobial activities in Ganoderma lucidum, Fomes fomentarius and Ganoderma tsugae. J. Sci. 5, 116–123 (2015).
Kolundžíć, M. et al. Antibacterial and cytotoxic activities of wild mushroom Fomes fomentarius (L.) Fr., Polyporaceae. Ind. Crop. Prod. 79, 110–115 (2016).
Zhao, J.-Y. et al. Three new phenyl-ethanediols from the fruiting bodies of the mushroom Fomes fomentarius. J. Asian Nat. Prod. Res. 15, 310–314 (2013).
Seniuk, O. F. et al. Anti-infective properties of the melanin-glucan complex obtained from medicinal tinder bracket mushroom, Fomes fomentarius (L.: Fr.) Fr. (Aphyllophoromycetideae). Int. J. Med. Mushrooms 13, 7–18 (2011).
Alves, M. J. et al. Antimicrobial activity of phenolic compounds identified in wild mushrooms, SAR analysis and docking studies. J. Appl. Microbiol. 115, 346–357 (2013).
Zhu, F., Du, B., Bian, Z. & Xu, B. Beta-glucans from edible and medicinal mushrooms: Characteristics, physicochemical and biological activities. J. Food Compos. Anal. 41, 165–173 (2015).
Putman, M., van Veen, H. W. & Konings, W. N. Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. 64, 672–93 (2000).
Grienke, U., Zöll, M., Peintner, U. & Rollinger, J. M. European medicinal polypores - A modern view on traditional uses. J. Ethnopharmacol. 154, 564–583 (2014).
Zjawiony, J. K. Biologically Active Compounds from Aphyllophorales (Polypore) Fungi⊥. J. Nat. Prod. 67, 300–310 (2004).
Rojas-Jiménez, K. & Hernández, M. Isolation of fungi and bacteria associated with the guts of tropical wood-feeding coleoptera and determination of their lignocellulolytic activities. Int. J. Microbiol. 2015 (2015).
Ishak, H. D. et al. Microbiomes of ant castes implicate new microbial roles in the fungus-growing ant Trachymyrmex septentrionalis. Sci. Rep. 1, 204 (2011).
Parshetti, G. K., Parshetti, S., Kalyani, D. C., Doong, R. & Govindwar, S. P. Industrial dye decolorizing lignin peroxidase from Kocuria rosea MTCC 1532. Ann. Microbiol. 62, 217–223 (2012).
Bugg, T. D., Ahmad, M., Hardiman, E. M. & Singh, R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr. Opin. Biotechnol. 22, 394–400 (2011).
Calderón-Cortés, N., Quesada, M., Watanabe, H., Cano-Camacho, H. & Oyama, K. Endogenous Plant Cell Wall Digestion: A Key Mechanism in Insect Evolution. Annu. Rev. Ecol. Evol. Syst. 43, 45–71 (2012).
Gai, C. S. et al. Transmission of Methylobacterium mesophilicum by Bucephalogonia xanthophis for paratransgenic control strategy of Citrus variegated chlorosis. J. Microbiol., https://doi.org/10.1007/s12275-008-0303-z (2009).
Patt, T. E., Cole, G. C. & Hanson, R. S. Methylobacterium, a New Genus of Facultatively Methylotrophic Bacteria. Int. J. Syst. Evol. Microbiol. 26, 226–229 (1976).
Esposti, M. D. & Romero, E. M. The functional microbiome of arthropods. Plos One 12, e0176573 (2017).
Kumm, S. & Moritz, G. First Detection of Wolbachia in Arrhenotokous Populations of Thrips Species (Thysanoptera: Thripidae and Phlaeothripidae) and Its Role in Reproduction. Environ. Entomol. 37, 1422–1428 (2008).
Saurav, G. K., Daimei, G., Rana, V. S., Popli, S. & Rajagopal, R. Detection and Localization of Wolbachia in Thrips palmi Karny (Thysanoptera: Thripidae). Indian J. Microbiol. 56, 167–171 (2016).
Nguyen, D. T., Spooner-Hart, R. N. & Riegler, M. Loss of Wolbachia but not Cardinium in the invasive range of the Australian thrips species, Pezothrips kellyanus. Biol. Invasions 18, 197–214 (2016).
Arakaki, N., Miyoshi, T. & Noda, H. Wolbachia-mediated parthenogenesis in the predatory thrips Franklinothrips vespiformis (Thysanoptera: Insecta). Proc. R. Soc. B. Biol. Sci. 268, 1011–1016 (2001).
Correa, C. C. & Ballard, J. W. O. Wolbachia Associations withInsects: Winning or Losing Against a Master Manipulator. Front. Ecol. Evol. 3, 1–18 (2016).
Charlat, S., Hurst, G. D. D. & Merçot, H. Evolutionary consequences of Wolbachia infections. Trends Genet. 19, 217–223 (2003).
Nault, B. A. et al. Reproductive Modes in Onion Thrips (Thysanoptera: Thripidae) Populations from New York Onion Fields. Environ. Entomol. 35, 1264–1271 (2006).
Nguyen, D. T., Spooner-Hart, R. N. & Riegler, M. Polyploidy versus endosymbionts in obligately thelytokous thrips. BMC Evol. Biol. 15, 23 (2015).
Mee, P. T., Weeks, A. R., Walker, P. J., Hoffmann, A. A. & Duchemin, J.-B. Detection of Low-Level Cardinium and Wolbachia Infections in Culicoides. Appl. Environ. Microbiol. 81, 6177–6188 (2015).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, 1–11 (2013).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410 (1990).
Ye, J., Mcginnis, S. & Madden, T. L. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 34, W6–W9 (2006).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G. & Naufeld, J. D. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13, 31 (2012).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. Peer J 4, e2584 (2016).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
DeSantis, T. Z. et al. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267 (2010).
McDonald, D. et al. The Biological Observation Matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. Gigascience 1, 1–6 (2012).
Lemos, L. N., Fulthorpe, R. R., Triplett, E. W. & Roesch, L. F. W. Rethinking microbial diversity analysis in the high throughput sequencing era. J. Microbiol. Methods 86, 42–51 (2011).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Env. Microb 71, 8228–8235 (2005).
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A. & Knight, R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 26, 2–16 (2013).
Wang, Y., Xu, L., Gu, Y. Q. & Coleman-Derr, D. MetaCoMET: a web platform for discovery and visualization of the core microbiome. Bioinformatics 32, btw507 (2016).
Clarke, K. R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143 (1993).
Ryan, P. D., Hammer, Ø., Harper, D. A. & Paul Ryan, D. D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 178 kb. T. Harper. Geol. Museum 4, 5–7 (2001).
Hood, G. M. PopTools version 3.2.5. Available on the internet, http://www.poptools.org (2010).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences HHS Public Access. Nat. Biotechnol. 31, 814–821 (2013).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 (2014).
Acknowledgements
We are very thankful to the Center for Medical Genomics – OMICRON for their assistance in the course of this project, especially to Pawel Wolkow, the Center Director. The authors declare that they have no competing interests, financial or otherwise.
Author information
Authors and Affiliations
Contributions
A.K., H.K. and S.Z. designed the study and H.K. and M.K. did the fieldwork. A.K. carried out the laboratory work and data analyses with contributions from P.K. and S.Z. A.K., H.K. and S.Z. drafted the manuscript and M.K., P.K. and J.S. helped completing the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaczmarczyk, A., Kucharczyk, H., Kucharczyk, M. et al. First insight into microbiome profile of fungivorous thrips Hoplothrips carpathicus (Insecta: Thysanoptera) at different developmental stages: molecular evidence of Wolbachia endosymbiosis. Sci Rep 8, 14376 (2018). https://doi.org/10.1038/s41598-018-32747-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32747-x
Keywords
This article is cited by
-
Stage correlation of symbiotic bacterial community and function in the development of litchi bugs (Hemiptera: Tessaratomidae)
Antonie van Leeuwenhoek (2022)
-
Unraveling bacterial diversity of the Indian Lac Insect Kerria lacca (Kerr) using next generation sequencing
International Journal of Tropical Insect Science (2022)
-
High Taxonomic and Functional Diversity of Bacterial Communities Associated with Melon Fly, Zeugodacus cucurbitae (Diptera: Tephritidae)
Current Microbiology (2021)
-
Two New Strains of Wolbachia Affecting Natural Avocado Thrips
Indian Journal of Microbiology (2021)
-
Bacterial analysis in the early developmental stages of the black tiger shrimp (Penaeus monodon)
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
