
Abstract
Waterborne pathogens have attracted a great deal of attention in the public health sector over the last several decades. However, little is known about the pathogenic microorganisms in urban water systems. In this study, the bacterial community structure of 16 typical surface waters in the city of Beijing were analyzed using Illumina MiSeq high-throughput sequencing based on 16S rRNA gene. The results showed that Bacteroidetes, Proteobacteria and Actinobacteria were the dominant groups in 16 surface water samples, and Betaproteobacteria, Alphaproteobacteria, Flavobacteriia, Sphingobacteriia and Actinobacteria were the most dominant classes. The dominant genus across all samples was Flavobacterium. In addition, fifteen genus level groups of potentialy pathogenic bacteria were detected within the 16 water samples, with Pseudomonas and Aeromonas the most frequently identified. Spearman correlation analysis demonstrated that richness estimators (OTUs and Chao1) were correlated with water temperature, nitrate and total nitrogen (p < 0.05), while ammonia-nitrogen and total nitrogen were significantly correlated with the percent of total potential pathogens (p ≤ 0.05). These results could provide insight into the ecological function and health risks of surface water bacterial communities during the process of urbanization.
Similar content being viewed by others

Introduction
Surface freshwater ecosystems including lakes, rivers and streams, play an important role in the recycling of nutrients, biogeochemical cycling, and energy flows. In these ecosystems a diverse range of microorganisms play critical roles in the functioning of these cycles, which is associated with physicochemical parameters of water1. In addition, the use of surface water for human recreation and consumption has increased with the pace and extent of urbanization, making microbial analysis of these water resources an essential part of water quality and human health.
A number of investigations into surface water microbial diversity have been performed2,3. However, the microbial communities inhabiting urban surface waters are still relatively unknown. Beijing, as one of the largest cities in China, with a population of over 20 million, contains a number of surface water ecosystems, including river, reservoir, stream and lakes, all of which are closely tied to people’s lives. A better understanding of the scale spatial characteristics of microbial population (distribution, abundance and structure, etc.). The influence of environmental factors is essential to understanding the microbial processes underlying the urban surface ecosystem and is of particular importance to water management and public health. In previous studies, Araya et al. studied bacterial community composition in an urban river using fluorescent in situ hybridization (FISH) and denaturing gradient gel electrophoresis (DGGE), and showed that stream water was dominated by β-Proteobacteria and the Cytophaga-Flavobacterium cluster4. Zhang et al. investigated the bacterial community structure of surface water in three rivers located in the city of Shanghai over a one-year period using DGGE, showing that temperature was the major driver of bacterial community composition5. Unfortunately, water microbial communities are particularly difficult to study by traditional PCR-cloning based approaches (DGGE, clone library and FISH, etc.). Recently developed high-throughput next-generation sequencing methods, such as MiSeq pyrosequencing of the 16S rRNA gene, have provided more comprehensive descriptions of bacterial communities in various environments due to the increased number of sequence reads that can be obtained. However, there is very little published research on bacterial diversity and community composition of the surface waters in Beijing. In 2015, Wei et al. reported the core microbial community from five surface water samples was composed of 13 phyla and 40 genera6. However, their correlation with physical and chemical factors in the environment remains unknown.
The objective of this study was to examine and compare the microbial diversity and abundance of sixteen surface waters in Beijing, using the Illumina MiSeq system. Meanwhile, multivariate statistical analysis was also performed to evaluate the relationships between microbial communities and environmental factors. Furthermore, the presence of potential pathogens in these water samples was also evaluated. Our results provide more detailed information on the characteristics of urban surface water microbial communities, as well as for risk management of potential bacterial pathogens in urban surface waters.
Results
Physicochemical characteristics of 16 surface water samples
The temperature of sampling sites ranged from 9.0 to 25.0 °C with an average of 17.65 °C. Plate counts showed the highest concentrations of total bacteria were in sample HL (3.83 × 106 CFU/mL), which also exhibited the highest turbidity value, followed by sample QR and GBD, with the values of 3.0 × 104 and 2.8 × 104 CFU/mL, respectively, while sample MY contained the lowest number (3.0 × 102 CFU/mL). The highest conductivity was found in sample QR. Total nitrogen ranged from 1.38 to 19.0 mg/L and nitrate-nitrogen from 0.8 to 17.3 mg/L. The highest of total phosphorus was observed in sample HL, and the lowest in sample CQ. The properties of 16 surface water samples are listed in Table 1.
Diversity analysis using operational taxonomic units
In total, Illumina MiSeq sequencing generated clean reads of 585,728 high quality 16S rRNA gene sequences with an average of 36,608 sequences per sample. After trimming, alignment, and chimera removal using the standard operating procedure for Mothur, the number of sequences of each sample ranged from 17,519 (QR) to 36,002 (WY). All samples were randomly resampled to 17,519 sequences, with 280,304 rarefied sequences for the 16 samples retained and were subjected to further statistical analyses.
Biodiversity of the 16 surface water samples was investigated by analyzing OTUs, Chao1, and Shannon indices at cut-off levels of 3% (Table 2). Slight differences in biodiversity were observed among the different sites. For example, the library derived from sampling site CQ had the greatest number of OTUs (1,483), followed by sample GBD (1,424) and QR (1,406), while sample ZGC had the smallest (393). Chao1 numbers were considerably higher than OTU numbers, suggesting that more OTUs may exist in these bacterial communities. However, the Good’s coverage value of all samples was above 95.14%, suggesting that the observed sequences could function as a good representation of the bacterial communities present in the 16 samples. Additionally, the Shannon diversity index was also generated for each sample, with the results supporting that sample CQ had the highest bacterial diversity of the 16 samples.
In order to test the interaction between biodiversity indices and water quality indices, Spearman’s rank correlation was calculated to compare the correlation between diversity indices (OTUs, Chao1 and Shannon diversity index) and water temperature as well as other qualitative parameters. As indicated by Spearman’s rank correlation, water temperature, nitrate, and TN had significant correlations (p < 0.05) with OTUs and Chao1 (Table 3). Among all the water quality indices, only the TN was significantly positively correlated with Chao1 at p ≤ 0.01 level. It should be noted that no significant correlation was found between Shannon index and any water physicochemical parameter.
Taxonomic distributions among samples
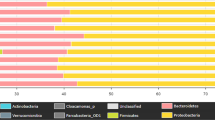
Surface water OTUs were classified to 43 bacterial phyla the most predominant phyla being Proteobacteria, Bacteroidetes and Actinobacteria. Based on relative abundances, the dominant groups (greater than 1% abundance) of each sample are displayed in Fig. 1. As shown, Proteobacteria and Bacteroidetes were the most commonly detected phyla in all the samples, accounting for 21.9–78.5% and 19.1–74.7% of sequences, respectively. The sample QR contained the most Proteobacteria (78.5%), while sample ZGC contained the most Bacteroidetes (74.7%). At the class level, the dominant groups for the 16 water samples varied. For instance, Betaproteobacteria were dominant in the majority of water samples including BJ, GBD, HR, NT, QH, QL, QR, RM, WY and YX, while in samples HL, NH, YD and ZGC, Flavobacteriia were the dominant group. It should be noted that only sample CQ was dominated by the Actinobacteria class, and Sphingobacteriia was dominant in only sample MY. Of the 16 samples, only in MY were there OTUs (2.97%) that could not be assigned to any known, which may indicate a novel uncharacterized phylum present at that site.

Bacterial community compositions at the phylum level as revealed by MiSeq sequencing. Phyla making up less than 1% of total composition were classified as “others” in the 16 samples.
The abundance of the top 30 genera are shown in Fig. 2. The most predominant genera among Proteobacterial sequences were unclassified_Comamonadaceae, Limnohabitans, Polynucleobacter, Rhodoferax, Hydrogenophaga, Candidatus_Aquirestis and Polaromonas. Flavobacterium was the predominant Bacteroidetes genus and was the most predominant genus in 12 of 16 surface water samples. The exceptions being CQ, GBD, QR and WY.

Heat map of the top 30 genera present in the sixteen surface water samples.
Principal coordinate analysis was used to analyze the bacterial community dissimilarity. As shown in Fig. 3, significant differences in bacterial community composition were found among most samples. However, similar patterns of bacterial community composition were observed for few samples, as exemplified by samples MY and RM.

The plots of principal coordinate analysis illustrating community dissimilarities based on weighted UniFrac distances metric.
To test possible linkages between the microbial communities and surface water quality parameters, canonical correspondence analysis (CCA) was performed (Fig. 4) and the results showed that bacterial community composition was significantly correlated to turbidity (r2 = 0.56; p = 0.01). CCA axis 1 was positively correlated with water temperature, pH, EC, nitrate, chloride, turbidity, AN, TP and TN, but was negatively correlated with TB. The first CCA dimension explained 8.51% of the variation of bacterial communities, and the second explained 8.42%.

Canonical correspondence analysis (CCA) biplot of bacterial communities and environmental parameters.
Detection of potential waterborne pathogens
Of the 66 potential waterborne pathogens recommend by USA EPA7, a total of 15 were detected across all of the samples. Only two types of potential pathogenic bacteria were found in sample ZGC, while 12 were detected in sample NH, the most of any sample. However, the greatest abundance of potential waterborne pathogens was observed in sample QR, accounting for 10.39% of all the OTUs, followed by sample HL (5.33%), QL (3.85%), GBD (1.94%), NH (1.26%), WY (1.34%), YX (1.07%), MY (0.96%), RM (0.94%), NT (0.80%), HR (0.56%), CQ (0.43%), YD (0.43%), QH (0.31%), BJ (0.23%) and ZGC (0.11%). At the phylum level, Proteobacteria contributed the highest proportion of bacterial richness that may be caused diseases in humans, accounting for 24.48–100% in all the potential pathogenic bacteria composition. The next most dominant phylum was the Firmicutes, followed by Actinobacteria, Spirochaetae and Tenericutes. The distribution of potential pathogenic bacteria at genus level is shown in Fig. 5.

Potential pathogenic bacteria diversity and abundance at genus level in total pathogen.
Among the potential bacterial pathogens, the genus Pseudomonas was dominant in all the samples expect for sample YD, which was dominated by the genus Mycobacterium. The next most dominant flora in each sample was different. For instance, Aeromonas was the second richest genus of potential pathogenic bacteria in samples CQ, GBD, HL, NT, QH, QL and YX. While in samples NH, QR, RM and WY, Arcobacter was the second most abundant genus of potential pathogenic bacteria among all the OTUs. Additionally, the relationship between water parameters and the percent of total potential pathogens was assessed using Spearman correlation coefficient analysis (Table 4). The results indicated that both AN and TN were significantly correlated with the percent of total potential pathogens (p ≤ 0.05). No significant links were found between the percent of total potential pathogens and the remaining water parameters.
Discussion
Surface water, which consists of both natural and man-made water bodies (rivers, lakes, reservoirs, wetlands, parks, etc.) plays a vital role in urban ecosystem services8,9, and remains an important source for drinking water, irrigation, and recreation, etc. Changes in urban surface water may result in fluctuation of microbial community, with consequences to water quality and water-borne pathogen contamination. Assessing and understanding the microbial ecology of urban surface water from different regions is essential for making water quality management plans. In our study, high-throughput amplicon sequencing was used to characterize bacterial community structure and diversity of 16 surface water samples collected from various locations in the city of Beijing. The total number of phyla in each of the 16 samples ranged from 9 to 37, while 20 to 27 phyla have been observed in five surface water samples collected from other areas of Beijing6. These results suggest that the bacterial community composition of Beijing surface waters was significantly different from each other. The dominant phyla also differed among the 16 surface water samples. Proteobacteria (21–53%) and Bacteroidetes (11–64%) were the dominant phyla, which is in agreement with the findings of Bai et al.10, who examined the bacterial community structures in wastewater treatment bioreactors via high-throughput sequencing. However, Actinobacteria (41.17% of total sequences) and Proteobacteria (31.80%) were the dominant phyla in the Ganjiang River basin11, similar results have been found in other Beijing urban surface water bodies6.
In previous studies, the factors that could influence the bacterial community in surface water were well understood. For example, the bacterial community composition in the Danube River was determined by denaturing gradient gel electrophoresis analysis, and results indicated its shifts were related to changes in environmental conditions12. In 2015, Zhong et al. reported that the composition and diversity of the prokaryotic communities were significantly related to the concentrations of salinity in lakes of the Tibetan Plateau13. Recently, Ibekwe et al. reported that microbial community structure of an urban river was significantly shaped by several key physical and chemical variables for water, including NO2, pH, and NO314. Temperature was found to be the most influential factor in determining bacterial community composition of Ganjiang River11. In contrast to these studies, we found turbidity significantly affected bacterial community. This may be because the 16 water samples showed a slightly different turbidity.
In many countries, especially in the developing world, surface waters are severely contaminated with pathogens leading to various waterborne disease outbreaks15. However, few studies concerning waterborne pathogens in urban surface waters have utilized high throughput sequencing. In the present study, it was found that potentially pathogenic bacteria were ubiquitous across all of the sampled surface waters. Among the potential waterborne pathogens, Pseudomonas spp. were the most prevalent at all sites; followed by Aeromonas. Several other potentially pathogenic genera, such as Arcobacter, Legionella, Enterobacter, Bacillus, Serratia, Enterococcus, Mycobacterium, Leptospira, Stenotrophomonas, Clostridium, Mycoplasma, Corynebacterium and Neisseria were also detected in various samples. Examples of how potentially pathogenic these organisms include the following examples: Between 2007 and 2009, 21% of the waterborne outbreaks caused by bacteria was reported in the United States, the leading etiologic pathogen bacteria including E. coli O157: H7, Shigella sonnei, Pseudomonas spp., and Legionella spp16. Among species of Legionella, a majority are considered pathogenic, with L. pneumophila the leading cause of severe pneumonia worldwide17. Members of the Aeromonas genus are common aquatic microorganisms and a number of them have been associated with human diseases18. Several species of Mycobacteria are also pathogenic to humans or animals, causing pulmonary and cutaneous disease, lymphadenitis, and disseminated infections, especially in the growing immunodeficient population19. In addition, three waterborne outbreaks associated with Arcobacter have been previously reported20.
It should be noted that both AN and TN correlated strongly with the percent of total potential pathogens (p ≤ 0.05), and potentially pathogenic bacteria were present at relatively high concentrations in samples QR (10.39%), HL (5.33%) and QL (3.85%). As the short reads generated by Illumina MiSeq platform cannot be accurately classified to the species level nor can they provide information on the amount of total or viable cells in the water samples. This limitation can lead to overestimation of the abundance of potential pathogens. Even so, more attention should be paid to health risks from surface waters in urban areas.
Conclusion
In conclusion, microbial community characteristics of 16 surface water samples in the Beijing area were studied using 16S rRNA gene amplicon sequencing. Bacteroidetes, Proteobacteria and Actinobacteria were the predominant phyla across all the samples. Principal coordinate analysis indicated significant differences in surface water bacterial community composition. Meanwhile, our results demonstrated the presence of potential waterborne pathogens in all of the surface waters sampled and that Pseudomonas spp. are ubiquitous bacteria in a wide variety of surface waters. Spearman correlation analysis showed a positive correlation between water quality properties (AN and TN) and total percent of the potential bacterial pathogens. These results give a better understanding of the urban surface waters ecology.
Materials and Methods
Study areas and sample collection
From March 26th to April 17th in 2014, samples from the 0–15 cm depth were taken from the lakes, rivers, or reservoir banks using ethanol-disinfected core tubes and stored in sterile Nalgene sampling bottles at 4 °C until processed within 24 h. These 16 surface waters are distributed across Beijing and consequently are located in different districts (Fig. 6), i.e., Bajia country park (BJ), Qing river (QR), Zhongguancun forest park (ZGC), Wenyu river (WY), Rome lake (RM), Huairou reservoir (HR), Hongluo lake (HL), Yanxi lake (YX), Qinglong lake (QL), Miyun reservoir (MY), Yongding river (YD), Gaobeidian reservoir (GBD), Chongqing reservoir (CQ), Niantan reservoir (NT), Nanhucheng river (NH) and Qianhai park (QH). All samples were collected in triplicate and then combined to create a composite sample. The temperature (Temp), electrical conductivity (EC) and pH of the samples were determined on-site using a Multi-parameter water quality meter (YSI, USA). Other analyses included assessment of turbidity, chloride, nitrate, ammonia nitrogen, total phosphorus, and total nitrogen were performed according to Chinese national standards (GB: 5749-2006) by Beijing ZKLH Analyzing & Testing Center (Beijing, China). For determination of total bacterial count, microbial biomass was harvested from approximately volume water samples using 0.45 µm polycarbonate membranes by means of a hand-operated pump, and then placed on aerobic count chromogenic plate (Qingdao Hope Bio-Technology Co., Ltd., Shandong, China). Typical colonies were counted and results expressed as colony forming units (CFU) ml−1 after incubated at 37 °C for 48 h. The experiment was repeated in triplicate.

Map of sampling site locations in the Beijing city. The map was generated using Esri ArcGIS v10.1 (Redlands, CA).
DNA extraction from water samples and sequencing
Total bacterial DNA was extracted from suitable volumes of water samples from each site using Power Soil and Water DNA kits (MO BIO, Inc., Solana Beach, CA), according to the manufacturer’s protocol. The quality and quantity of the isolated nucleic acids were determined using the Nanodrop ND1000 (NanoDrop Technologies, Delaware, USA) and adjusted to 20–30 ng/µL with sterilized ultra-pure water. The V4-V5 regions of the bacterial 16S rRNA gene were amplified by PCR using primers 515F (5′-GTGCCAGCMGCCGCGG-3′) and 907R (5′- CCGTCAATTCMTTTRAGTTT-3′). PCR reactions were performed using 50 μL mixtures containing 5 μL of 10× PCR Buffer, 4 μL of 2.5 mM dNTPs, 1.5 μL of each primer (5 μM), 0.5 μL of Taq DNA polymerase, and 1 μL of template DNA, and sterilized ultrapure water to bring volume up to 50 μL. A negative control containing all reagents except the template DNA was included with each set of reaction mixtures. PCR conditions were as follows: initial denaturation at 94 °C for 5 min; followed by 35 cycles of 94 °C for 30 s, annealing at 52 °C for 30 s, and extension at 72 °C for 1 min; and a final extension at 72 °C for 10 min. PCR products were extracted from 1% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, USA) according to the manufacturer’s instructions and quantified using QuantiFluorTM-ST (Promega, USA). All PCR products were sequenced using the Illumina MiSeq platform by the Shanghai Majorbio Bio-pharmTechnology Co., Ltd., China.
Data analysis
Sequences obtained from the Illumina sequencing platform were strictly filtered to obtained clean data and each sample was split by Qiime (V1.7.0, http://qiime.org/index.html). Low quality sequences were identified by denoising and filtered out of the dataset. The data generated by Illumina MiSeq was analyzed using the quantitative insights into microbial ecology (QIIME 1.7) toolkit21. Chimeric sequences were removed using the Uchime function with the sequence collection as its own database in the Mothur program. The effective sequences were then clustered into operational taxonomic units (OTU) at 97% sequence similarity with standard UCLUST method22. Finally, the RDP classifier was used to assign representative sequence to the microbial taxa23. Phylogenetic trees were then built from all representative sequences using the FASTTREE algorithm24. Heatmaps were drawn using gplots25 in R. The differences in overall community composition between each pair of samples were determined using the weighted and unweighted UNIFRAC metric26. For the statistical analysis, we used a randomly selected subset of 17,519 sequences per water sample to compare relative differences between samples. Normalized libraries were used to calculate the rarefaction curves and phylogenetic diversity. Rarefaction curves are a result of repeated randomizations of the observed species accumulation curve, allowing the observed richness to be compared among samples27. The Good’s coverage estimator was used to estimate the percent of the total species represented in a sample. Canonical correspondence analysis (CCA) was used to evaluate the relationship between the bacterial diversity revealed by pyrosequencing of 16S rRNA genes and water properties. The data of total bacteria was standardized using ln (CFU/mL) transformed before the analysis. CCA were performed using R (2.14.0, http://www.r-project.org/) with the community ecology package “vegan (2.0-4)”28. Spearman correlation was applied to identify the associations between water parameters and the percent of total potential pathogens, p-values < 0.05 were considered significant.
Nucleotide sequence accession numbers
The sequences obtained in this study are available in the NCBI Nucleotide Archive database under project identification numbers SRR5405060-62, SRR5405064-65, SRR5405069-70, SRR5405072-75, SRR5420965-68 and SRR5429450.
References
Wu, C. et al. Characterization of coastal urban watershed bacterial communities leads to alternative community-based indicators. PLoS ONE 5, e11285 (2010).
Ibekwe, A. M., Leddy, M. B., Bold, R. M. & Graves, A. K. Bacterial community composition in low-flowing river water with different sources of pollutants. FEMS Microbiol Ecol 79, 155–166 (2012).
Bryant, J. A. et al. Wind and sunlight shape microbial diversity in surface waters of the North Pacific Subtropical Gyre. ISME J 10, 1308–1322 (2016).
Araya, R., Tani, K., Takagi, T., Yamaguchi, N. & Nasu, M. Bacterial activity and community composition in stream water and biofilm from an urban river determined by fluorescent in situ hybridization and DGGE analysis. FEMS Microbiol Ecol 43, 111–119 (2003).
Zhang, M. L. et al. Structure and seasonal dynamics of bacterial communities in three urban rivers in China. Aquat Sci 74, 113–120 (2012).
Wei, Y. M. et al. Bacterial communities of Beijing surface waters as revealed by 454 pyrosequencing of the 16S rRNA gene. Environ Sci Pollut Res 22, 12605–12614 (2015).
USEPA. Workshop on Emerging Infectious Disease Agents and Associated with Animal Manures, Biosolids and Other Similar By-products USEPA National Risk Management Research Laboratory, Cincinnati, OH. (2001).
Rietveld, L. C. et al. Improving health in cities through systems approaches for urban water management. Environ Health 15(Suppl 1), 31 (2016).
Garcia, X. et al. Placing ecosystem services at the heart of urban water systems management. Sci Total Environ 563–564, 1078–1085 (2016).
Bai, Y. H., Qi, W. X., Liang, J. S. & Qu, J. H. Using high-throughput sequencing to assess the impacts of treated and untreated wastewater discharge on prokaryotic communities in an urban river. Appl Microbiol Biotechnol 98(4), 1841–1851 (2014).
Wang, P., Chen, B., Yuan, R. Q., Li, C. Q. & Li, Y. Characteristics of aquatic bacterial community and the influencing factors in an urban river. Sci Total Environ 569-570, 382–389 (2016).
Winter, C., Hein, T., Kavka, G., Mach, R. L. & Farnleitner, A. H. Longitudinal changes in the bacterial community composition of the Danube River: a whole-river approach. Appl Environ Microb 73, 421–431 (2007).
Zhong, Z. P. et al. Prokaryotic community structure driven by salinity and ionic concentrations in Plateau lakes of the Tibetan Plateau. Appl Environ Microbiol 82, 1846–1858 (2016).
Ibekwe, A. M., Ma, J. & Murinda, S. E. Bacterial community composition and structure in an Urban River impacted by different pollutant sources. Sci Total Environ 566–567, 1176–1185 (2016).
Patel, C. B., Shanker, R., Gupta, V. K. & Upadhyay, R. S. Q-PCR based culture-independent enumeration and detection of Enterobacter: An emerging environmental human pathogen in riverine systems and potable water. Front Microbiol 7, 172 (2016).
Hlavsa, M. C. et al. Surveillance for waterborne disease outbreaks and other health events associated with recreational water-United States, 2007–08. MMWR Surveill Summ 60, 1–32 (2011).
Berjeaud, J. M. et al. Legionella pneumophila: The paradox of a highly sensitive opportunistic waterborne pathogen able to persist in the environment. Front Microbiol 7, 486 (2016).
Pianetti, A. et al. Determination of the viability of Aeromonas hydrophila in different types of water by flow cytometry, and comparison with classical methods. Appl Environ Microbiol 71, 7948–7954 (2005).
Le Dantec, C. et al. Occurrence of Mycobacteria in water treatment lines and in water distribution systems. Appl Environ Microbiol 68, 5318–5325 (2002).
Collado, L. et al. Occurrence and diversity of Arcobacter spp. along the Llobregat River catchment, at sewage effluents and in a drinking water treatment plant. Water Res 44, 3696–3702 (2010).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624 (2012).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73, 5261–5267 (2007).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26, 1641–1650 (2009).
Warnes, G.R. et al. gplots: Various R Programming Tools for Plotting Data R package version 301. https://CRANR-projectorg/package=gplots. (2015).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235 (2005).
Hughes, T. R. et al. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol 19, 342–347 (2001).
Oksanen, J. Multivariate analysis of ecological communities in R: vegan tutorial R package version 20-1 (2011).
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Nos 31500083, 31570494 and 41561096), the National Water Pollution Control and Treatment Science and Technology Major Project (Nos 2014ZX07204-005 and 2015ZX07206-006).
Author information
Authors and Affiliations
Contributions
D.C.J. performed all the experiments with the help of X.K., B.J.C., Y.F.X. and X.R.W. D.C.J. wrote the manuscript. Y.D. and D.C.J. designed the experiment. All the authors contributed to the revision of the manuscript and approved the version to be published.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jin, D., Kong, X., Cui, B. et al. Bacterial communities and potential waterborne pathogens within the typical urban surface waters. Sci Rep 8, 13368 (2018). https://doi.org/10.1038/s41598-018-31706-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-31706-w
This article is cited by
-
Microbial hitchhikers harbouring antimicrobial-resistance genes in the riverine plastisphere
Microbiome (2023)
-
Investigating bio-remediation capabilities of a constructed wetland through spatial successional study of the sediment microbiome
npj Clean Water (2023)
-
Distribution of Antibiotic Resistance Genes and Their Associations with Bacterial Communities and Water Quality in Freshwater Lakes
Water, Air, & Soil Pollution (2023)
-
Bacterial diversity and predicted enzymatic function in a multipurpose surface water system – from wastewater effluent discharges to drinking water production
Environmental Microbiome (2021)
-
Proteomic profiling of clinical and environmental strains of Pseudomonas aeruginosa
Molecular Biology Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
