
Abstract
The polymerization of nitrogen can be used as high energy density materials. The crystal structures of Li2N2 at high pressures are explored by using the first-principles method combined with evolutionary algorithm. The phase transitions Pmmm → Immm → Pnma → Cmcm-1 → I41/acd are predicted in the pressure range of 0–300 GPa. Enthalpy calculations reveal that the tetragonal phase I41/acd containing the spiral nitrogen chains is stable above 242 GPa, indicating that the polymerization of nitrogen is realized in Li2N2 under pressure.
Similar content being viewed by others

Introduction
Nitrogen-related compounds attract great interesting for its potential application in high energy density materials (HEDM). The bond energies of the N-N single bond (160 kJ/mol) and double bond (418 kJ/mol) are much less than that of the triple bond (954 kJ/mol) of N2. The polymeric nitrogen, in which nitrogen atoms form polymer chains and three-dimensional networks, contains the N-N single and N=N double bonds. Therefore, huge energy will be released when the polymeric nitrogen transformations to N2 with the triple bond without producing pollutants. For decades, the polymeric nitrogen was an interesting subject for both experimental and theoretical researchers1,2,3,4,5,6. The monatomic nitrogen was firstly predicted by McMahan et al.1. The cubic gauche structure (cg-N) with N-N single bonds was successfully synthesized above 110 GPa and 2000 K2, which prompts the further study on nitrogen-related materials. Recently, alkali metal-nitrogen systems are extensively studied and new compositions are reported theoretically7,8,9,10,11.
As the lightest element in alkali metal, lithium can form different nitrides with nitrogen including Li3N, Li2N2, and LiN3, which had been synthesized successfully. Particularly, Lithium-to-nitrogen (Li:N) ratio equals one in Li2N2 which is neither Li-rich compound nor N-rich compound. In N-rich compound of LiN3, the polymerization of nitrogen was predicted above 375 GPa12. It is also expected that the polymerization of nitrogen forms in Li2N2 at high pressure. Li2N2 is obtained and crystallizes in Immm phase at high pressure and high temperature by decomposition of LiN313. The nitrogen atoms form \({{\rm{N}}}_{2}^{2-}\) ions with the N=N double bonds in the orthogonal phase which is the first alkali diazenide. The compound containing the N=N double bonds is proposed as the precursor of the polymeric nitrogen, which may facilitate the formation of polymeric nitrogen under pressure. Also, it is possible to enhance the stability by introducing the metal element. Theoretically, Li2N2 consisting \({{\rm{N}}}_{2}^{2-}\) ion is proved to be one of the stable stoichiometries in Li-N system under the pressure range from 0 to 100 GPa7,8. Moreover, to the best of our knowledge, the polymerization of Li2N2 is not reported yet. Therefore, in this work, we systematically illustrate the phase transitions and bond features of Li2N2 at 0–300 GPa. Our results show that the polymerization of nitrogen in Li2N2 will occur at the pressure higher than 242 GPa.
Methods
The crystal structures are the key to analyzing the properties of materials at high pressures. Here, the evolutionary algorithm (USPEX)14,15 combined with the first-principles calculations are adopted to search the structures of Li2N2. The fix-composition simulations are performed in the pressure range from 0 to 300 GPa. In the structural searches, the Perdew-Burke-Ernzerhof (PBE) exchange correlation functional16 and projector augmented wave (PAW) method17 are used as implemented in VASP code18. For Li and N atoms, 1s22s1 and 2s22p3 are treated as the valence electrons, respectively. After the structural searches are completed, the structures with the lowest enthalpies are re-relaxed with higher accuracy by employing the hard PAW pseudopotential of N atoms. The convergence tests are described in Fig. S1. The energy cutoff is set to be 1000 eV, and the k-points with 2π × 0.025 Å−1 resolution is automatically generated by Monkhorst-Pack scheme, at which the total energies are well converged. The relaxation is stopped until the force and the energy are converged to 1.0 × 10−3 eV/Å and 1.0 × 10−6 eV, respectively. To check the dynamically stability of the structures we obtained, the phonon calculation is carried out by Quantum ESPRESSO code19 based on density functional perturbation theory20.
Results
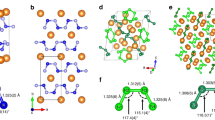
The structure searches are performed at 0, 5, 50, 100, 150, 200, and 300 GPa. The calculated enthalpies of candidate structures are shown in Fig. 1. At ambient pressure, the Pmmm structure of Li2N2 is the most stable phase, which is the same as that of Na2N221. In our calculations, the energy difference between Pmmm and Immm is very small (2.8 meV/Li2N2) at 9 GPa, at which Immm structure was synthesized experimentally13. By considering the temperature effects in the experiment, Immm may become more favorable than Pmmm phase. As shown in Fig. 2, the difference of the two structures is the arrangement of N atoms in the frame formed by Li atoms. Pnma structure is the favorable phase above 9 GPa, and it transforms to Cmcm-1 structure with almost the same energy as Pnma above 60 GPa. In Pnma structure, the Li atoms are compressed into the same plane with the nitrogen atoms as increasing pressure, and the Pnma structure becomes Cmcm-1 structure finally (see Fig. 3). The phase transition order Pmmm → Immm → Pnma → Cmcm-1 agrees with the previous theoretical result at the pressure range of 0–100 GPa8, where the polymerization of nitrogen is not observed. In the following, we will discuss the behaviors of Li2N2 under the pressure higher than 100 GPa.

The enthalpy-pressure curve of the candidate structures refered to Cmcm-1 structure. There are two structures with the same symmetry Cmcm among all of the structures. In order to distinguish them, they are denoted as Cmcm-1 and Cmcm-2. The inset shows the enthalpies for Immm and Pnma structures relative to Pmmm structure at small pressure range.

Crystal structures of Li2N2. In order to compare with Immm structure, the 1 × 1 × 4 supercell is used for Pmmm structure. The I41/acd structure is viewed along b axis (1 × 1 × 2 supercell). Perspective view of the I41/acd structure is also shown, looking down the nitrogen chains along c axis. The green and grey spheres represent Li and N atoms, respectively.

The evolutionary of Pnma structure at high pressures. When all of the atoms move along the direction 1/4b-1/4c, the equivalent unit cell of Pnma structure is obtained. It is easier to see the similarity between this Pnma structure and the Cmcm-1 structure shown in Fig. 2.
Our results show that Cmcm-1 structure transforms into the tetragonal phase I41/acd at 242 GPa. Interestingly, the polymerization of nitrogen is observed in the I41/acd phase, which contains the infinite N-chains with spiral structure (see Fig. 2). The polymerization pressure of 242 GPa in Li2N2 is much lower than that of LiN3 (375 GPa)12. At 300 GPa, the lattice parameters of I41/acd structure are a = b = 6.16 Å and c = 3.10 Å with Li atoms at Wyckoff positions 16 f (0.39865, 0.64865, 0.125) and N atoms at Wyckoff positions 16 f (0.16073, 0.41073, 0.125). Besides, the results of the standard N pseudopotential are compared with that of the hard N pseudopotential. As shown in Figs 1, S2 and S3, the same order of phase transitions is obtained in the calculations by using both the standard N pseudopotential and the hard N pseudopotential. The pressure of Cmcm-1 → I41/acd transition is 237 GPa in the calculations with the standard N pseudopotential, which is lower than the transition pressure obtained with the hard N pseudopotential. The small difference has marginal effect on the description of the phase transitions. Additionally, we consider the decomposition formula of Li2N2 → 2Li + N2, Li2N2 → 2/3LiN3 + 4/3Li, and Li2N2 → 2/3Li3N + 2/3N2, in which Fm \(\bar{3}\) m, I \(\bar{4}\) 3d, Aba2, and Cmca structures for Li22,23, P41212, I213, Pba2, and I \(\bar{4}\) 3m structures for N23,4, C2/m and P6/m structures for LiN312, P6/mmm, P63/mmc, and Fm \(\bar{3}\) m structures for Li3N7,8, are adopted. As shown in Fig. S4, the decomposition enthalpy is always higher than the lowest enthalpy curve, indicating that the Li2N2 is stable against decomposition.
The dependence of bond length on pressure is shown in Fig. 4. At ambient pressure, for Pmmm, Immm, Pnma, Cmcm-1, the N-N bond lengths are 1.258 Å, 1.268 Å, 1.271 Å, 1.274 Å, respectively, which are comparable to the N=N bond length (1.25 Å), indicating the existence of the N=N double bond in these phases. With increasing pressure, the N-N bond length is decreased sluggishly. Specially, the N-N bond length of Pnma is smaller than that of Cmcm-1 below 40 GPa, while above 60 GPa, the bond length of the two structures becomes the same, as indicated by an arrow in Fig. 4. This implies that the Pnma phase transforms to Cmcm-1 with increasing pressure, as shown in Fig. 3. In the I41/acd structure, the nitrogen atoms form infinite spiral chains, and the bond length is 1.535 Å at 0 GPa which is bigger than the length of single bond (1.45 Å). The bond length decreases with increasing pressure. At 300 GPa, the N-N bond length becomes 1.346 Å, and the bond order24 is calculated to be 1.17, which is slightly larger than 1.0, confirming the single bonding nature. All of the N-N bond length has the same value in the spiral nitrogen chain, implying that the nitrogen atoms are equivalent. Furthermore, the equal bonding nature of all N atoms in a nitrogen chain is confirmed by the electron localization function (ELF) as shown in Fig. 5. Each N atom in the chains exhibits two N-N σ bonds and two lone pairs.

The variation of N-N bond length with pressure.

ELF plots of I41/acd structure at 300 GPa. The isosurface value is set as 0.8.
The phase I41/acd with polymeric chains is observed for the first time in Li2N2. The dynamical stability of I41/acd structure is examined by phonon spectra using density functional perturbation theory. As shown in Fig. 6, there is no imaginary frequency in the whole Brillouin zone at 300 GPa, indicating the dynamical stability of I41/acd structure. Since the primitive cell of I41/acd has 16 atoms, the phonon dispersion curves consist of 3 acoustic branches and 45 optic branches. At Γ point, the frequencies of optic modes are 12 to 45 THz. When the pressure is decreased, the imaginary frequencies appear below 40 GPa, as shown in Fig. S5, which indicates that the structure is dynamically unstable at ambient pressure. Moreover, by using the substrate or nanostructured confinement25,26, the new polymeric phase may be stable at ambient condition. The calculated electronic band structure and density of states (DOS) for I41/acd at 300 GPa are shown in Fig. 7, indicating that the I41/acd is an insulator with the band gap of 2.7 eV at 300 GPa. The conduction band minimum locates at Z, while the valence band maximum appears along N-T. Therefore, the tetragonal phase with I41/acd symmetry is descried as an indirect insulator. The electrons near Fermi level is mainly contributed by N 2p orbital and the contribution from Li is away from the Fermi level, indicating that Li2N2 is indeed a nitrogen dominated materials. The Bader charge analysis27 reveals that the charge transfer from Li atom to N atom is 0.8 e, suggesting the Li-N ionic bonding characteristic.

The phonon spectra of I41/acd structure at 300 GPa.

The electronic structure of I41/acd phase at 300 GPa. The dotted line is the Fermi level.
Summary
In summary, the crystal structures of Li2N2 under high pressure are predicted by using evolutionary algorithm. Interestingly, the tetragonal phase I41/acd featuring the polymeric nitrogen chains is identified for the first time in Li2N2. The transformation from \({{\rm{N}}}_{2}^{2-}\) ions to N chains is driven by pressure, implying that the N-N bonding pattern evolves from the double bond to the single bond. The infinite nitrogen chain is stabilized by introducing metal element, which transfers electrons to the nitrogen atoms. Also, it provides a suitable starting material for pursuit of nitrogen chain synthesis. This newly predicted tetragonal phase is energetically favorable above 242 GPa, which adds the understanding to the energetic nitrogen compounds under pressure.
References
McMahan, A. K. & LeSar, R. Pressure dissociation of solid nitrogen under 1Mbar. Phys. Rev. Lett. 54, 1929–1932 (1985).
Eremets, M. I., Gavriliuk, A. G., Trojan, I. A., Dzivenko, D. A. & Boehler, R. Single bonded cubic form of nitrogen. Nat. Mater. 3, 558–563 (2004).
Pickard, C. J. & Needs, R. J. High-pressure phases of nitrogen. Phys. Rev. Lett. 102, 125702 (2009).
Wang, X. et al. Cagelike diamondoid nitrogen at high pressures. Phys. Rev. Lett. 109, 175502 (2012).
Tomasino, D., Kim, M., Smith, J. & Yoo, C.-S. Pressure-induced symmetry-lowering transition in dense nitrogen to layered polymeric nitrogen (LP-N) with colossal raman intensity. Phys. Rev. Lett. 113, 205502 (2014).
Eremets, M. I. et al. Polymerization of nitrogen in sodium azide. J. Chem. Phys. 120, 10618–10623 (2004).
Peng, F., Yao, Y., Liu, H. & Ma, Y. Crystalline LiN5 predicted from first-principles as a possible high-energy material. J. Phys. Chem. Lett. 6, 2363–2366 (2015).
Shen, Y. et al. Novel lithium-nitrogen compounds at ambient and high pressures. Sci. Rep. 5, 14204 (2015).
Steele, B. A. & Oleynik, I. I. Sodium pentazolate: A nitrogen rich high energy density material. Chem. Phys. Lett. 643, 21–26 (2016).
Peng, F., Han, Y., Liu, H. & Yao, Y. Exotic stable cesium polynitrides at high pressure. Sci. Rep. 5, 16902 (2015).
Steele, B. A. et al. High-pressure synthesis of a pentazolate salt. Chem. Mater. 29, 735–741 (2017).
Wang, X. et al. Polymerization of nitrogen in lithium azide. J. Chem. Phys. 139, 164710 (2013).
Schneider, S. B., Frankovsky, R. & Schnick, W. High-pressure synthesis and characterization of the alkali diazenide Li2N2. Angew. Chem. Int. Ed. 51, 1873–1875 (2012).
Oganov, A. R. & Glass, C. W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 124, 244704 (2006).
Glass, C. W., Oganov, A. R. & Hansen, N. USPEX-Evolutionary crystal structure prediction. Comp. Phys. Comm. 175, 713–720 (2006).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21, 395502 (2009).
Baroni, S., Gironcoli, S. d., Corso, A. D. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001).
Zhang, X., Zunger, A. & Trimarchi, G. Structure prediction and targeted synthesis: A new NanN2 diazenide crystalline structure. J. Chem. Phys. 133, 194504 (2010).
Hanfand, M., Syassen, K., Christensen, N. E. & Novikov, D. L. New high-pressure phases of lithium. Nature 408, 174–178 (2000).
Lv, J., Wang, Y., Zhu, L. & Ma, Y. Predicted novel high-pressure phases of lithium. Phys. Rev. Lett. 106, 015503 (2011).
Manz, T. A. Introducing DDEC6 atomic population analysis: part 3. Comprehensive method to compute bond orders. RSC Adv. 7, 45552–45581 (2017).
Abou-Rachid, H., Hu, A., Timoshevskii, V., Song, Y. & Lussier, L.-S. Nanoscale high energetic materials: A polymeric nitrogen chain N8 confined inside a carbon nanotube. Phys. Rev. Lett. 100, 196401 (2008).
Timoshevskii, V., Ji, W., Abou-Rachid, H., Lussier, L.-S. & Guo, H. Polymeric nitrogen in a graphene matrix: An ab initio study. Phys. Rev. B 80, 115409 (2009).
Henkelman, G., Arnaldsson, A. & Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36, 354–360 (2006).
Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant Nos 11504381, 11534012, 11674329 and Science Challenge Project No. TZ2016001. The calculations were performed in Center for Computational Science of CASHIPS, the ScGrid of Supercomputing Center and Computer Network Information Center of Chinese Academy of Sciences. This research work is supported by a Tianhe-2JK computing time award at the Beijing Computational Science Research Center (CSRC).
Author information
Authors and Affiliations
Contributions
J.Z. and Z.Z. conceived the research. J.Z. carried out the calculations. J.Z., X.W., K.Y., Y.C. and Z.Z. analyzed the data. J.Z. and Z.Z. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Wang, X., Yang, K. et al. The polymerization of nitrogen in Li2N2 at high pressures. Sci Rep 8, 13144 (2018). https://doi.org/10.1038/s41598-018-31355-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-31355-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
