
Abstract
Telomere length (TL) is associated with numerous disease states and is affected by genetic and environmental factors. However, TL has been mostly studied in adult populations of European or Asian ancestry. These studies have identified 34 TL-associated genetic variants recently used as genetic proxies for TL. The generalizability of these associations to pediatric populations and racially diverse populations, specifically of African ancestry, remains unclear. Furthermore, six novel variants associated with TL in a population of European children have been identified but not validated. We measured TL from whole blood samples of 492 healthy African American youth (children and adolescents between 8 and 20 years old) and performed the first genome-wide association study of TL in this population. We were unable to replicate neither the 34 reported genetic associations found in adults nor the six genetic associations found in European children. However, we discovered a novel genome-wide significant association between TL and rs1483898 on chromosome 14. Our results underscore the importance of examining genetic associations with TL in diverse pediatric populations such as African Americans.
Similar content being viewed by others
Introduction
Telomeres are DNA-protein structures composed of tandem hexamer repeat sequences (TTAGGGn) that cap the ends of each chromosome1. Telomeres play a vital role in maintaining DNA stability and integrity, and are therefore, critical for preserving genomic information2,3. With each mitotic division, a portion of telomeric DNA is lost. The cell enters senescence upon reaching a critical telomere length (TL) threshold4. TL has thus become an important biomarker of aging and overall health5,6,7,8. A complex interaction between genetic9 and non-genetic factors10 affects TL. While heritability estimates of TL range from 36% to 82%11, much is still unknown about genetic factors leading to variation in TL12,13.
Although epidemiological research in pediatric populations has linked TL to early life adversity14 and environmental exposures15,16, few studies have focused on the genetic determinants of TL in pediatric populations. In contrast, several genetic studies of TL in European and Asian adults have identified and replicated 34 genetic variants associated with TL17,18,19,20,21,22,23,24,25,26. Over 30 studies have used these variants as genetic proxies for TL through Mendelian randomization approaches to address reverse causation when examining association between TL and disease in diseased patients17,27,28. However, recent studies in Chinese newborns and European children have failed to replicate these variants, suggesting that they are not generalizable across age groups29,30. One study, by Stathopoulou et al., reported six novel genetic variants associated with TL in European children (age 4–18 years) not previously discovered in adult telomere studies30. Replication of these six genetic variants has not yet been attempted. Given that adult TL appears determined prior to adulthood31, further research in diverse pediatric populations is necessary to validate the existence of genetic effects on TL early in life.
Previous genetic studies of TL have been done almost exclusively in populations of European ancestry32, yet there is evidence that TL varies by race/ethnicity32,33,34. African Americans have been shown to have longer telomeres throughout life34,35,36 and a greater rate of telomere attrition than populations of European ancestry37. Population-specific differences in genetic variants have previously been shown across the genome38. Thus, it is possible that population-specific variation of genetic factors contributing to TL influences the difference in TL observed between populations of African and European ancestries33.
To further understand the relationship between genetic variants and TL, we performed the first large-scale genetic study of TL in African American children and adolescents (n = 492) from the Study of African Americans, Asthma, Genes and Environments (SAGE). Herein, we analyze genome-wide genetic data to attempt validation of previously reported genetic associations with TL and identify genetic variants influencing TL in African American children and adolescents.
Results
Study Population
Demographic information for the study population (n = 492) is presented in Table 1. The age of participants ranged from 8 to 20 with a median age of 15.8 (IQR = 12.4, 18.3; Table 1). Median African ancestry was 0.81 (IQR = 0.74, 0.85; Table 1); additionally, we observed a subtle, but positive, correlation between African Ancestry and TL (β = 0.333, P = 0.022, Supplementary Fig. S1 and Supplementary Table S1). While individuals with public health insurance had significantly longer TL than individuals with private health insurance (P = 1.84 × 10−4), there was no significant association of age or maternal education with TL (Supplementary Table S1).
Evaluation of Previous Variants
We evaluated 40 variants, 34 from adult studies (Table 2) and six from a pediatric study (Table 3), for an association with log-transformed TL. None of the variants from either the adult or pediatric studies were significantly associated with TL in our study population (P > 0.05). To determine whether the combined effect of the six previously discovered pediatric variants was associated with TL in our study population, we calculated a weighted genetic prediction score (GPS) by aggregating the allele associated with longer TL in European children weighted by the published β-coefficient30. There was no significant association between the GPS and TL in our study population of African American children and adolescents (β = 0.377, P = 0.150, Fig. 1).

Adjusted association between log-transformed TL and GPS in healthy African American children and adolescents in SAGE: San Francisco Bay Area, 2006–2015. Regression association adjusted for sex, age, genetic ancestry, maternal educational attainment, health insurance type and batch effects.
Discovery Genome-Wide Association Study
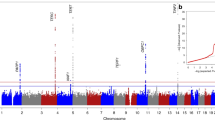
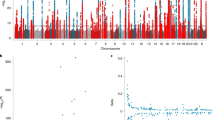
We performed a discovery GWAS to identify significant and suggestive associations between common genetic variants and TL in our study population. We identified a novel association between rs1483898 and TL that reached genome-wide significance (P = 7.86 × 10−8, Fig. 2). Rs1483898 is an intergenic single nucleotide polymorphism (SNP) located proximal to the LRFN5 gene on chromosome 14. An increase in copies of the rs1483898 A allele was significantly associated with longer TL (β = 0.148, P = 7.86 × 10−8, Fig. 3) and rs1483898 had a minor allele frequency (MAF) of 0.236 in our study population. We also discovered 41 suggestive associations between common variants and TL (P < 2.32 × 10−6, Supplementary Table S2). Of particular note were rs9675924 (β = −0.171, P = 2.27 × 10−6, Supplementary Table S2) located in CABLES1 and rs4305653 (β = 0.167, P = 1.81 × 10−6, Supplementary Table S2) located in TTC37. These genes have been previously associated with telomere biology39,40.

Results of GWAS for TL in healthy African American children and adolescents in SAGE: San Francisco Bay Area, 2006–2015. (A) Manhattan plot of the GWAS of TL with three SNPs relevant to telomere biology highlighted. Genome-wide significance threshold is indicated as red line (P = 1.2 × 10−7) and suggestive significance threshold is indicated as blue line (P = 2.3 × 10−6). (B) Expansion of 1 Mb flanking region around the top hit (rs1483898) with surrounding SNPs colored by amount of linkage disequilibrium with the top SNP, indicated by pairwise r2 values from hg19/November 2014 1000 Genomes AFR.

Comparison of mean TL between rs1483898 genotypes in healthy African American children and adolescents in SAGE: San Francisco Bay Area, 2006–2015.
Discussion
In this study, we contribute to the nascent body of research on genetic determinants of TL by assessing the generalizability of genetic markers of TL to African American children and adolescents. Our results are consistent with recent studies in pediatric populations29,30 that did not replicate variants associated with TL in adults17,18,19,20,21,22,23,24,25,26, suggesting that these variants may not play a significant role in the regulation of TL during the first two decades of growth and development. However, we were also unable to replicate genetic variants associated with TL in a population of European children30, highlighting potential population-specific effects of genetic associations with TL. Lastly, we identified a genome-wide significant variant, rs1483898, and 41 suggestively associated variants within genes relevant to telomere biology in a GWAS for TL in African American children and adolescents.
Genetic determinants of TL are critical to understanding inter-individual variation in TL. However, most studies of TL have been performed in adults, after the developmental time window when age-dependent telomere shortening may have already occurred32. Studies conducted among adults have identified and replicated 34 variants that have been used in recent years as proxies of TL in studies of disease risk17,27. We did not replicate these genetic associations in our study population of African American children and adolescents. Pediatric studies of TL by other groups29,30 have also been unable to replicate associations found among adults, suggesting that the genetic components influencing TL may differ between adult and pediatric populations. It is possible that variants identified in adults relate to telomere maintenance in adulthood but do not regulate TL during earlier developmental windows. For example, resistance to telomere shortening during childhood may be influenced by genetic factors impacting telomerase, a critical enzyme in telomere elongation41 that is influenced by genetic loci42 and shows age-related reduction in activity43. TL is determined prior to adulthood dependent on the TL setting at birth and the rates of shortening and elongating during the first two decades of life10. These factors have genetic influences that have yet to be fully characterized1,10.
We attempted replication of six TL-associated genetic variants discovered in healthy children of European ancestry30. We found no significant association with TL among the six variants independently or in a weighted GPS, which tests cumulative variation at multiple genetic loci. Heritability estimates of TL range from 36% to 82%11, yet has only been reported in populations of European ancestry and may not be generalizable to other populations. Similarly, genetic determinants of TL have primarily been studied in populations of European or Asian descent. Recent studies attempting to replicate and/or identify genetic associations with TL in non-European populations, including Punjabi Sikh24, Han Chinese26,44 and Bangladeshi45, have had mixed success. Among the limited set of studies assessing TL-associated genetic variants in populations of African ancestry, all have been performed in adult populations. One study discovered genetic variants associated with TL in adults of European ancestry that were not associated in an adult population of African Americans22. Another study in adult African Americans was only able to replicate the effects of variants in TERC, the gene encoding the enzyme telomerase, that had been identified in populations of European ancestry46. We found a subtle positive association between the proportion of African ancestry and TL in African American children and adolescents, which is consistent with research among adults34. Considering TL dynamics vary by race/ethnicity32,33,34, our study augments the current literature by demonstrating that TL-associated genetic variants differ between ancestral populations in the pediatric age range. Ancestry-specific genetic associations with a phenotypic trait have been demonstrated previously47, thus, the difference we observed may result from population-specific effects impacting genetic regulation of TL. It is worth noting regional variation in environmental and social exposures between SAGE’s urban San Francisco Bay Area and the more rural Nancy, France of the Stathopoulou et al. study48 as potential factors effecting the association between genetic variants and TL and possibly precluding replication of the Stathopoulou et al. study.
We identified associations with TL in biologically relevant pathways relating to apoptosis, cell senescence and telomere replication. The most significant association, reaching genome-wide significance, was for rs1483898. Rs1483898 is located on the q21.1 arm of chromosome 14 in the regulatory region of LRFN5, a neuronal transmembrane protein. In our population of African American children and adolescents, increasing copies of the A allele of rs1483898 associated with longer TL. The A allele of rs1483898 has allele frequencies of 0.74, 0.86 and 0.45 in the African, European and East Asian populations of 1000Genomes, respectively49.
We identified 41 variants that were suggestively associated with TL. The A allele of rs9675924, located within an intron of cell cycle regulator CABLES1 on chromosome 18, is associated with shorter TL in our study population. CABLES1 is co-expressed with protein kinase CDK5, a known contributor to apoptosis in certain neuronal diseases39. CABLES1 has also been shown to inhibit cell proliferation and induce cell senescence in umbilical endothelial cells40. The C allele of rs4305653 associated with longer telomeres. This variant is located on chromosome 5 within an intron of TTC37, a component of the SKI complex which mediates protein-protein interactions. TTC37 is co-expressed with apoptosis-promoting protein APAF1 as well as with TEP1, a protein that binds to the RNA subunit of telomerase. There is evidence that TEP1 is involved in telomere replication but the nature of that relationship in humans remains unclear39. Ultimately, co-expression is only a proxy for co-regulation50; replication and further investigation of our results are needed to better characterize relevant associations between these genetic loci and telomere biology.
Our study lacked an independent replication cohort to assess the reproducibility of our genetic associations due to the unique characteristics of our study population (African American children and adolescents with genetic and TL data). Measurement of TL in our study population provided a snapshot of TL at a specific point in the life course. Longitudinal studies of TL are required to understand changes in TL over the life course. The major advantages of our study are that (1) it provides novel information about the genetic determinants of TL in a non-European pediatric population, and (2) our depth of phenotype data allowed us to adjust for social, environmental and genetic covariates (Supplementary Table S1).
It is important to note that while our study was well powered to detect moderate to strong effects (f2 = 0.15 and 0.35, respectively) in common variants with a MAF of 0.05 or higher in our study population, we were not powered to detect weak effects (f2 < 0.02)51. It is possible that our inability to replicate the reported variants may be explained by limited statistical power; however, this would only be the case if these variants had effect sizes in our population that were significantly lower than the strong effects reported in previous studies. All but two of the variants of the 40 variants we attempted to replicate were common (MAF > 0.05) in both the populations in which they were discovered and in our study population.
In summary, the paucity of research on factors affecting TL in pediatric and non-European populations creates a knowledge gap in the scientific understanding of gene-environment interactions regulating telomeres. Epidemiological studies reporting associations between TL and disease risk are potentially biased by the disease itself or exposures relating to treatment. Genetic proxies for TL have recently been employed to overcome these and other potential biases, such as social and environmental exposures. A critical assumption when using genetic proxies for TL is that they are generalizable across age and racial/ethnic groups. However, we were unable to replicate previous findings of TL-associated variants in our study population. We also identified novel genetic associations with TL that have not been identified in previous studies in pediatric or adult populations. Further telomere research in pediatric populations from diverse ancestral backgrounds is required to fully understand the impacts of age- and population-effects on the genetic regulation of TL.
Methods
Ethics statement
This study has been approved by the institutional review boards of University of California San Francisco, Kaiser Permanente and Children’s Hospital Oakland. Written informed consent was obtained from all subjects or from their appropriate surrogates for participants under 18 years old. All methods were performed in accordance with the relevant guidelines and regulations for human subject research.
Study population
Our study included 492 healthy controls from the Study of African Americans, Asthma, Genes and Environments (SAGE). SAGE is one of the largest ongoing gene-environment interaction studies of asthma in African American children and adolescents in the USA. SAGE includes detailed clinical, social, and environmental data on asthma and asthma-related conditions. Full details of the SAGE study protocols have been described in detail elsewhere52,53,54. Briefly, SAGE was initiated in 2006 and recruited participants with and without asthma through a combination of clinic- and community-based recruitment centers in the San Francisco Bay Area. Recruitment for SAGE ended in 2015. All participants in SAGE self-identified as African American and self-reported that all four grandparents were African American.
After all quality control procedures relating to TL measurement, TL was computed for 596 healthy controls in SAGE from whole blood. There were 495 healthy controls with complete sex, age, African ancestry, maternal educational attainment and health insurance information. Three individuals showed extreme outlier measurements for TL (three times the interquartile range) and were thus removed.
Covariates
Maternal educational attainment and health insurance type were used as proxies of SES55,56,57. Maternal educational attainment was dichotomized based on whether a participant’s mother had pursued education beyond high school (i.e., ≤12 versus >12 years of education). Health insurance type was defined as private versus public insurance. The genetic ancestry of each participant was determined using the ADMIXTURE software package58 with the supervised learning mode assuming two ancestral populations (African and European) using HapMap Phase III data from the YRI and CEU populations as references59.
Variant selection and genotyping
TL-associated variants were selected for replication using criteria set a priori. We only tested genetic associations if the (i) published association reaches genome-wide significance (P ≤ 5 × 10−8) on NHGRI-EBI GWAS Catalog by October 26, 2017; (ii) variant used as genetic proxy of TL in at least one study; (iii) variant reaches suggestive genome-wide significance (P ≤ 5 × 10−5) in a novel GWAS of TL in children; (iv) variant has a minor allele frequency (MAF) of at least 1% in our study population. We identified variants from 11 studies17,18,19,20,21,22,23,24,25,26,30. Ten of the 11 studies were performed in adult populations and nine of the 11 studies were performed in populations of European descent, with the remaining two performed in Punjab Sikh24 and Han Chinese26 populations. In total, we identified 40 variants from the literature, of which 12 were genotyped and 28 were imputed. The 28 imputed SNPs had r2 (squared correlation between imputed and expected genotypes) ranging from 0.88 to 1.00.
DNA was isolated from whole blood collected from SAGE participants at the time of study enrollment using the Wizard® Genomic DNA Purification kits (Promega, Fitchburg, WI). Samples were genotyped with the Affymetrix Axiom® LAT1 array (World Array 4, Affymetrix, Santa Clara, CA), which covers 817,810 SNPs. This array was optimized to capture genetic variation in African-descent populations such as African Americans and Latinos60. Genotype call accuracy and Axiom array-specific quality control metrics were assessed and applied according to the protocol described in further detail in Online Resource 1. Data was submitted to the Michigan Imputation Server and phased using EAGLE v2.3 and imputed from the Haplotype Reference Consortium r1.1 reference panel using Minimac361. Imputed SNPs were included if they had an r2 higher than 0.3. Quality control inclusion criteria consisted of individual genotyping efficiency >95%, Hardy-Weinberg Equilibrium (HWE) P > 10−4, and MAF > 5%. Cryptic relatedness was also assessed to ensure that samples were effectively unrelated. Samples with an estimate of genetic relatedness greater than 0.025 were excluded. After quality control procedures, 7,519,176 imputed and genotyped SNPs were available for analysis.
Telomere length measurement
DNA isolation and quantification
Genomic DNA was isolated from whole blood according to manufacturer’s recommendation using Wizard® Genomic DNA Purification Kits (Promega, Fitchburg, WI). A NanoDrop® ND-1000 spectrophotometer (Thermo Scientific) was used to assess DNA quality and quantity. All samples assayed had absorbance ratios (260/280) between 1.8 and 2.0.
Determination of Relative Telomere Length
Relative TL for each sample was determined using the quantitative real time PCR (qPCR) method first described by Cawthon et al., which quantified TL in terms of telomere/single copy gene (T/S) expression ratios62. This protocol was modified with regard to data processing and control samples as previously published by O’Callaghan et al. and described in further detail in Supplemental Methods63. In brief, relative TL for each sample was calculated using the delta-delta CT (2−∆∆Ct) formula62. Using this formula, the TL computed for each SAGE sample is proportional to the T/S ratio of that sample normalized to the T/S ratio of the PCR plate positive DNA control sample62,64,65. Inter- and intra-experimental coefficients of variation for our internal control (1301 cell line DNA) were 3% and 4.25%, respectively. Average amplification efficiency across plates was ≥90% for telomere and 36B4 assays. As TL was not normally distributed in our study population, we performed all parametric tests on a log-transformation of TL.
Replication analysis
Genotypes for all 40 previously published SNP’s in adults and children were tested for association with log-transformed TL in a multivariable linear regression analysis. Regression analyses were run separately for each SNP under an additive model to calculate the individual effect of the SNP on TL. Each regression analysis was adjusted for biological, environmental and social factors that may impact TL including sex, age, African ancestry, maternal education, and health insurance type. We adjusted for qPCR plate ID in all regression analyses to ensure that our results were not impacted by qPCR batch effects. To ensure direct comparison of results between previous studies and our current study we coded the effect alleles in our analysis to be the same as those used in previous studies.
Genetic Prediction Score construction
Recent research suggests the cumulative effect of multiple genetic markers may be a stronger predictor of a quantitative phenotype than the individual markers66,67. We therefore constructed a weighted GPS based on the six variants from Stathopoulou et al. to test their cumulative effect on TL30. We calculated each subject’s weighted GPS by summing the number of alleles (0, 1 or 2) associated with longer telomeres after weighting the allele count by the reported β-coefficient from the literature. We assumed that an effect allele having a positive β-coefficient meant that each additional copy of that allele was positively associated with TL. We used the GPS as a predictor in a linear regression against log-transformed TL controlling for sex, age, genetic ancestry, maternal educational attainment, health insurance type and batch effects. We were unable to calculate a weighted GPS based on the 34 variants in adult studies because the effect size could not be standardized across the studies.
Calculation of population-specific genome-wide significance threshold
The standard GWAS threshold for statistical significance is 5 × 10−8. This number was derived by applying a Bonferroni correction for multiple testing to a dataset of one million independent markers/SNPs. However, in many cases, this threshold is overly conservative and can be inappropriate when (1) a smaller number of markers is genotyped, and (2) the assumption of independence of tests is violated.
In order to adjust the Bonferroni correction based on the actual number of independent test performed on our dataset, we determined the number of independent tests using the protocol published by Sobota et al.68. This method estimates the effective number of independent tests in a genetic dataset after accounting for linkage disequilibrium (LD) between SNPs using the LD pruning function in the PLINK 1.9 software package69. The following parameters were used in PLINK 1.9 as advised by the authors: 100 SNP sliding window, step size of 5 base pairs, and a variance inflation factor of 1.25. Applying this method on 7,519,176 genotyped and imputed SNPs yielded 431,896 independent tests, which was then used to calculate the genome-wide significance threshold (Bonferroni correction 0.05/431,896 = 1.2 × 10−7). A suggestive threshold was set at 2.3 × 10−6 for association results based on the widely used formula: 1/(effective number of tests)70.
Discovery Genome-Wide Association Study
We performed a genome-wide association study (GWAS) using 7,519,176 genotyped and imputed SNPs to assess the relationship between SNP genotype and log-transformed TL. The GWAS linear regression model adjusted for sex, age, African ancestry, maternal educational attainment, health insurance type and batch effects. All testing was performed using PLINK1.969. Manhattan plots (Fig. 2A,B) were generated using the qqman package71 in the R statistical software environment (R Development Core Team 2010) and LocusZoom72. Curated protein-protein interactions were extracted using the STRING database39. An integrated confidence score for the interaction ranges from 0.5 (medium confidence) to 1 (high confidence).
References
Blackburn, E. H., Epel, E. S. & Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 350, 1193–1198 (2015).
Armanios, M. Telomeres and age-related disease: how telomere biology informs clinical paradigms. J. Clin. Invest. 123, 996–1002 (2013).
Calado, R. T. & Young, N. S. Telomere Diseases. N. Engl. J. Med. 361, 2353–2365 (2009).
Tümpel, S. & Rudolph, K. L. The role of telomere shortening in somatic stem cells and tissue aging: lessons from telomerase model systems. Ann. N. Y. Acad. Sci. 1266, 28–39 (2012).
Bekaert, S., Meyer, T. D. & Oostveldt, P. V. Telomere Attrition as Ageing Biomarker. Anticancer Res. 25, 3011–3021 (2005).
Mather, K. A., Jorm, A. F., Parslow, R. A. & Christensen, H. Is Telomere Length a Biomarker of Aging? A Review. J. Gerontol. Ser. A 66A, 202–213 (2011).
Monaghan, P. Telomeres and life histories: the long and the short of it. Ann. N. Y. Acad. Sci. 1206, 130–142 (2010).
Heidinger, B. J. et al. Telomere length in early life predicts lifespan. Proc. Natl. Acad. Sci. 109, 1743–1748 (2012).
Slagboom, P. E., Droog, S. & Boomsma, D. I. Genetic determination of telomere size in humans: a twin study of three age groups. Am. J. Hum. Genet. 55, 876–882 (1994).
Dugdale, H. L. & Richardson, D. S. Heritability of telomere variation: it is all about the environment! Phil Trans R Soc B 373, 20160450 (2018).
Hjelmborg, J. B. et al. The heritability of leucocyte telomere length dynamics. J. Med. Genet. 52, 297–302 (2015).
Monaghan, P., Eisenberg, D. T. A., Harrington, L. & Nussey, D. Understanding diversity in telomere dynamics. Phil Trans R Soc B 373, 20160435 (2018).
Lin, J., Epel, E. & Blackburn, E. Telomeres and lifestyle factors: Roles in cellular aging. Mutat. Res. Mol. Mech. Mutagen. 730, 85–89 (2012).
Coimbra, B. M., Carvalho, C. M., Moretti, P. N., Mello, M. F. & Belangero, S. I. Stress-related telomere length in children: A systematic review. J. Psychiatr. Res. 92, 47–54 (2017).
Martens, D. S. et al. Prenatal Air Pollution and Newborns’ Predisposition to Accelerated Biological Aging. JAMA Pediatr. 171, 1160–1167 (2017).
Lee, E. Y. et al. Traffic-Related Air Pollution and Telomere Length in Children and Adolescents Living in Fresno, CA: A Pilot Study. J. Occup. Environ. Med. 59, 446–452 (2017).
Codd, V. et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat. Genet. 45, 422–427e2 (2013).
Pooley, K. A. et al. A genome-wide association scan (GWAS) for mean telomere length within the COGS project: identified loci show little association with hormone-related cancer risk. Hum. Mol. Genet. 22, 5056–5064 (2013).
Mangino, M. et al. Genome-wide meta-analysis points to CTC1 and ZNF676 as genes regulating telomere homeostasis in humans. Hum. Mol. Genet. 21, 5385–5394 (2012).
Mangino, M. et al. A genome-wide association study identifies a novel locus on chromosome 18q12.2 influencing white cell telomere length. J. Med. Genet. 46, 451–454 (2009).
Prescott, J. et al. Genome-Wide Association Study of Relative Telomere Length. Plos One 6, e19635 (2011).
Levy, D. et al. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proc. Natl. Acad. Sci. 107, 9293–9298 (2010).
Lee, J. H. et al. Genome wide association and linkage analyses identified three loci—4q25, 17q23.2, and 10q11.21—associated with variation in leukocyte telomere length: the Long Life Family Study. Front. Genet. 4 (2014).
Saxena, R. et al. Genome-Wide Association Study Identifies Variants in Casein Kinase II (CSNK2A2) to be Associated With Leukocyte Telomere Length in a Punjabi Sikh Diabetic Cohort Clinical Perspective. Circ. Genomic Precis. Med. 7, 287–295 (2014).
Gu, J. et al. A Genome-Wide Association Study Identifies a Locus on Chromosome 14q21 as a Predictor of Leukocyte Telomere Length and as a Marker of Susceptibility for Bladder Cancer. Cancer Prev. Res. (Phila. Pa.) 4, 514–521 (2011).
Liu, Y. et al. A Genome-Wide Association Study Identifies a Locus on TERT for Mean Telomere Length in Han Chinese. Plos One 9, e85043 (2014).
Machiela, M. J. et al. Genetically predicted longer telomere length is associated with increased risk of B-cell lymphoma subtypes. Hum. Mol. Genet. 25, 1663–1676 (2016).
Davey Smith, G. & Hemani, G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23, R89–98 (2014).
Weng, Q. et al. The known genetic loci for telomere length may be involved in the modification of telomeres length after birth. Sci. Rep. 6, 38729 (2016).
Stathopoulou, M. G. et al. Genetic Determinants of Leucocyte Telomere Length in Children: a Neglected and Challenging Field. Paediatr. Perinat. Epidemiol. 29, 146–150 (2015).
Benetos, A. et al. Tracking and fixed ranking of leukocyte telomere length across the adult life course. Aging Cell 12, 615–621 (2013).
Aviv, A. & Shay, J. W. Reflections on telomere dynamics and ageing-related diseases in humans. Philos. Trans. R. Soc. B Biol. Sci. 373 (2018).
Hansen, M. E. B. et al. Shorter telomere length in Europeans than in Africans due to polygenetic adaptation. Hum. Mol. Genet. 25, 2324–2330 (2016).
Hunt, S. C. et al. Leukocyte telomeres are longer in African Americans than in whites: the National Heart, Lung, and Blood Institute Family Heart Study and the Bogalusa Heart Study. Aging Cell 7, 451–458 (2008).
Drury, S. S. et al. Setting the Trajectory: Racial Disparities in Newborn Telomere Length. J. Pediatr. 166, 1181–1186 (2015).
Rewak, M. et al. Race-Related Health Disparities and Biological Aging: Does Rate of Telomere Shortening Differ Across Blacks and Whites? Biol. Psychol. 99, 92–99 (2014).
Aviv, A. et al. Leukocyte Telomere Dynamics: Longitudinal Findings Among Young Adults in the Bogalusa Heart Study. Am. J. Epidemiol. 169, 323–329 (2009).
Jorgenson, E. et al. Ethnicity and Human Genetic Linkage Maps. Am. J. Hum. Genet. 76, 276–290 (2005).
von Mering, C. et al. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 (2003).
Pu, Z. et al. Cables1 Inhibits Proliferation and Induces Senescence by Angiotensin II via a p21-Dependent Pathway in Human Umbilical Vein Endothelial Cells. J. Vasc. Res. 54, 13–21 (2017).
Blackburn, E. H., Greider, C. W. & Szostak, J. W. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nature Medicine, https://doi.org/10.1038/nm1006-1133 (2006).
Atzmon, G. et al. Genetic variation in human telomerase is associated with telomere length in Ashkenazi centenarians. Proc. Natl. Acad. Sci. 107, 1710–1717 (2010).
Iwama, H. et al. Telomeric length and telomerase activity vary with age in peripheral blood cells obtained from normal individuals. Hum. Genet. 102, 397–402 (1998).
Ding, H. et al. Association between previously identified loci affecting telomere length and coronary heart disease (CHD) in Han Chinese population. Clinical Interventions in Aging, https://doi.org/10.2147/CIA.S60760 (2014).
Delgado, D. A. et al. Genome-wide association study of telomere length among South Asians identifies a second RTEL1 association signal. J. Med. Genet. 55, 64–71 (2018).
Hamad, R., Tuljapurkar, S. & Rehkopf, D. H. Racial and Socioeconomic Variation in Genetic Markers of Telomere Length: A Cross-Sectional Study of U.S. Older Adults. EBioMedicine 11, 296–301 (2016).
Torgerson, D. G. et al. Meta-analysis of Genome-wide Association Studies of Asthma In Ethnically Diverse North American Populations. Nat. Genet. 43, 887–892 (2011).
Billon, S. et al. Family resemblance in breakfast energy intake: the Stanislas Family Study. Eur. J. Clin. Nutr. 56, 1011–1019 (2002).
1000 Genomes Project Consortium. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. 95, 14863–14868 (1998).
Cohen, J. Statistical power analysis for the behavioral sciences. 2nd (1988).
Borrell, L. N. et al. Childhood Obesity and Asthma Control in the GALA II and SAGE II Studies. Am. J. Respir. Crit. Care Med. 187, 697–702 (2013).
Nishimura, K. K. et al. Early-Life Air Pollution and Asthma Risk in Minority Children. The GALA II and SAGE II Studies. Am. J. Respir. Crit. Care Med. 188, 309–318 (2013).
Thakur, N. et al. Socioeconomic Status and Childhood Asthma in Urban Minority Youths. The GALA II and SAGE II Studies. Am. J. Respir. Crit. Care Med. 188, 1202–1209 (2013).
Drury, S. et al. Telomere length and early severe social deprivation: linking early adversity and cellular aging. Mol. Psychiatry 17, 719–727 (2012).
Mitchell, C. et al. Social disadvantage, genetic sensitivity, and children’s telomere length. Proc. Natl. Acad. Sci. 111, 5944–5949 (2014).
Needham, B. L., Fernandez, J. R., Lin, J., Epel, E. S. & Blackburn, E. H. Socioeconomic status and cell aging in children. Soc. Sci. Med. 74, 1948–1951 (2012).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
The International HapMap 3 Consortium. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010).
Hoffmann, T. J. et al. Design and coverage of high throughput genotyping arrays optimized for individuals of East Asian, African American, and Latino race/ethnicity using imputation and a novel hybrid SNP selection algorithm. Genomics 98, 422–430 (2011).
Loh, P.-R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Cawthon, R. M. Telomere measurement by quantitative PCR. Nucleic Acids Res. 30, e47 (2002).
O’Callaghan, N. J. & Fenech, M. A quantitative PCR method for measuring absolute telomere length. Biol. Proced. Online 13, 3 (2011).
Raschenberger, J. et al. The association of relative telomere length with symptomatic peripheral arterial disease: Results from the CAVASIC study. Atherosclerosis 229, 469–474 (2013).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 29, e45 (2001).
Dudbridge, F. Power and Predictive Accuracy of Polygenic Risk Scores. Plos Genet. 9 (2013).
Talmud, P. J. et al. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. BMJ 340, b4838 (2010).
Sobota, R. S. et al. Addressing Population-Specific Multiple Testing Burdens in Genetic Association Studies. Ann. Hum. Genet. 79, 136–147 (2015).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Duggal, P., Gillanders, E. M., Holmes, T. N. & Bailey-Wilson, J. E. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics 9, 516 (2008).
Turner, S. D. Qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv 005165, https://doi.org/10.1101/005165 (2014).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinforma. Oxf. Engl. 26, 2336–2337 (2010).
Acknowledgements
The authors acknowledge the patients, families, recruiters, health care providers and community clinics for their participation. In particular, the authors thank clinical recruiters Lisa Caine, RT, Elizabeth Castellanos and Shahdad Saeedi. This work was supported in part by the Sandler Family Foundation, the American Asthma Foundation, the RWJF Amos Medical Faculty Development Program, the Harry Wm. and Diana V. Hind Distinguished Professor in Pharmaceutical Sciences II, the National Heart, Lung, and Blood Institute (NHLBI) R01HL117004, R01HL128439, R01HL135156, X01HL134589, R01HL141992, the National Institute of Environmental Health Sciences R01ES015794, R21ES24844, the National Institute on Minority Health and Health Disparities P60MD006902, R01MD010443, RL5GM118984 and the Tobacco-Related Disease Research Program under Award Numbers 24RT-0025, 27IR-0030. M.J.W. was supported by a diversity supplement of NHLBI R01HL117004, an Institutional Research and Academic Career Development Award K12GM081266, and a NHLBI Research Career Development (K) Award K01HL140218. K.L.K. was supported by a diversity supplement of NHLBI R01HL135156. E.Y.L and L.A.S were supported by a National Institute of General Medical Sciences Award T32GM007546. Research reported in this article was funded by the National Institutes of Health Common Fund and Office of Scientific Workforce Diversity under three linked awards RL5GM118984, TL4GM118986, 1UL1GM118985 administered by the National Institute of General Medical Sciences.
Author information
Authors and Affiliations
Contributions
A.M.Z., M.J.W., J.W., S.S.O. and E.G.B. were involved in the conception and design of the study. A.M.Z., M.J.W., S.S.O., E.Y.L., J.W., P.C.G., J.R.L., A.C.Y.M., C.E., D.H., S.H., M.G.C., L.A.S., K.L.K., O.R.A., J.M. and E.G.B. were involved in the analysis and interpretation of data. S.S., C.E., A.D., K.M., E.B.B., M.A.L., H.J.F., K.B.D., L.N.B. and E.G.B. planned and supervised the collection of data. C.E., O.R.A., M.G.C. and M.J.W. generated telomere length data. All authors provided revisions and approval of the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeiger, A.M., White, M.J., Eng, C. et al. Genetic Determinants of Telomere Length in African American Youth. Sci Rep 8, 13265 (2018). https://doi.org/10.1038/s41598-018-31238-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-31238-3
This article is cited by
-
Low frequency variants associated with leukocyte telomere length in the Singapore Chinese population
Communications Biology (2021)
-
A different view on fine-scale population structure in Western African populations
Human Genetics (2020)
-
Association between common telomere length genetic variants and telomere length in an African population and impacts of HIV and TB
Journal of Human Genetics (2019)
-
Loci for human leukocyte telomere length in the Singaporean Chinese population and trans-ethnic genetic studies
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
