
Abstract
Congenital hand malformations is rare and characterized by hand deformities. It is highly heterogeneous, both clinically and genetically, which complicates the identification of causative genes and mutations. Recently, targeted next-generation (NGS) sequencing has been successfully used for the detection of heterogeneous diseases, and the use of NGS also has contributed significantly in evaluating the etiology of heterogeneous disease. Here, we employed targeted NGS to screen 248 genes involved in genetic skeletal disorders, including congenital hand malformations. Three pathogenic mutations located in the GJA1, ROR2 and TBX5 genes were detected in three large Chinese families with congenital hand malformations. Two novel mutations were reported, and a known causative mutation was verified in this Chinese population. This is also the first report that the same panel of targeted NGS was employed to perform molecular diagnosis of different subtypes of congenital hand malformations. Our study supported the application of a targeted NGS panel as an effective tool to detect the genetic cause for heterogeneous diseases in clinical diagnosis.
Similar content being viewed by others

Introduction
Congenital hand malformations characterized by hand deformities is rare and occurs in approximately 23 of 10,000 total births1,2. It is also a highly clinically heterogeneous disease with different phenotypes, including syndactyly (SD), brachydactyly (BD), and ectrodactyly. Moreover, a particular phenotype also contains several subtypes, such as brachydactyly (BD), a common subtype of congenital hand deformity and characterized by short fingers and toes3. BD contains five subtypes (Type A-E), and Type A can further be classified as five subtypes (Type A1, A2, A3, A4, and A5)4,5. Because of the complexity of phenotypes congenital hand malformations, an accurate diagnosis of the congenital hand malformations is usually difficult and delayed. Thus, more diagnosis methods are required.
In addition to the variable phenotypes of congenital hand malformations, its genetic heterogeneity creates significant challenges for diagnosis. Syndactyly, a common disorder of congenital hand malformations, can be inherited as an autosomal dominant (AD), autosomal recessive (AR), and X-linked recessive (XR) model6,7. In addition, the number of identified genes associated with congenital hand malformations has been growing, and research involving the etiology of congenital hand malformations has been flourishing in the past decade. Although the functions of some of these genes have been extensively studied, it is still difficult to establish a precise genotype-phenotype correlation.
Genetic variations play an important role in congenital hand malformations. Determining the causative variants in patients affected by congenital hand malformations provides them with a number of benefits; for example, it can supply appropriate genetic counseling about recurrent risk and an accurate prognosis of the clinical course of the disease8. It is also critical for prenatal diagnosis because serious fetal hand deformities can have a strong impact on family and social life, and a necessary treatment plan should be undertaken before birth2,9. Moreover, the molecular diagnosis of patients evaluates the etiology of congenital hand malformations in clinics and lays the foundation for future inclusion in clinical trials based on gene therapy.
In recent years, the development of next-generation sequencing (NGS) has allowed to screen a large number of genes with a high sensitivity. Compared with the time-consuming, expensive, and gene-by-gene traditional analysis method, NGS is a faster, cheaper, and more efficient method for mutation screening. Recent studies have demonstrated that NGS has been successfully used to diagnose heterogeneous diseases8,10,11,12,13.
In this study, we applied a targeted NGS to screen 248 genes known to be associated with genetic skeletal disorders in three Chinese families with congenital hand malformations. We identified three pathogenic mutations in the GJA1, ROR2 and TBX5 genes. Our results support the application of targeted NGS in clinical diagnosis.
Results
Clinical description
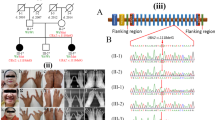
Three patients with congenital hand malformations and their families were recruited from Henan Provincial People’s Hospital, Henan, China. All of the patients were characterized by visible hand deformities such as SD, BD, and ektrodactylia (Fig. 1A–C, and Supplementary Figs S1 and S2). Pedigrees of the cases recruited in this study are shown in Fig. 1D–F. Available family members were recruited to assist in the analysis of pathogenic variations.

Clinical phenotypes and pedigrees of the cases. (A) The proband showed bilateral syndactyly between third and fourth fingers in Case 1. (B) The proband of Case 2 showed bilateral hypoplasia of the distal and middle phalanges of fingers 2–5, nail hypoplasia and the absence of nails on some fingers. (C) Proband of Case 3 was afflicted with an absent thumb. (D–F) Pedigrees of Case1, Case2 and Case3. Solid boxes and circles indicate affected individuals. The proband is marked with a black arrow. Symbols +/− and −/− represent heterozygous mutation and wild-type genotype respectively.
Identification of candidate mutations by targeted NGS
A total of 248 genes known to be associated with genetic skeletal disorders were the target genes in this study, including genes involved in hand malformations (Supplementary Table S1). All exons, splicing sites, and the immediately adjacent introns of these genes were included in the NGS panel.
In general, at least 500 Mb of clean data was obtained from the raw data for each patient. The average sequencing depth was more than 246X, and the mean coverage of target regions was greater than 99.6%. Coverage of the target base for the N10 and N20 was more than 96.2% and 93.0%, respectively. An overview of the NGS data is listed in Supplementary Table S2.
In order to avoid false negatives in clinical diagnosis, we filter and retain the variants with allele frequency less than 5%. After annotation and filtration, there were three candidates of pathogenic mutations, including a missense mutation, a nonsense mutation and a splice-site mutation. The heterozygous mutation-c.388 A > T (p.I130F)-of the GJA1 gene, the heterozygous mutation-c.2247 G > A (p.W749X)-of the ROR2 gene, and the heterozygous splice-site mutation-c.663 + 1 G > C- of the TBX5 gene were found in Case 1, Case 2, and Case 3, respectively (Supplementary Fig. S3).
To validate the positive correlation of these mutations with diseases, Sanger sequencing was employed to detect mutations in patients and their available family members. The results showed three mutations in different genes with the mode of autosomal dominant inheritance (Fig. 1).
Characterization of candidate mutations
The only one missense mutation of c.388 A > T (p.I130F) in GJA1 gene was predicted pathogenic mutation by some of the software tools (Supplementary Table S4). The missense mutation was located in exon 2 and resulted in the replacement of isoleucine with phenylalanine. Conservative analysis of the protein sequence among various species showed the region, including the mutation p.I130F, was highly conserved (Fig. 2A). The SIFT, PoluPhen, and MutationTaster predicted that the c.388 A > T (p.I130F) mutation was a pathogenic mutation, with values of 0.08, 0.824, and 1, respectively. Although the c.388 A > T (p.I130F) mutation was not reported in several databases (Human Gene Mutation Database (HGMD), the Exome Aggregation Consortium (ExAC) database, and the 1000 Genomes database), the c.389 T > C (p.I130T) mutation was reported to be a pathogenic mutation in the HGMD database14. Taken together, the c.388 A > T (p.I130F) mutation was considered pathogenic in Case 1.

Mutation analysis of c.388 A > T (p.I130F) in the GJA1 gene, c.2247 G > A (p.W749X) in the ROR2 gene and c.663 + 1 G > C in the TBX5 gene. (A) Domain structure of Connexin 43 (Cx43) and protein conservation analysis of the mutations across multiple species. The Cx43 mutations indicated as a red dot in the intracellular loop domain. (B) Diagram of the ROR2 molecule and protein conservation analysis of the mutations across multiple species. (C) Schematic representation of the TBX5 protein and protein conservation analysis. NLS1 = nuclear localization segment 1, NLS2 = nuclear localization segment 2, NES = nuclear export segment.
In Case 2, a heterozygous nonsense mutation, c.2247 G > A (p.W749X), was detected in ROR2 gene. The mutation caused a premature stop codon and resulted in the loss of 195 amino acids. Conservative analysis of the protein sequence showed the amino acid (Tryptophan) was highly conserved among various species (Fig. 2B). Moreover, both the c.2247 G > A (p.W749X) and c.2246 G > A (p.W749X) mutations had been reported as pathogenic mutations in the HGMD database15,16.
A splice-site mutation of the TBX5 gene was detected in Case 3. The c.663 + 1 G > C mutation was a canonical splice site mutation, located in sixth intron. Conservative analysis of the genomic DNA sequencing showed that the base, located in a splice-site, was highly conserved. To predict whether the mutation changed the transcriptional profile, NetGene 2 was employed to evaluate the effect of the mutation. The result showed that the mutation was caused a loss of splice-site. MutationTaster predicted that the mutation was pathogenic, with a value of 1. Moreover, Sanger sequencing results also showed that the mutation had a mode of autosomal dominant inheritance. In summary, these experiments strongly suggest the splice-site of c.663 + 1 G > C in the TBX5 gene was the causative mutation in Case 3.
Discussion
There are many syndromes that are associated with congenital hand malformations17. The significant phenotype and genetic heterogeneity of congenital hand malformations is a great challenge for clinical diagnosis and genetic counseling. The traditional detection methods for individual genes are time-consuming and expensive. In this study, targeted NGS was performed to screen 248 genes involved in genetic skeletal disorders, including genes associated with congenital hand malformations. We identified pathogenic mutations in three large Chinese families with congenital hand malformations, and these pathogenic mutations belonged to known hand deformity genes, including GJA1, ROR2 and TBX5.
The missense mutation of c.388 A > T (p.I130F) found in Case 1 is located in the GJA1 gene, which is a member of the connexin gene family. GJA1 encodes the gap junction protein alpha-1 (Connexin 43, Cx43), which is a major component of gap junctions in osteoblasts, osteocytes, osteoclasts and chondrocytes18. Syndactyly type III (SD3) is characterized by bilateral complete syndactyly between the fourth and fifth fingers, with occasional involvement of the third fingers19. In Case 1, the proband (III 1) showed bilateral syndactyly between the third and fourth fingers; however, the proband’s mother (II 2) showed bilateral syndactyly between the fourth and fifth fingers (Fig. 1A and Supplementary Fig. S1). SD3 is inherited in an autosomal dominant pattern with incomplete penetrance7. The Sanger sequencing results showed that the c.388 A > T(p.I130F) mutation was inherited with an autosomal dominant model in Case 1. Although the phenotype and genetic results suggested the disease was SD3, another syndrome, oculodentodigital dysplasia (ODDD), was considered the clinical diagnosis of this case. ODDD is a rare genetic disorder usually inherited in an autosomal dominant manner, and SD3 has been reported to occur as a part of ODDD20,21. In addition to the phenotypes of SD3, the affected members showed camptodactyly, short and narrow palpebral fissure, a narrow nose, and hypoplastic enamel (Supplementary Fig. S1), which are characteristics of ODDD22. According to the molecular results and phenotype, we reclassified the syndrome of Case1 as ODDD. The reclassification according to molecular diagnosis is crucial for accurate genetic counseling and evaluates the knowledge in the clinic.
Brachydactyly type B (BDB), one type of BD, is the most severe of the brachydactylies and is characterized by hypoplasi of distal phalanges and nails23,24 (Fig. 1B and Supplementary Fig. S2). The c.2247 G > A (p.W749X) mutation of the ROR2 gene in Case 2 was the pathogenic mutation of brachydactyly tape B1 (BDB1), which is a subtype of BDB. ROR2, located on chromosome 9q22, encodes the orphan receptor tyrosine kinase (RTK)-like orphan receptor 2 (ROR2). The structure of ROR2 is composed of an extracellular region, a transmembrane domain, and an intracellular domain. The extracellular region associated with protein-protein interaction, which contains an immunoglobulin-like domain (Ig), a cysteine-rich domain (CRD), and a kringle domain (KD). The intracellular region includes a trysine kinase domain (TK), two serine/threonine-rich domains (ST), and a proline-rich domain (PR)16,25,26. The nonsense mutation found in Case 2 resulted in a premature stop codon and caused the defects of the PR domain and two ST domains (Fig. 2B). Oldridge M et al. reported that the deletion of ROR gene on an allele could not cause BDB1; however, the intragenic mutations resulted in BDB116. In this case, the truncated protein was the causative etiology.
Mutations of the TBX5 gene caused Holt-Oram syndrome (HOS), which is a rare syndrome and is characterized by a malformation of the upper limbs and variable cardiac defects27,28. The mutation of c.663 + 1 G > C found in Case 3 was a splice-site mutation, which resulted in the incorrect transcription of the TBX5 protein and caused the changes of the second nuclear localization segment (NLS), a transactivation domain, and the C-terminal region29 (Fig. 2C). The truncated protein is unable to perform accurate nuclear localization and is unable to bind with other proteins to active downstream signals. According to the molecular diagnosis results, we also supplied a prognosis for this family. The proband (II 1) of Case 3 is not only affected with an absent thumb but also has an atrial septal defect, while his 3-year-old son (III 1) presented with a triphalangeal thumb. During genetic counseling, we recommended a heart test to be performed on the proband’s son. The results revealed that the proband’s son is also affected with a mild atrial septal defect, and pediatric cardiothoracic surgeons guided further treatments. In this case, we were not only able to detect the pathogenic mutation but also predicted the unknown phenotype due to accurate molecular diagnosis.
Accurate molecular diagnosis is crucial for classification in clinical diagnosis and is also important for individual treatment. Recently, NGS was widely used in diagnosis of single gene inheritance diseases, heterogeneous diseases and cancers30,31,32. In this study, targeted NGS was successfully used to detect the causative variations in three large Chinese families with congenital hand malformations, which are highly phenotypic and genetically heterogeneous. However, it is important to be aware of NGS limitations, such as miss detections of small insertions/deletions, trinucleotide repeats and CNV variations. Targeted gene panels are typically less expensive than whole exome sequencing, but go out of date as new causal genes are discovered for relevant phenotypes. Whole exome sequencing covers all annotated coding exons and will be widely used in future clinical diagnostics.
In conclusion, targeted NGS was employed to screen 248 genes involved in genetic skeletal disorders, and three pathogenic mutations located in GJA1, ROR2 and TBX5 were detected in three large Chinese families with congenital hand malformations. To our knowledge, this is the first report of the c.388 A > T (p.I130F) mutation in the GJA1 gene and the c.663 + 1 G > C mutation in the TBX5 gene, while the c.2247 G > A (p.W749X) mutation in the ROR2 gene was verified as a pathogenic mutation in Chinese population. These findings expand the mutation spectrum and the population specific variants frequency. This is also the first report that the same panel of targeted NGS was employed to perform molecular diagnosis of different subtypes of congenital hand malformations. Our study supported the application of a targeted NGS panel as an effective tool to detect the genetic cause for heterogeneous diseases in clinical diagnosis.
Methods
Targeted NGS approach
The probes used in this study were RNA and is about 120 bp. Targeted genes were chosen according to OMIM database and were designed by the MyGenostics company (Beijing, China).Genomic DNA was extracted from the peripheral blood lymphocytes of probands and their family members using the Qiagen genomic DNA isolation kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The genomic DNA of the probands was quantified by NanoDrop (Thermo, Waltham, USA). Informed consent was obtained from all subjects. This study was approved by the Ethics Committee of Henan Provincial People’s Hospital and conducted in accordance with relevant guidelines and regulations.
Libraries of probands used in targeted NGS were prepared as described previously33. 3 µg of genomic DNA of each proband was fragmented by nebulization, end-repaired, an ‘A’ was ligated to the 3′-end, the fragments were marked by Illumina adapters, and the size selected was performed aiming for a 350–400 base pair product. Then, the final products were amplified by PCR and validated using the Agilent Bioanalyzer.
The amplified DNA was captured with Target Enrichment System (MyGenostics, Beijing, China) according to the manufacturer’s protocol. To perform the hybridization, 1 µg of DNA library was mixed with Buffer BL and GenCap probe (MyGenotics, Beijing, China) to perform pre-hybridization, and Buffer HY (MyGenotics, Beijing, China) was added to the mixture for further hybridization. After hybridization, the target DNA was bound to MyOne beads (Thermo, Waltham, USA) and eluted with Buffer Elute. Then, the eluted DNA received final PCR and purification. The enrichment libraries were sequenced on Illumina HiSeq. 2000 sequencer (Illumina, San Diego, USA) for paired read 100 bp according to the manufacturer’s protocol.
Bioinformatics analysis
Clean reads were generated from raw data by removing the adapters and the low quality reads. The BWA program package was used to align the filtered reads to the human reference genome, and the GATK software package was employed to recalibrate quality scores and realign to reference. Sequence Alignment/Map tools 3 (SAMtools 3) was used to remove the duplicated reads. The uniquely mapped reads were used for variation detection.
Single nucleotide variants (SNVs) were detected with the GATK UnifiedGenotyper, and the variants were filtered by GATK VariantFiltration. Indels (insertions and deletions) were called by GATKIndelGenotyper V2 Annotation. Variants were annotated by an in-house developed bioinformatics tool with RefSeq (hg19, from UCSC) and UCSC annotation.
Several databases (dbSNP138, 1000 Genomes, and an in-house Asia database) were used to filter the SNPs/indels, which showed >5% frequency in these databases. Several bioinformatic software tools (SIFT, PoluPhen and MutationTaster) and several online websites (InterVar and Clinvar) were employed to predict the probable pathogenic mutation.
Sanger sequencing
Sanger sequencing was used to confirm these candidate pathogenic mutations. Primers were designed to amplify the mutations and flanking sequences (Supplementary Table S3). PCR products were purified with a gel extraction kit (Promega, Madison, USA) according to the manufacturer’s protocol, and sequenced on an ABI 3130 sequencer (Thermo, Waltham, USA). The sequencing results were analyzed by sequence analysis software.
References
Bates, S. J., Hansen, S. L. & Jones, N. F. Reconstruction of congenital differences of the hand. Plastic and reconstructive surgery 124, 128e–143e, https://doi.org/10.1097/PRS.0b013e3181a80777 (2009).
Yesilada, A. K., Sevim, K. Z., Sucu, D. O. & Kilinc, L. Congenital hand deformities - a clinical report of 191 patients. Acta chirurgiae plasticae 55, 10–15 (2013).
Thomas-Teinturier, C. et al. Report of two novel mutations in PTHLH associated with brachydactyly type E and literature review. American journal of medical genetics. Part A 170, 734–742, https://doi.org/10.1002/ajmg.a.37490 (2016).
Temtamy, S. A. & McKusick, V. A. The genetics of hand malformations. Birth defects original article series 14(i-xviii), 1–619 (1978).
Fitch, N. Classification and identification of inherited brachydactylies. Journal of medical genetics 16, 36–44 (1979).
Jordan, D., Hindocha, S., Dhital, M., Saleh, M. & Khan, W. The epidemiology, genetics and future management of syndactyly. The open orthopaedics journal 6, 14–27, https://doi.org/10.2174/1874325001206010014 (2012).
Malik, S. Syndactyly: phenotypes, genetics and current classification. European journal of human genetics: EJHG 20, 817–824, https://doi.org/10.1038/ejhg.2012.14 (2012).
Perez-Carro, R. et al. Panel-based NGS Reveals Novel Pathogenic Mutations in Autosomal Recessive Retinitis Pigmentosa. Scientific reports 6, 19531, https://doi.org/10.1038/srep19531 (2016).
Zhou, L. et al. Metacarpal Bone Plane Examination by Ultrasonography for the Diagnosis of Fetal Forearm and Hand Deformity. Scientific reports 7, 42161, https://doi.org/10.1038/srep42161 (2017).
Wang, Y. et al. Genetic Variants Identified from Epilepsy of Unknown Etiology in Chinese Children by Targeted Exome Sequencing. Scientific reports 7, 40319, https://doi.org/10.1038/srep40319 (2017).
Huang, H. et al. Targeted next generation sequencing identified novel mutations in RPGRIP1 associated with both retinitis pigmentosa and Leber’s congenital amaurosis in unrelated Chinese patients. Oncotarget 8, 35176–35183, https://doi.org/10.18632/oncotarget.17052 (2017).
Zhang, W. et al. Identification and functional analysis of a novel LHX1 mutation associated with congenital absence of the uterus and vagina. Oncotarget 8, 8785–8790, https://doi.org/10.18632/oncotarget.14455 (2017).
Santasree Banerjee, J. Y., Zhang, X., Niu, J. & Chen, Z. Next generation sequencing identified novel heterozygous nonsense mutation in CNGB1 gene associated with retinitis pigmentosa in a Chinese patient. Oncotarget 8 (2017).
Paznekas, W. A. et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. American journal of human genetics 72, 408–418, https://doi.org/10.1086/346090 (2003).
Bacchelli, C., Wilson, L. C., Cook, J. A., Winter, R. M. & Goodman, F. R. ROR2 is mutated in hereditary brachydactyly with nail dysplasia, but not in Sorsby syndrome. Clinical genetics 64, 263–265 (2003).
Oldridge, M. et al. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B. Nature genetics 24, 275–278, https://doi.org/10.1038/73495 (2000).
Ashbaugh, H. & Gellman, H. Congenital thumb deformities and associated syndromes. The Journal of craniofacial surgery 20, 1039–1044, https://doi.org/10.1097/SCS.0b013e3181abb1d8 (2009).
Hu, Y. et al. A novel autosomal recessive GJA1 missense mutation linked to Craniometaphyseal dysplasia. PloS one 8, e73576, https://doi.org/10.1371/journal.pone.0073576 (2013).
Gladwin, A. et al. Localization of a gene for oculodentodigital syndrome to human chromosome 6q22-q24. Human molecular genetics 6, 123–127 (1997).
You, G. et al. A novel GJA1 mutation identified by whole exome sequencing in a Chinese family with autosomal dominant syndactyly. Clinica chimica acta; international journal of clinical chemistry 459, 73–78, https://doi.org/10.1016/j.cca.2016.05.024 (2016).
Schrander-Stumpel, C. T., De Groot-Wijnands, J. B., De Die-Smulders, C. & Fryns, J. P. Type III syndactyly and oculodentodigital dysplasia: a clinical spectrum. Genetic counseling (Geneva, Switzerland) 4, 271–276 (1993).
Laird, D. W. Syndromic and non-syndromic disease-linked Cx43 mutations. FEBS letters 588, 1339–1348, https://doi.org/10.1016/j.febslet.2013.12.022 (2014).
Houlston, R. S. & Temple, I. K. Characteristic facies in type B brachydactyly? Clinical dysmorphology 3, 224–227 (1994).
Huang, D. et al. A new mutation in the gene ROR2 causes brachydactyly type B1. Gene 547, 106–110, https://doi.org/10.1016/j.gene.2014.06.035 (2014).
Afzal, A. R. & Jeffery, S. One gene, two phenotypes: ROR2 mutations in autosomal recessive Robinow syndrome and autosomal dominant brachydactyly type B. Human mutation 22, 1–11, https://doi.org/10.1002/humu.10233 (2003).
Masiakowski, P. & Carroll, R. D. A novel family of cell surface receptors with tyrosine kinase-like domain. The Journal of biological chemistry 267, 26181–26190 (1992).
Al-Qattan, M. M. & Abou Al-Shaar, H. Molecular basis of the clinical features of Holt-Oram syndrome resulting from missense and extended protein mutations of the TBX5 gene as well as TBX5 intragenic duplications. Gene 560, 129–136, https://doi.org/10.1016/j.gene.2015.02.017 (2015).
Basson, C. T. et al. Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proceedings of the National Academy of Sciences of the United States of America 96, 2919–2924 (1999).
Zaragoza, M. V. et al. Identification of the TBX5 transactivating domain and the nuclear localization signal. Gene 330, 9–18, https://doi.org/10.1016/j.gene.2004.01.017 (2004).
Shao, D. et al. A targeted next-generation sequencing method for identifying clinically relevant mutation profiles in lung adenocarcinoma. Scientific reports 6, 22338, https://doi.org/10.1038/srep22338 (2016).
Taniguchi-Ikeda, M. et al. Next-generation sequencing discloses a nonsense mutation in the dystrophin gene from long preserved dried umbilical cord and low-level somatic mosaicism in the proband mother. Journal of human genetics 61, 351–355, https://doi.org/10.1038/jhg.2015.157 (2016).
Wang, Y. et al. Next-generation sequencing-based molecular diagnosis of neonatal hypotonia in Chinese Population. Scientific reports 6, 29088, https://doi.org/10.1038/srep29088 (2016).
Qin, L. et al. A novel MIP mutation in familial congenital nuclear cataracts. European journal of medical genetics 59, 488–491, https://doi.org/10.1016/j.ejmg.2016.07.002 (2016).
Acknowledgements
We are very appreciative of the families’ participation in this study. This work was supported by the National Nature Science Foundation of China (grants 81650010 to S.L., 81270488 to G.Y., 81472681 to X.L.).
Author information
Authors and Affiliations
Contributions
X.L., S.L. and L.Q. designed the study. L.G., L.W., Q.H. and H.L. recruited patients and worked on the clinical study. L.Q., G.L., Y.Z. and H.W. performed the molecular diagnosis. X.L., S.L. and L.Q. wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, L., Lou, G., Guo, L. et al. Targeted next-generation sequencing-based molecular diagnosis of congenital hand malformations in Chinese population. Sci Rep 8, 12721 (2018). https://doi.org/10.1038/s41598-018-30940-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-30940-6
This article is cited by
-
A novel variant in the ROR2 gene underlying brachydactyly type B: a case report
BMC Pediatrics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
