
Abstract
Pseudomonas aeruginosa is a prevalent opportunistic pathogen that causes fatal infections in immunocompromised individuals. Quorum sensing (QS) is a cell-to-cell communication process that controls virulence gene expression and biofilm formation in P. aeruginosa. Here, the QS systems and the relevant virulence traits in clinical P. aeruginosa isolates were characterized. Eleven out of the ninety-four P. aeruginosa isolates exhibited a biofilm-deficient phenotype. Two biofilm-deficient isolates, one from blood and the one from pleural effusion, appeared to carry a same mutation in lasR. These two isolates differed in the ability to produce QS-regulated virulence factors, but contained the same functionally deficient LasR with the truncated C-terminal domains and belonged to the same multilocus sequence type (ST227). Chromosomal lasR complementation in these lasR mutants verified that lasR inactivation was the sole cause of las deficiency. LasR was not absolutely required for rhl signal in these lasR mutants, suggesting the presence of lasR-independent QS systems. We provided evidence that the virulence gene expression are not regulated in the same manner in these isolates. These results support the hypothesis that conventional QS hierarchy can be smashed by naturally occurring lasR mutation in clinical P. aeruginosa isolates and that complex QS hierarchy may play a role in maintaining infection of this opportunistic pathogen.
Similar content being viewed by others

Introduction
Pseudomonas aeruginosa is a notoriously opportunistic pathogen that causes considerable morbidity and mortality in immunocompromised patients1,2. It is particularly dangerous for patients with severe wounds, cystic fibrosis (CF), and cancer. The infection strategy of P. aeruginosa hinges on the production of numerous cell-associated and secreted molecules, including various proteases and toxins3,4,5,6. P. aeruginosa can also form a biofilm that prevents host defenses and increases 10–1000 fold resistance to antimicrobial treatment compared to the same strains in planktonic culture7,8.
P. aeruginosa uses intertwined cell-to-cell signaling or quorum sensing (QS) to sense bacterial cell density, regulate the expression of ~10% of its transcriptome9,10, and adapt to biofilm lifestyle7,8. Much of our understanding of P. aeruginosa QS comes from the analysis of laboratory strains, such as PAO1 and PA149,11. Three major QS systems are hierarchically arranged in P. aeruginosa12,13,14,15. At the top of the signaling cascade is the acyl-homoserine lactone (AHL)-based las system which contains the signal receptor, LasR, its cognate signal molecule or autoinducer 3-oxo-C12-homoserine lactone (OdDHL), and the OdDHL synthase, LasI. The second system is the AHL-based rhl system which contains RhlR, its autoinducer C4-homoserine lactone (C4HSL), and the C4HSL synthase, RhlI. The third QS system is the Pseudomonas quinolone signal (PQS) involving the binding of the receptor, PqsR, to 2-heptyl-3-hydroxy-4-quinolone14,16. The OdDHL-bound-LasR complex serves as a transcriptional regulator to activate the expression of QS genes, including lasR, lasI, rhlR, rhlI, and pqsR. It also positively regulates the production of virulence factors, such as elastase LasB17, exotoxin A18, pyocyanin19, and extracellular polymeric substrates (EPS)20. The latter of which is an important component of biofilm. PQS and the rhl system regulate each other21,22. Thus, the QS systems control the virulence gene transcription mainly via the master regulator, LasR, and the subordinate regulator, RhlR in P. aeruginosa.
Bacterial pathogens often require QS to establish and promote infection23. Several animal studies have shown the importance of QS systems in the pathogenesis of P. aeruginosa acute and chronic infections24,25,26,27. The significance of QS in human P. aeruginosa infection is less clear, because QS-deficient strains are frequently identified in respiratory acute and chronic infections28,29,30,31, which raises interesting questions about the necessity of QS in infection. Feltner et al.28 have reported that 22% of P. aeruginosa isolates from the lungs of CF patients are mutants and contain polypeptides that differed from LasR in laboratory strains. The presence of lasR mutants has been considered to be an adaptation to specific environments, such as the airways of CF patients. An increasing amount of clinical studies has linked lasR mutants to worsening disease progression of chronic and acute infections29,32. Therefore, better understanding the mechanisms underlying QS-mediated virulence in clinical P. aeruginosa isolates may aid in full elucidation of the P. aeruginosa pathogenesis.
The present study aimed at exploring the relationship between QS and the relevant virulence traits in clinical P. aeruginosa isolates derived from Chinese patients. Specially, two isolates, one from bloodstream and other one from pleural effusion, drew our interest, because they appeared to carry the same mutation in lasR and exhibited different virulence properties. Using molecular approaches and biological assays, these lasR mutants were characterized in comparison with the well-studied reference P. aeruginosa strain, PAO1. Our results explained the phenotypic variation observed in these lasR mutants and demonstrated that lasR deficiency can occur naturally in P. aeruginosa isolates. It is likely that complex QS hierarchy plays a role in maintaining the infection of this opportunistic pathogen.
Results
Phenotypic variation in clinical P. aeruginosa isolates
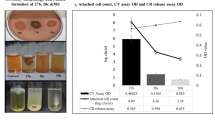
With the aim to characterize the QS-regulated virulence properties of clinical P. aeruginosa isolates, we, first, examined the ability to form biofilm using static biofilm assays33. A total of ninety-four P. aeruginosa isolates derived from varying cases of infections (Supplemental Table S1), including sepsis, ventilation-associated pneumonia, and wound infections, were analyzed. Each isolate was from a single specimen of an individual patient. We found that 83 out of 94 (88.3%) P. aeruginosa isolates formed a biofilm similar to or thicker than that of PAO1 (Fig. 1a). In contrast, 11 isolates formed defective or no biofilms (Fig. 1b).

Phenotypic analysis of the clinical P. aeruginosa isolates. (a) Biofilm forming proficiency of isolates from different sources (see Supplemental Table S1 for details of all clinical isolates). (b) Representative results of biofilm forming assays. The values of OD570 are presented as means ± standard deviations (SDs) of triplicate assays. The break line indicates OD570 value equal to 1/2 of that of PAO1. The isolates had the OD570 values ≤ 1/2 of that in PAO1 are defined as the biofilm-deficient. (c) Analysis of proteolytic activity using the milk plate assays. Measurement of the radius of the proteolytic zones after incubation at 37 °C for 48 h. Data are the radius values from clinical isolates (gray bar) reported as the mean ± SDs from three individual experiments with triplicates in each experiment. The proteolytic zones produced by strains, PAO1 (red bar) and the isogenic mutants deficient in elastase and/or four proteases (Supplemental Table S2) (black bar) were used as the controls.
We next assessed production of protease, which plays an important role in virulence, in these biofilm-deficient isolates by measuring the proteolytic activity on the milk plates17. P. aeruginosa PAO1 and its isogenic strains deficient in alkaline protease, protease IV, and/or elastase (Supplemental Table S2) were used as the controls. Three different patterns were observed (Fig. 1c): five isolates (cq94, bj4, bj8, Hz8, and Hz32) with strong proteolytic activity, three isolates (cq9, cq12, and bj13) with reduced activity, and three others (bj3, bj14, and Hz28) lacking proteolytic activity. The protease-absence was not fully correlated to the biofilm deficiency. This could be explained by the complex regulatory pathways governing of biofilm formation and/or protease production. Beside QS, other signaling pathways, such as two-component systems, also contribute to biofilm formation. Collectively, survey of the biofilm formation and proteolytic activity revealed the repertoire of phenotypic variation in these clinical isolates, reflecting changes in their biology.
Genomic variation in QS related genes
We asked whether the biofilm-deficiency phenotype was related to malfunction of the QS. The possession of QS genes, lasI, lasR, rhlI, and rhlR, was determined by PCR and DNA sequencing. All biofilm-deficient isolates produced PCR fragments with the predicted molecular sizes (Supplemental Fig. S1). DNA sequencing results showed that the intact lasI and rhlR genes, same as those in PAO1, were present in all isolates. A few point mutations in rhlI were found to alter amino acid residue (s): D83E (in all isolates), S62G (in six isolates), V37A in cq94, and P159S in Hz28 (Fig. 2a). The sequence conflicts with RhlI D83E and S62G in different stock of PAO1 have been reported previously11,34. It has been reported the mutation of D83E is unaffected on rhlI and its encoded product, C4HSL, in P. aeruginosa isolates from CF patient35. The mutants V37A/D83E or S62G/D83E/P159S might be disruptive to RhlI functionality because at least one of these residues was located in the predicted functional region in RhlI. Moreover, all isolates carried an intact lasR gene same as that of PAO1 with the exception of bj13 (from pleural effusion) and bj14 (from bloodstream). Both bj13 and bj14 carried a single mutation at nucleotide 180 (g → a) in lasR (Fig. 2b), leading to the change from a tryptophan codon (TGG) to a stop codon (TGA) and premature termination of LasR translation at 60 amino acids. This mutant was designated as LasRW60*.

Genotyping of clinical P. aeruginosa isolates that displayed a weakened biofilm formation. (a) Mutations in rhlI gene that resulted in changes in amino acid residues in biofilm-deficient isolates. (b) Mutation in lasR genes in two isolates, bj13 and bj14. The red box show the changes in the 180th nucleotide in lasR leading to LasRW60* mutation. All sequencing results were compared to reference PAO1 genome sequences. (c) PFGE pattern of clinical isolates. Genomic DNA extracted from the isolates were digested with SpeI. PAO1 genomic DNA digested with SpeI was used as a control.
The analysis of multiple locus sequencing typing (MLST) revealed that bj13 and bj14 had the same allelic profile of 39-5-9-11-27-5-2, corresponding to ST227, a rare ST clone in China. To further distinguish these and other biofilm-deficient isolates, pulsed-field gel electrophoresis (PFGE)36 was performed. All biofilm-deficient isolates were successfully typed by PFGE (Fig. 2c). Eight isolates, including bj13 and bj14, displayed distinct PFGE profiles, indicating the different clonality. Three isolates (bj8, Hz8, and Hz28) displayed a similar PFGE pattern. However, these isolates were from different patients and displayed different proteolytic activity (Fig. 1). Thus, each of the isolates was distinct.
Because the P. aeruginosa isolates were analyzed shortly after isolation without prolonged subculturing, the genotypic and phenotypic variations observed might emerge in vivo. With intact lasI, rhlR and rhlI, bj13 and bj14 appeared to have the same LasRW60* mutant and different proteolytic activity. These isolates may have evolved distinct QS circuits to regulate virulence factor production. To test this possibility, bj13 and bj14 were chosen for further study.
Impaired las signal in bj13 and bj14
A functional LasR contained the N-terminal autoinducer binding-dimerization domains and a C-terminal DNA binding domain37,38. To address whether the LasRW60* mutant was, indeed, a loss-of-function variant, the levels of OdDHL produced by LasI in bj13 and bj14 were assessed using reporter assays39. While PAO1 generated a high level of OdDHL, neither bj13 nor bj14 produced detectable OdDHL, the observation similar to the PAO1-derived lasR knockout, ΔlasR (Fig. 3b). To confirm that the decreased OdDHL was solely caused by the lack of LasR, we introduced wild-type lasR from PAO1 into the chromosome of bj13, bj14, and ΔlasR (for a control), respectively. We observed that the OdDHL levels were fully restored in the complemented strains, bj13lasR, bj14lasR, and ΔlasR+(Fig. 3b). These results are consistent with the intact lasI that produces OdDHL in the presence of LasR. Thus, loss-of-lasR-function was the cause of las deficiency in bj13 and bj14.

LasR dependence of OdDHL production. (a) Schematic diagram shows domain structure of LasR and the location of W60* mutation. (b) OdDHL levels assessed by a reporter assay. Extracts of autoinducer from the supernatant of bacterial culture grown for 18 hours were added into the agar plates containing the reporter strains. After incubation at 30 °C for 24 hours, the gray values were measured. The LasR mutants (bj13 and bj14) and the relevant LasR complemented strains (bj13lasR & bj14lasR) are shown for comparison. PAO1, ΔlasR, and the ΔlasR+were used as the controls. The gray values are presented as the mean ± SDs of tests performed in triplicates in three different occasions. ****P<0.0001; P > 0.05, ns (no significance). P values were obtained by one way ANOVA with Bonferroni test.
Altered rhl signal and QS-regulated gene expression in bj13 and bj14
We sought to assess the rhl signal in these lasR mutants by measuring the levels of C4HSL produced by RhlI using reporter assays39. It was found that the C4HSL levels in bj13 and bj14 were 39% and 44% of that detected in PAO1 (Fig. 4a), respectively, indicating that RhlI partially functioned in the absence of LasR in these two isolates. Chromosomal complementation with LasR had little effects on C4HSL product in bj14 or PAO1 background. However, the levels of C4HSL increased in strain bj13lasR compared to bj13, suggesting a role of LasR in Rhl activation. The failure of restoring for rhl signal in strains, bj14lasR and ΔlasR+, was not due to the poor expression of LasR, as the OdDHL production was restored in the same strains (Fig. 3). These results suggest that the rhl signal is not entirely relies on LasR dependent on different strains, consistent with the report by Dekimpe and Déziel16.

Difference in rhl-related gene expression in P. aeruginosa isolates. (a) C4HSL levels assessed by a reporter assay. The supernatants of P. aeruginosa culture grown for 18 hours were assayed for C4HSL levels. (b–d) The relative mRNA levels of rhlR,rhlA and pqsA in P. aeruginosa. The total RNAs from P. aeruginosa at the stationary phase were used for real time qRT-PCR analysis. Relative mRNA levels were obtained by normalizing the level of rhlR, rhlA and pqsA to that of the rplS. The representative data from the strains with backgrounds of PAO1 (b), bj13 (c), or bj14 (d) were reported as mean ± SD of quadruplicates in an experiment. Experiments were repeated for three times. ****P < 0.0001; P > 0.05, ns (no significance). P values were obtained by one way ANOVA with Bonferroni test.
It is not easy to explain the disparity in C4HSL results observed in strains bj13lasR and bj14lasR given that both have the intact rhlI and rhlR. To test whether rhl-mediated QS acts differently in bj13 and bj14, real-time qRT-PCR was performed to measure the mRNA levels of rhlR and its regulated rhlA, in the presence or absence of lasR. The rhlA is the first gene in rhlAB operon encoding rhamnosyl transferases. The rplS coding for the 50S ribosomal protein L19 was used as an internal control for quantitation of mRNA levels. We evaluated the growth curve of the isolates compared to PAO1 and found all strains entered into the stationary phase after growing for 10 h and longer in Luria-Bertani (LB) broth (Supplemental Fig. S2). For comparison, mRNA extracted from P. aeruginosa organisms grown for 6, 12, or 18 h were measured. Both bj13 and bj14 expressed a low level of rhlR regardless of the growth phase (Fig. 4 and Supplemental Fig. S3). There was no differences in rhlR levels between bj13 and bj13lasR or between bj14 and bj14lasR, implying little impact of LasR on rhlR expression in these strains. Whereas bj13 expressed a higher level of rlhA than bj14, the levels of rhlA were repressed in bj13lasR but increased in bi14lasR (Fig. 4 and Supplemental Fig. S3). This change in rhlA levels occurred only at the stationary phase, consistent with that QS-based transcriptional responses in P. aeruginosa increase at high bacterial density.
Curiously, PAO1 assumed as QS proficient expressed little rhlR and rhlA. Deleting lasR caused an increase in rhlA and unchanged rhlR levels in PAO1 at the stationary phase (Fig. 4b and Supplemental Fig. S3). Neither rhlA nor rhlR expression was altered in ΔlasR+ expressing LasR. Previous studies have shown that, in addition to interlined las/rhl, PQS can regulate rhlAB transcription in PAO116. To confirm this and to explore whether PQS was active in bj13 and bj14, we assessed the expression of pqsA, the first gene in the pqsABCDE operon necessary for PQS production. Indeed, the pqsA levels were increased in ΔlasR lacking LasR coupled to the upregulation of rhlA at the stationary phase, suggesting the role of PQS in the activation of rhlR in PAO1. However, we failed to detect the similar correlations between rhlA and pqsA in isolates, bj13 and bj14. Seemingly, PQS did not activate the rhl system in bj13 and bj14 background. These results verify that the QS circuits regulating rhl in bj13 and bj14 are different from those in PAO1.
Differences in the production of LasB, exotoxin A, and pyocynain in bj13 and bj14
To address whether QS-controlled virulence gene products are expressed differently in bj13 and bi14, we assessed the bacterial secreted LasB, pyocynain, and exotoxin A in the presence or absence of LasR. These virulence factors affect a variety of P. aeruginosa-host interactions. While LasB damages host tissue and promotes invasion17,40, exotoxin A causes host cell death by blocking protein synthesis6. Pyocynain affects the electron transport chain and interferes with host cell growth19,41.
Whereas the ability to produce LasB and pyocyanin was impaired in all lasR mutants, the activity was restored to a certain degree in lasR-complemented strains (Fig. 5a–c). The weak LasB elastinolytic activity in the culture supernatants from bj13 and bj14 (Fig. 5a) was not due to lasB mutation in these isolates. Despite two nucleotide mutations in lasB resulting in S241G and S435L in bj13 and bj14, none of these changes were at the active site for LasB function as reported previously42 (Supplemental Fig. S4). LasB with natural variant S241G have been found in P. aeruginosa strains, PA103 and N-10. Additionally, we noted that the lasR-induced increase in pyocyanin levels in bj13lasR was much higher than that in bj14lasR (fold changes ~5 vs 1.8 times) (Fig. 5c), indicating that regulation of pyocyanin production was different in these isolates. Moreover, a full length exotoxin A (69 kDa) band was readily detected form the cultural supernatant of bj13 and bj14 using immunoblotting analysis (Fig. 5d). Interestingly, in the presence of LasR, this exotoxin A band was less intense coupled to appearance of several faint, smaller antibody reactive bands. At the same time, more intense mature LasB band was detected in the same sample. Exotoxin A was also easily detected from strain, ΔlasR lacking lasR, but little was detected in PAO1 and ΔlasR+. It was unclear if these faint bands were the degraded product of the exotoxin A in the presence of the high levels of proteases, such as LasB. Adding bacterial protease inhibitors did not prevent the appearance of these faint bands. Together with the transcriptional studies described in Fig. 4, these data support that virulence factor production is regulated in different manners in isolates, bj14 and bj13.

Effects of LasR on production of virulence factors in P. aeruginosa isolates. (a) Complementation of a lasR mutant with the lasR restored proteolytic activity. Data represent the lysis zones on milk plate after 24 h of P. aeruginosa growth at 37 °C. (b) Analysis of LasB activity using ECR assays. Cell-free supernatant from bacterial culture grown for 18 hours were used. (c) Levels of pyocynain. Extraction of pyocyanin from the supernatant fraction of bacterial cultures grown for 48 hours were measured at 520 nm. Values of each experiment are presented as the mean ± SDs of tests performed in triplicate. Experiments were performed in three different occasions. ****P<0.0001; *P < 0.05. P values were obtained by one way ANOVA with Bonferroni test. (d) Immunoblotting analysis of secreted exotoxin A (ETA) and LasB. Equal amounts of proteins were loaded and subjected to SDS-PAGE. ETA and LasB were disclosed by anti-ETA antibody and anti-LasB antibody. Arrows show unknown small protein bands crossreacted with anti-ETA antibody. The ratios of ETA levels to LasB levels were shown underneath the blots. The blot of LasB and ExoA in strains PAO1, ΔlasR, and ΔlasR+were in the same gel. Strains bj13, bj13lasR, bj14 and bj14lasR were in the same gel and the full-length blots were shown in Supplemental Figure S5.
Discussion
To study QS and its regulated virulence factor expression, we started with survey of biofilm formation and protease production with clinical P. aeruginosa isolates. The screened biofilm-deficient isolates were then analyzed for the possession of QS-related genes and the genotypes. Finally, two unusual isolates, bj13 from pleural effusion and bj14 from bloodstream, were selected because of carrying the same LasRW60* mutant and intact lasI, rhlI, and rhlR genes. Despite the same lasR allele and the identical MLST, bj13 and bj14 exhibited clearly different PFGE patterns and virulence traits. We reasoned that bj13 was dissimilar to bj14. Their virulence properties were further characterized.
Of importance, we found that the virulence gene expressions at transcriptional level is regulated in the different manner in these isolates. Consistent with these observations, there were notable differences in the virulence products (i.e., protease, LasB, pyocyanin, and exotoxin A). Although LasR is known as a master positive virulence regulator in P. aeruginosa12,13,14,15, crossingregulation between the las and rhl systems were observed in these isolates. For example, the production of low but detectable C4HSL in the absence of LasR; as such, detection of C4HSL alone is not a reliable proxy for RhlR activity. Additionally, the lack of correlation between pqsA and rhlA levels suggest that PQS is unlikely the major regulator of the rhl system in these isolates. These observations reinforce that the complex QS circuits important for virulence factor expression in these isolates are different from that in PAO1. Previous studies of clinical isolate have demonstrated the las system is not necessarily crucial in some occasions28,29,32,35,43,44. The LasR-deficient P. aeruginosa isolates are prevalent in the lung of CF patients28 and the individuals with acute ventilation-associated pneumonia45. It is known that P. aeruginosa can genetically adjust the QS hierarchy and the relevant lifestyle when they colonize and infect at the epithelial cells28,46 or shift from acute to chronic mode of infections44. Loss of lasR control might give P. aeruginosa a selective advantage in pathogenic interactions with the hosts and minimize fitness cost at a bacterial population level. Deficiency in lasR may also have been offset by other highly interactive QS systems and/or global regulators, such as Vfr, VqsR, MvaT, GacA, RpoN, RpoS, and RsmA12. The presence of rhl signal without las signal in clinical isolates suggest that the use of las-independent QS activity may play a role in maintaining P. aeruginosa infection.
Many factors could directly contribute to multicellular adaptation and diversities in bacteria47. For example, patient immune response, antibiotic therapy, bacterial interspecies or interkingdom competition in microbiome, and the duration of bacterial infections. Selections of lasR-inactivation associated with the chronic CF airway infections28,35 and the more acute ventilator-associated pneumonia30 have been well studied. It is unknown what selection force are for losing lasR in bj13 and bj14. Since these isolates are originated from bloodstream and inflammatory pleural fluid, it is possible that exposure to host immunity and environmental factor (such as antibiotics) could be involved. Clinical studies have shown that P. aeruginosa may enhance the expression of some virulence genes and reduces the expression of others as part of its survival strategy. Hammond et al.32 reported that lasR inactivation was associated with the increased production of CupA fimbriae in the isolates from the patients with acute keratitis. We observed the decreased production of the total protease and LasB coupled to the high steady-state-levels of exotoxin A in lasR mutants. Thus, exploring whether the presence of steady-state-levels of exotoxin A in the lasR mutants are relevant in P. aeruginosa pathogenesis in vivo will be an interesting future study. Kruczek et al.9 revealed that P. aeruginosa strain, PA14, cultured with blood from severe burned patients displayed an altered transcriptome. Transcripts from QS genes and QS-regulated virulence factors were significantly downregulated. Conversely, genes encoding the type III secretion (T3S) system were increased, consistent with the previous reports indicating that the T3S effector is more important in the pathogenesis of the acute infections48. Whether bj13 and bj14 carrying lasR mutants utilize T3S to compensate for the reduced virulence factors during infection in vivo remains to be determined.
Our work is limited in scope but the findings suggest that additional investigation of the important QS networks in clinical isolates may produce fruitful advancements towards helping understanding P. aeruginosa pathogenesis. Central to P. aeruginosa’s success as an opportunistic pathogen is the genetic plasticity and the great metabolic capabilities provided by its large genome. To thoroughly identify the pathogenic determinants in bj13 and bj14, characterization of their genetic natures by whole genome sequencing is warranted. Our data did not reveal whether specific lasR mutants were associated with a particular patient population or type of diseases. Large sampling, prospective, longitudinal studies of the clinical isolates are needed to gain deeper insights into the evolution dynamics of P. aeruginosa. Nevertheless, the emergency of the lasR mutant in clinical isolates suggests that the las system, while important, is not absolutely essential for the establishment of P. aeruginosa infection in vivo. It also calls into question the previously accepted hierarchical understanding of the QS systems which apparently are much multifaceted in the clinical contexts.
Methods
Ethics Statement
This study was approved by the Institutional Review Board of Children’s Hospital, Chongqing Medical University (Approval No. 2017-61-1). Given that the data were collected and interpreted anonymously, the need for written informed consent from patients or their legal guardians was waived by the ethics committee.
Bacteria organisms
A total of 94 P. aeruginosa strains were isolated from patients admitted for treatment from 2011 to 2013 to three university teaching hospitals located in different cities. These isolates represented one-time isolates from individual patients receiving antimicrobial therapy. The source of the isolates are listed in Supplemental Table S1. They included 48 from sputa, 28 from blood, 11 from pus or skin wounds, 2 from peritoneal fluid, 2 from gall gladder effusion, 1 from pleural effusion, 1 from synovial fluid, and 1 from urine. Broth cultures of isolates were aliquoted to avoid repeated sub-culturing and then were stored at −80 °C. The two main clinical P. aeruginosa isolates, bj13 and bj14, and other bacteria strains used for detailed molecular analysis were list in Supplemental Table S2. P. aeruginosa strain, PAO140 and the isogenic strains deficient in alkaline protease, protease IV, and/or elastase were used for controls. Competent Escherichia coli DH5α cells were used as the host cells for molecular cloning. Bacteria were cultured in LB broth or LB agar plate at 37 °C supplemented with antimicrobials when appropriate. For selection of P. aeruginosa transformants, chloramphenicol 600 μg/ml, spectinomycin 600 μg/ml, or tetracycline 600 μg/ml were used. For other experiments, we used carbenicillin 100 μg/ml, tetracycline 30 μg/ml, chloramphenicol 25 μg/ml, and spectinomycin 50 μg/ml.
Plasmid, mutant construction, and complementation of mutants
The plasmids and primers used are listed in Supplemental Tables 2 and 3, respectively. To create plasmid pUCPRedCm, the Ahd I-Nar I fragment of pARC49 containing the promoter region and the coding region of chloramphenicol acetyltransferase was cloned into pUCP-Red at Ahd I-Nar I sites. Plasmids were transformed into P. aeruginosa by electroporation39. The bacteriophage lambda (λ) Red recombinase system50 was used for lasR mutant creation and complementation. To generate a lasR knockout in PAO1 background, the DNA fragment containing tetracycline resistant gene (tet) flanked by the 50 bp of homology regions to the target sequence was obtained by polymerase chain reaction (PCR) with primers DLRF/DLRR using plasmid pBR322 as the template. The purified DNA fragment was electroporated into PAO1, bj13 and bj14 carrying pUCPRedCm, resulting in strain ΔlasR, bj13ΔlasR and bj14ΔlasR. For chromosomal lasR complementation studies, a DNA fragment containing the cassette of lasR-aadA (conferring resistance to streptomycin and spectinomycin) flanked by 50 bp of homology regions to the target sequence was generated by overlap extension PCR with multiple steps. The lasR-containing DNA fragment was obtained from PAO1 genomic DNA with primers CLRF/CLRR. The aadA containing fragment was amplified from pCDFScc451 with primers CSPF/CSPR. Following the annealing of these two DNA fragments, the full-length lasR-aadA cassette was amplified with primers CLRF/CSPR, purified, and electroporated into strain ΔlasR, bj13ΔlasR and bj14ΔlasR; each of which carried the pUCPRedCm. The resultant complemented strains were designated as ΔlasR+, bj13lasR or bj14lasR. All gene regions of interest in the constructs were confirmed by DNA sequencing with primers PGDF and PGFR.
Bacterial genomic analysis
The QS-related genes, lasB, lasR, lasI, rhlR, and rhlI, were amplified by PCR with bacterial genomic DNA as the templates using appropriate primers (Supplemental Table S3). The purified PCR products were sequenced. Multilocus sequence typing (MLST) was conducted as proposed previously52. The fragments of the seven different central housekeeping genes, acsA, aroE, guaA, mutL, nuoD, ppsA, and trpE, were amplified by PCR using appropriate primers (Supplemental Table S3). The resulting PCR products were purified and subjected to DNA sequencing. The database at http://pubmlst.org was used to assign numbers to particular alleles and to identify sequence types. Pulsed-field gel electrophoresis (PFGE) was performed using P. aeruginosa genomic DNA digested with restriction enzyme, Spe I, as described previously36. All sequence comparison and primers design were based on the PAO1 genome sequence (www.pseudomonas.com).
Biofilm formation assays
A static microplate biofilm assay33 was performed in a polystyrene 96-well plate (Corning). Overnight cultures of P. aeruginosa were standardized to be diluted to OD600 = 0.5 in fresh LB to ensure that equal numbers of bacteria were present in each well. Bacteria were grown in triplicate in a 96-well plate at 37 °C for 48 hours without agitation. P. aeruginosa strain PAO1 was inoculated on each plate as a positive control and LB served as a blank. Plates were washed three times in distilled water to remove unattached bacteria. Wells were stained with 0.05% crystal violet (Sigma) for 10 min, then washed twice with water. Adherent stain was dissolved in 30% acetic acid for 15 min and then measured at 570 nm. The values of biofilm staining was determined by (OD570 sample−OD570 LB blank).
Proteolytic activity, LasB, and pyocyanin assays
Total proteolytic activity was determined using milk plate assays17. The LasB activity was measured by an Elastin Congo red (ECR) assay41. Bacteria cells were grown in LB broth at 37 °C for 18 h and removed by centrifugation. The supernatant fractions passed through a 0. 22 mm filter were subjected to ECR assay. The reaction liquids were centrifuged and then measured at 495 nm. Pyocyanin was extracted from the supernatant fraction of bacterial cultures grown for 48 hours and measured at 520 nm53.
Autoinducer measurement
The bioluminescence-based reporter assays were performed as described previously39. E. coli reporter strains, JM109/pSB1075 and JM109/pSB53654 (Supplemental Table S2) were used to measure the levels of OdDHL and C4HSL, respectively. The supernatant of P. aeruginosa culture grown for 18 hours was obtained for autoinducer extraction with ethyl acetate (acidified with 0.5% formic acid). The autoinducer containing extracts were dissolved in 50% methanol. Then, the 1:10 dilution of extracts in methanol was pipetted into LB agar plate containing reporter strains. After incubation at 30 °C for 24 h, the gray value formed were measured.
Real-time reverse transcription quantitative PCR (RT-qPCR)
P. aeruginosa grown in LB broth were collected after 6, 12, and 18 hours of growth. Total RNA was isolated using the RNA MiniPrep Kit (Zymo) according to the manufacturer’s instructions. DNA-free RNA (1 μg) was reverse transcribed into cDNA with 20 ng random hexamer primers using the high capacity cDNA reverse transcription Kit (Applied Biosystems). Amplification and quantification of cDNA were performed using the VeriQuest Fast SYBR Green qPCR Master Mix (USB). Primer pairs (Supplemental Table S3) with the amplicon sizes of 150 to 200 bp were designed for genes, rhlR, rhlA, pqsA, and rplS (as an internal control) using the Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3.cgi/primer3_www.cgi). The qPCR cycle conditions were as follows: 50 °C for 2 min, 95 °C for 2 min, 95 °C for 30 s, 59 °C for 1 min. The latter two steps were repeated for 30 cycles, and fluorescence was detected at the end of each cycle. The 2−ΔΔCT threshold cycle (CT) method was used to calculate the relative transcript levels of genes studied compared to rplS transcripts. Note, qPCR reactions were setup quadruplicates and the experiments were performed three times.
Western blot analysis
The supernatants of P. aeruginosa culture grown for 18 h (stationary phase) were obtained by centrifugation. The total proteins in supernatants were determined using BCA assays (ThermoFisher). Protein samples in SDS loading buffer containing β-mercaptoethanol were boiled at 98 °C for 5 minutes. Equal amounts of protein were loaded and separated by 10% SDS-PAGE, followed by transferring to a PVDF membrane. A rabbit polyclonal antibody specific to Pseudomonas exotoxin A (Sigma, catalog # P2318) or LasB55 were used to probe exotoxin A or LasB, respectively. The probed membranes were incubated with anti-rabbit horseradish peroxidase-conjugated IgG at room temperature for 1 hour and 30 min and developed using SuperSignal West Pico chemiluminescent substrate (Pierce).
Statistical analysis
Statistical analysis of data was conducted using GraphPad Prism 5.0 (La Jolla, CA, USA). A P value < 0.05 was considered significant.
References
Gellatly, S. L. & Hancock, R. E. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathogens and disease 67, 159–173, https://doi.org/10.1111/2049-632x.12033 (2013).
Young, L. S. Problems of studying infections in the compromised host. Reviews of infectious diseases 8(Suppl 3), S341 (1986).
Jimenez, P. N. et al. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiology and molecular biology reviews: MMBR 76, 46–65, https://doi.org/10.1128/mmbr.05007-11 (2012).
Moradali, M. F., Ghods, S. & Rehm, B. H. A. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Frontiers in Cellular and Infection Microbiology 7, 39, https://doi.org/10.3389/fcimb.2017.00039 (2017).
Nicas, T. I. & Iglewski, B. H. The contribution of exoproducts to virulence of Pseudomonas aeruginosa. Canadian journal of microbiology 31, 387–392 (1985).
Pollack, M. Pseudomonas aeruginosa exotoxin A. The New England journal of medicine 302, 1360–1362, https://doi.org/10.1056/nejm198006123022410 (1980).
Davies, D. Understanding biofilm resistance to antibacterial agents. Nature reviews Drug discovery 2, 114–122, https://doi.org/10.1038/nrd1008 (2003).
De Kievit, T. R. Quorum sensing in Pseudomonas aeruginosa biofilms. Environmental Microbiology 11, 279–288, https://doi.org/10.1111/j.1462-2920.2008.01792.x (2009).
Kruczek, C. et al. Major Transcriptome Changes Accompany the Growth of Pseudomonas aeruginosa in Blood from Patients with Severe Thermal Injuries. PloS one 11, e0149229, https://doi.org/10.1371/journal.pone.0149229 (2016).
Schuster, M., Lostroh, C. P., Ogi, T. & Greenberg, E. P. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. Journal of bacteriology 185, 2066–2079, https://doi.org/10.1128/jb.185.7.2066-2079.2003 (2003).
Latifi, A. et al. Multiple homologues of LuxR and LuxI control expression of virulence determinants and secondary metabolites through quorum sensing in Pseudomonas aeruginosa PAO1. Molecular microbiology 17, 333–343 (1995).
Balasubramanian, D., Schneper, L., Kumari, H. & Mathee, K. A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Research 41, 1–20, https://doi.org/10.1093/nar/gks1039 (2013).
Gambello, M. J. & Iglewski, B. H. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. Journal of bacteriology 173, 3000–3009 (1991).
Pesci, E. C. et al. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences of the United States of America 96, 11229–11234 (1999).
Pesci, E. C., Pearson, J. P., Seed, P. C. & Iglewski, B. H. Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. Journal of bacteriology 179, 3127–3132 (1997).
Dekimpe, V. & Déziel, E. Revisiting the quorum-sensing hierarchy in Pseudomonas aeruginosa: the transcriptional regulator RhlR regulates LasR-specific factors. Microbiology (Reading, England) 155, 712–723 (2009).
Hamood, A. N., Griswold, J. & Colmer, J. Characterization of elastase-deficient clinical isolates of Pseudomonas aeruginosa. Infection and immunity 64, 3154–3160 (1996).
Gambello, M. J., Kaye, S. & Iglewski, B. H. LasR of Pseudomonas aeruginosa is a transcriptional activator of the alkaline protease gene (apr) and an enhancer of exotoxin A expression. Infection and immunity 61, 1180–1184 (1993).
Mavrodi, D. V. et al. Functional Analysis of Genes for Biosynthesis of Pyocyanin and Phenazine-1-Carboxamide from Pseudomonas aeruginosa PAO1. Journal of bacteriology 183, 6454–6465, https://doi.org/10.1128/jb.183.21.6454-6465.2001 (2001).
Sakuragi, Y. & Kolter, R. Quorum-Sensing Regulation of the Biofilm Matrix Genes (pel) of Pseudomonas aeruginosa. Journal of bacteriology 189, 5383–5386 (2007).
Mcknight, S. L., Iglewski, B. H. & Pesci, E. C. The Pseudomonas quinolone signal regulates rhl quorum sensing in Pseudomonas aeruginosa. Journal of bacteriology 182, 2702–2708 (2000).
Wade, D. S. et al. Regulation of Pseudomonas Quinolone Signal Synthesis in Pseudomonas aeruginosa. Journal of bacteriology 187, 4372 (2005).
Rutherford, S. T. & Bassler, B. L. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harbor Perspectives in Medicine 2, 705–709 (2012).
Rumbaugh, K. P., Griswold, J. A. & Hamood, A. N. Contribution of the regulatory gene lasR to the pathogenesis of Pseudomonas aeruginosa infection of burned mice. The Journal of burn care & rehabilitation 20, 42–49 (1999).
Bondi, R. et al. Affecting Pseudomonas aeruginosa phenotypic plasticity by quorum sensing dysregulation hampers pathogenicity in murine chronic lung infection. PloS one 9, e112105, https://doi.org/10.1371/journal.pone.0112105 (2014).
Yanagihara, K. et al. Role of elastase in a mouse model of chronic respiratory Pseudomonas aeruginosa infection that mimics diffuse panbronchiolitis. Journal of medical microbiology 52, 531–535, https://doi.org/10.1099/jmm.0.05154-0 (2003).
Lesprit, P. et al. Role of the quorum-sensing system in experimental pneumonia due to Pseudomonas aeruginosa in rats. Am J Respir Crit Care Med 167, 1478–1482 (2003).
Feltner, J. B. et al. LasR Variant Cystic Fibrosis Isolates Reveal an Adaptable Quorum-Sensing Hierarchy in Pseudomonas aeruginosa. mBio 7, https://doi.org/10.1128/mBio.01513-16 (2016).
Hoffman, L. R. et al. Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society 8, 66–70, https://doi.org/10.1016/j.jcf.2008.09.006 (2009).
Köhler, T., Guanella, R., Carlet, J. & van Delden, C. Quorum sensing-dependent virulence during Pseudomonas aeruginosa colonisation and pneumonia in mechanically ventilated patients. Thorax 65, 703–710, https://doi.org/10.1136/thx.2009.133082 (2010).
Kohler, T., Buckling, A. & van Delden, C. Cooperation and virulence of clinical Pseudomonas aeruginosa populations. Proceedings of the National Academy of Sciences of the United States of America 106, 6339–6344, https://doi.org/10.1073/pnas.0811741106 (2009).
Hammond, J. H. et al. Environmentally Endemic Pseudomonas aeruginosa Strains with Mutations in lasR Are Associated with Increased Disease Severity in Corneal Ulcers. mSphere 1, https://doi.org/10.1128/mSphere.00140-16 (2016).
Merritt, J. H., Kadouri, D. E. & O’Toole, G. A. Growing and Analyzing Static Biofilms. Current protocols in microbiology 01, Unit-1B.1, https://doi.org/10.1002/9780471729259.mc01b01s00 (2005).
Ochsner, U. A. & Reiser, J. Autoinducer-mediated regulation of rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences of the United States of America 92, 6424–6428 (1995).
Bjarnsholt, T. et al. Quorum sensing and virulence of Pseudomonas aeruginosa during lung infection of cystic fibrosis patients. PloS one 5, e10115, https://doi.org/10.1371/journal.pone.0010115 (2010).
Tenover, F. C. et al. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. Journal of clinical microbiology 33, 2233–2239 (1995).
Bottomley, M. J., Muraglia, E., Bazzo, R. & Carfì, A. Molecular Insights into Quorum Sensing in the Human Pathogen Pseudomonas aeruginosa from the Structure of the Virulence Regulator LasR Bound to Its Autoinducer. Journal of Biological Chemistry 282, 13592–13600, https://doi.org/10.1074/jbc.M700556200 (2007).
Kiratisin, P., Tucker, K. D. & Passador, L. LasR, a transcriptional activator of Pseudomonas aeruginosa virulence genes, functions as a multimer. Journal of bacteriology 184, 4912–4919 (2002).
Yu, H. et al. Ndk, a novel host-responsive regulator, negatively regulates bacterial virulence through quorum sensing in Pseudomonas aeruginosa. Scientific reports 6, 28684, https://doi.org/10.1038/srep28684 (2016).
Cowell, B. A., Twining, S. S., Hobden, J. A., Kwong, M. S. F. & Fleiszig, S. M. J. Mutation of lasA and lasB reduces Pseudomonas aeruginosa invasion of epithelial cells. Microbiology (Reading, England) 149, 2291–2299, https://doi.org/10.1099/mic.0.26280-0 (2003).
Lau, G. W., Hassett, D. J., Ran, H. & Kong, F. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends in molecular medicine 10, 599–606, https://doi.org/10.1016/j.molmed.2004.10.002 (2004).
Mciver, K. S., Kessler, E. & Ohman, D. E. Identification of residues in the Pseudomonas aeruginosa elastase propeptide required for chaperone and secretion activities. Microbiology (Reading, England) 150, 3969–3977 (2004).
D’Argenio, D. A. et al. Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Molecular microbiology 64, 512–533, https://doi.org/10.1111/j.1365-2958.2007.05678.x (2007).
Smith, E. E. et al. Genetic Adaptation by Pseudomonas aeruginosa to the Airways of Cystic Fibrosis Patients. Proceedings of the National Academy of Sciences of the United States of America 103, 8487–8492 (2006).
Kohler, T., Perron, G. G., Buckling, A. & van Delden, C. Quorum sensing inhibition selects for virulence and cooperation in Pseudomonas aeruginosa. PLoS pathogens 6, e1000883, https://doi.org/10.1371/journal.ppat.1000883 (2010).
Sousa, A. M. & Pereira, M. O. Pseudomonas aeruginosa Diversification during Infection Development in Cystic Fibrosis Lungs—A Review. Pathogens (Basel, Switzerland) 3, 680–703, https://doi.org/10.3390/pathogens3030680 (2014).
de la Fuente-Nunez, C., Reffuveille, F., Fernandez, L. & Hancock, R. E. Bacterial biofilm development as a multicellular adaptation: antibiotic resistance and new therapeutic strategies. Current opinion in microbiology 16, 580–589, https://doi.org/10.1016/j.mib.2013.06.013 (2013).
Hauser, A. R. The Type III Secretion System of Pseudomonas aeruginosa: Infection by Injection. Nature reviews. Microbiology 7, 654–665, https://doi.org/10.1038/nrmicro2199 (2009).
Rao, X. C. et al. A regulator from Chlamydia trachomatis modulates the activity of RNA polymerase through direct interaction with the β subunit and the primary σ subunit. Genes & Development 23, 1818 (2009).
Lesic, B. & Rahme, L. G. Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC molecular biology 9, 20–20, https://doi.org/10.1186/1471-2199-9-20 (2008).
Shen, L. et al. Multipart Chaperone-effector Recognition in the Type III Secretion System of Chlamydia trachomatis. Journal of Biological Chemistry 290, 28141–28155 (2015).
Curran, B., Jonas, D., Grundmann, H., Pitt, T. & Dowson, C. G. Development of a multilocus sequence typing scheme for the opportunistic pathogen Pseudomonas aeruginosa. J Clin Microbiol. Journal of clinical microbiology 42, 5644–5649 (2005).
Schaber, J. A. et al. Analysis of quorum sensing-deficient clinical isolates of Pseudomonas aeruginosa. Journal of medical microbiology 53, 841–853, https://doi.org/10.1099/jmm.0.45617-0 (2004).
Winson, M. K. et al. Construction and analysis of luxCDABE-based plasmid sensors for investigating N-acyl homoserine lactone-mediated quorum sensing. FEMS microbiology letters 163, 185–192 (1998).
Yu, H. et al. Elastase LasB of Pseudomonas aeruginosa promotes biofilm formation partly through rhamnolipid-mediated regulation. Canadian journal of microbiology 60, 227–235, https://doi.org/10.1139/cjm-2013-0667 (2014).
Acknowledgements
We thank Dr. Jeffery Hobden (Louisiana State University Health Sciences Center, New Orleans) for kindly providing P. aeruginosa reference strains and help with editing the manuscript. This work is supported by the funds from the Graduate Student Innovation Project in Chongqing, China, CYS14127, National Natural Science Foundation of China 81370777, and the Louisiana State University School of Medicine Dean’s Research Bridge Funding.
Author information
Authors and Affiliations
Contributions
Y.W. and L.G. contributed equally to the manuscript. Y.W. and L.S. designed the study and wrote the manuscript; Y.W., L.G., X.R., Jing W., J.J. and W.Z. performed experiments; Jing W., J.J., J.W., Y.X. collected the strains; Y.W., L.G., L.S., Z.H., and X.R. analyzed the data and interpreted the results; H.Y. and K.Z. provide reagents. Y.Z. and M.L. provided technical assistance. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Gao, L., Rao, X. et al. Characterization of lasR-deficient clinical isolates of Pseudomonas aeruginosa. Sci Rep 8, 13344 (2018). https://doi.org/10.1038/s41598-018-30813-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-30813-y
This article is cited by
-
Ketoprofen attenuates Las/Rhl quorum-sensing (QS) systems of Pseudomonas aeruginosa: molecular and docking studies
Molecular Biology Reports (2024)
-
Tackling recalcitrant Pseudomonas aeruginosa infections in critical illness via anti-virulence monotherapy
Nature Communications (2022)
-
Quorum sensing systems and related virulence factors in Pseudomonas aeruginosa isolated from chicken meat and ground beef
Scientific Reports (2021)
-
Iron limitation by transferrin promotes simultaneous cheating of pyoverdine and exoprotease in Pseudomonas aeruginosa
The ISME Journal (2021)
-
Quorum sensing systems, related virulence factors, and biofilm formation in Pseudomonas aeruginosa isolated from fish
Archives of Microbiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
