
Abstract
We have developed a series of yellow genetically encoded Ca2+ indicators for optical imaging (Y-GECOs) with inverted responses to Ca2+ and apparent dissociation constants (Kd′) ranging from 25 to 2400 nM. To demonstrate the utility of this affinity series of Ca2+ indicators, we expressed the four highest affinity variants (Kd′s = 25, 63, 121, and 190 nM) in the Drosophila medulla intrinsic neuron Mi1. Hyperpolarization of Mi1 by optogenetic stimulation of the laminar monopolar neuron L1 produced a decrease in intracellular Ca2+ in layers 8–10, and a corresponding increase in Y-GECO fluorescence. These experiments revealed that lower Kd′ was associated with greater increases in fluorescence, but longer delays to reach the maximum signal change due to slower off-rate kinetics.
Similar content being viewed by others


Introduction
Genetically encoded fluorescent protein (FP)-based calcium ion (Ca2+) indicators are widely used for non-invasive monitoring of intracellular signaling dynamics in systems ranging from cultured cells to live animals1,2,3,4. Directed protein evolution has proven to be a highly effective strategy to develop Ca2+ indicators with altered fluorescence hues5,6,7 or improved performance8,9,10.
We previously introduced a first-generation microfluidic fluorescence activated cell sorter (μFACS) platform for directed evolution of FP-based Ca2+ indicators with higher throughput than typical manual screening of bacterial colonies11. This platform was applied to the development of yellow genetically encoded Ca2+ indicators for optical imaging (Y-GECO, Fig. 1a) based on mPapaya12, a monomeric variant of the Zoanthus sp. yellow FP13. Among the inverse (becoming dimmer upon binding Ca2+) indicators, we identified variants with both fast (Y-GECO1f) and medium (Y-GECO1, designated as Y-GECO1m in the following discussions) dissociation kinetics11. The Y-GECO1f indicator exhibited fast Ca2+-dissociation kinetics with koff = 9.75 s−1, which compares favorably to Y-GECO1m (koff = 1.40 s−1) and GCaMP6f9 (koff = 2.32 s−1) measured under the same conditions. However, the improved kinetics were also associated with substantially decreased Ca2+ affinity (Kd′ = 2.5 μM for Y-GECO1f vs. 190 nM for Y-GECO1m), and Ca2+-dependent fluorescence change (i.e., the change for Y-GECO1f is ~32% that of Y-GECO1m). The inverse response of the Y-GECO series of indicators is similar to that of inverse-pericam (IP)4 and the recently reported IP2.0 (ref.14).

Development of new Y-GECO Ca2+ indicators. (a) Gene structure of Y-GECO. (b) Single mutation of M300I, Q276D, and L309F and combinations thereof were introduced into Y-GECO1m. The ratio of R526/416 in 10 mM EGTA to R526/416 in 141 nM Ca2+ was evaluated for Y-GECO1m variants. R526/416 is the ratio of fluorescence excited at 526 nm to fluorescence excited at 416 nm. (c) Ca2+ titrations for Y-GECO1m-derived variants. Data points were fit to Hill equation to determine Kd′. (d) Screening of Y-GECO2f [ref.15] variants with mutations at the same positions as in (b). The ratio of R526/416 in 10 mM EGTA to R526/416 in 1.27 μM Ca2+ was evaluated for all variants. The variant with all 3 mutations was designated as Y-GECO2m. (e) Ca2+ titration of Y-GECO2m. Y-GECO2f data from ref.15. Triplicate measurements were performed and the results were averaged. Error bar represents the standard deviation of each set of measurements.
To develop Ca2+ indicators tailored to specific applications in biology, we have used site-directed mutagenesis to modify the Ca2+ affinities and off-rate kinetics of Y-GECO variants. We demonstrate that these inverse response indicators are particularly well-suited for imaging of inhibitory neuronal signaling associated with transient decreases in intracellular Ca2+ concentration in neurons within the Drosophila visual system.
Results
Development of Y-GECO variants with higher affinity and slower kinetics
To expand the Y-GECO series of indicators to include variants with higher affinity for Ca2+, we explored the introduction of three previously reported mutations, M300I, Q276D, and L309F, in Y-GECO1m (Kd′ = 190 nM). One of these mutations (M300I) represents the reversion of a mutation in the first Ca2+-binding site of Y-GECO that was acquired during the earlier development of Y-GECO1f11. The other two mutations were reported to contribute to the increased Ca2+ affinity of jRGECO1a10. Individually, all three mutations increased the affinity to Ca2+ (Fig. 1b). We designated Y-GECO1m M300I as Y-GECO2s (Kd′ = 120 nM), Y-GECO1m Q276D M300I as Y-GECO2.1 s (Kd′ = 63 nM), and Y-GECO1m Q276D M300I L309F as Y-GECO2.2s (Kd′ = 25 nM) (Fig. 1c and Supplementary Tables 1–3). These variants are designated with an ‘s’ due to the slower kinetics (Supplementary Fig. 1a), which are a consequence of their higher Ca2+ affinities. These Y-GECO variants exhibited very similar pH dependence (Supplementary Fig. 2b–d).
Development of Y-GECO variants with lower affinity and faster kinetics
In an effort to identify variants with fast and large fluorescent responses, we developed and used a μFACS system based on a polydimethylsiloxane (PDMS) microchip with a two-point detection system (work described in ref.15). This system allowed us to screen libraries of randomly mutated Y-GECO1f variants expressed in Escherichia coli. Use of this system led to the identification of Y-GECO2f (Supplementary Table 1) that is 26% brighter in the Ca2+-free state, and exhibits a greater than 300% larger Ca2+-dependent fluorescence decrease relative to Y-GECO1f (Supplementary Table 2) while retaining a similar spectral profile. Y-GECO2f retained a similar Ca2+ affinity and slightly slower koff than Y-GECO1f (Supplementary Table 3).
While Y-GECO1f and Y-GECO2f exhibit relatively fast kinetics, they also share relatively high apparent dissociation constants of 2.5 μM and 2.2 μM, respectively. In an effort to lower the Kd′ to better match the typical concentrations of Ca2+ in cytoplasm (~0.1 to 1 μM), we introduced the three mutations discussed earlier (M300I, Q276D, and L309F) (Fig. 1d). Ultimately we found that the combination of all three mutations together gave the highest affinity, with a Kd′ of 204 nM (Fig. 1e). Accordingly, Y-GECO1f Q276D M300I L309F was designated as Y-GECO2m (Supplementary Table 1). As expected for a higher affinity variant, the Ca2+ dissociation kinetics of Y-GECO2m had slowed and were similar to Y-GECO1m (Supplementary Fig. 1b and Supplementary Table 3). Relative to Y-GECO1m, Y-GECO2m exhibits a larger Ca2+-dependent fluorescence change (over 200% increase) when excited at 526 nm (Supplementary Fig. 3), due to the shift of pKa in the presence of Ca2+ towards a higher value (Supplementary Fig. 2a and Supplementary Table 2).
Imaging of new Y-GECO variants in cultured cells
To demonstrate the performance of the most promising of the new Y-GECO variants for live cell imaging, genes were expressed in cultured HeLa cells and dissociated rat hippocampal cells, and fluorescence was imaged using wide-field illumination. With their near optimal Kd′s for detection of cytosolic Ca2+ concentration changes, and reasonable dissociation kinetics, Y-GECO1m (Kd′ = 190 nM) and Y-GECO2m (Kd′ = 204 nM) are promising indicators for imaging of Ca2+ dynamics in cultured cells. Expression in HeLa cells and treatment with histamine resulted in large oscillations in fluorescence intensity and excitation ratio. To determine the maximal changes, cells were treated with EGTA/ionomycin to deplete intracellular Ca2+ and then Ca2+/ionomycin to saturate the indicator. These treatments resulted in intensity changes of 10-fold for Y-GECO2m and 6-fold for Y-GECO1m (Fig. 2a and Supplementary Table 4). Ratiometric imaging with alternating 438 nm and 480 nm excitation revealed that Y-GECO2m gives ratiometric changes approximately 2-fold greater than that of Y-GECO1m (maximal ratio changes of 35-fold for Y-GECO2m vs. 18-fold for Y-GECO1m) (Fig. 2b,c and Supplementary Table 4). Analogous experiments with Y-GECO2s, another variant with an appropriate affinity (Kd′ = 121 nM) for imaging Ca2+ dynamics in cultured cells, revealed average intensity and ratio changes similar to those of Y-GECO2m (Supplementary Table 4). The intensiometric response of Y-GECO2m (10-fold) and Y-GECO2s (11-fold), when treated with EGTA/ionomycin followed by Ca2+/ionomycin, is smaller than that of the most widely used Ca2+ indicator GCaMP6s9, which we have previously determined to have a 20-fold change under similar conditions11. However, the ratiometric response of these Y-GECO variants does provide signal changes that are comparable to the intensiometric changes of GCaMP6s. Y-GECO2m also proved effective for imaging of slow Ca2+ waves when expressed in glial cells in dissociated rat hippocampal cultures (Fig. 2d).

Imaging of new Y-GECO2m. (a–c) Fluorescence images of HeLa cells expressing Y-GECO2m. For each panel, the right hand chart shows fluorescence signals for the cell enclosed with a dashed-line in response to histamine-induced Ca2+ oscillations. (a) Fluorescence with excitation at 480 nm. (b) Fluorescence with excitation at 440 nm. (c) Ratiometric response (excitation at 440 nm/excitation at 480 nm). (d) Fluorescent image of glial cells in dissociated rat hippocampal culture expressing Y-GECO2m. The fluorescence responses of selected regions to the spontaneous Ca2+ changes over time are demonstrated in the traces at the right side in the same color (excitation at 480 nm). Quantitative measurements of fluorescence responses in HeLa cells is provided in Supplementary Table 4.
Imaging of neuronal inhibition in Drosophila using the YGECO2s series
Since all Y-GECO variants exhibit an inverted response to Ca2+, we reasoned that they would be useful as positive indicator for detecting inhibitory (hyperpolarizing) responses, possibly enabling transient decreases in Ca2+ to be imaged with improved sensitivity. This application is similar to one reported by Hara-Kuge et al., who recently applied the IP2.0 inverse response indicator to visualize neuronal in the AWCON neurons of Caenorhabditis elegans14. To explore this possibility and to examine the dependence of the response on indicator Kd′, we expressed a series of Y-GECO variants (specifically, those with the highest Ca2+-binding affinities) in the Drosophila melanogaster Mi1 neuron. It has previously been demonstrated that the visual system of Drosophila is split into two pathways: the L1 “ON” pathway for light increment, and the L2 “OFF” pathway for light decrement16. L1 and L2 cells in the lamina serve as primary synaptic targets of photoreceptors and send input signals to neurons in medulla such as Mi1 (for L1) and Tm1 (for L2). The Mi1 neuron acts in the ON circuit, depolarizing when light increases and hyperpolarizing when light decreases17,18. Here, we mimic the light decrease by optogenetically stimulating the L1 neuron that makes an inhibitory synaptic connection to Mi119 (Fig. 3a). When inhibited, the free Ca2+ levels in Mi1 drop, a response thus far observed with Ca2+ indicators that increase fluorescence in response to increases in Ca2+ concentration19.

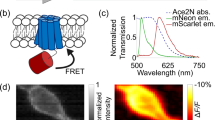
Imaging of Mi1 activation upon optogenetic activation of L1. (a) Schematic (modified from Strother et al.)18 featuring an L1 neuron (red) projecting from lamina to the medulla where it connects to an Mi1 neuron in layer 4. The Y-GECO response in Mi1 is measured in a region-of-interest (ROI) (faded yellow) spanning layers 8–10 (Supplementary Fig. 4b). (b) Y-GECO fluorescence was imaged using 2-photon excitation at 920 nm, while optogenetic activation of Chrimson was achieved using 1-photon excitation at 660 nm. (c) Image of L1 neurons labeled with Chrimson-tdTomato (red) and Mi neurons expressing Y-GECO1m (green). (d) Red light triggered Chrimson activation in L1 over durations spanning 1, 2, 4, and 8 seconds and presented here in the order they occurred during the protocol (Supplementary Fig. 4c). The median ∆F/F response for each time period is shown. Raw data is provided in Supplementary Fig. 5. (e,f) The response properties to the 1, 2, 4, and 8 second stimulations for the four Y-GECO variants are summarized. (e) The maximum ∆F/F reveals a trend for higher ∆F/F in inhibitory responses for indicators with a lower Kd. Kruskal-Wallis H-test found a significant difference (P = 0.0097) among the variants only over the 8 s time period. Post-hoc analysis using paired Wilcoxon ranksum between all variant combinations only found a significant difference between Y-GECO1m and Y-GECO2.2s (P = 0.0278, as indicated by *). (f) The delay to maximum signal. Kruskal-Wallis H-test found significant difference (P = 0.0033) among the variants only over the 8 s time period. Post-hoc analysis using paired Wilcoxon ranksum between all variant combinations only found significant differences between Y-GECO1m and Y-GECO2.1s (P = 0.0404) and Y-GECO1m vs Y-GECO2.2s (P = 0.0063). (g) The signal-to-noise ratio is the maximum ΔF/F signal divided by the baseline standard deviation (5 s period before simulation started). Lines for Y-GECO variants are colored as in (d). Numbers tested are: Y-GECO1m = 5, Y-GECO2s = 9, Y-GECO2.1 s = 8, and Y-GECO2.2s = 9.
We used the Gal4/UAS system to express Chrimson20 in L1 neurons and the LexA/LexAOP system to express Y-GECO1m (Kd′ = 190 nM), Y-GECO2s (Kd′ = 120 nM), Y-GECO2.1 s (Kd′ = 63 nM), or Y-GECO2.2s (Kd′ = 25 nM) in Mi1 neurons (Supplementary Fig. 4a). We chose to use Y-GECO1m, rather than Y-GECO2m, due to its higher Ca2+ affinity and faster koff. Chrimson activation with red light stimulation (660 nm, 1-photon) occurred every 30 s at a constant intensity of 0.24 mW/mm2 with duration increasing from 1, to 2, to 4, to 8 s, followed by a final 1 s pulse (Supplementary Fig. 4c). During activation, Y-GECO fluorescence was imaged using 2-photon excitation (920 nm) in ex vivo preparations, in an ROI spanning medulla layers 8–10 where the dendritic arbors from Mi1 provide the largest grouped area (Fig. 3a–c and Supplementary Fig. 4b).
These experiments revealed that fluorescence response (∆F/F) of a particular Y-GECO variant corresponded with the variant’s Kd′ and the length of the stimulation period (Fig. 3d,e). The correspondence between Kd′ and fluorescence response is best observed during the 8 s stimulation period. For this stimulation period, the highest affinity indicator, Y-GECO2.2s, exhibited the greatest ∆F/F, followed by Y-GECO2.1s, then Y-GECO2s, and finally Y-GECO1m. All variants exhibited an increasing delay to maximum signal with increasing stimulation period (Fig. 3f). Y-GECO2.2s exhibited the greatest signal to noise ratio (SNR) for the 8 s stimulation period but the two variants with intermediate Ca2+ affinity, Y-GECO2s and Y-GECO2.1 s, exhibited higher SNR at the 4 s stimulation, and similar SNR at 2 s and 1 s stimulation (Fig. 3g).
An effort was made to further characterize Y-GECO variants in vivo in Drosophila using visual stimulation. In this experiment, the cuticle was removed from the back of the head to allow the Mi1 neurons to be imaged. We placed a blue light LED in front of the fly eye and used 2-photon excitation (920 nm) to image fluorescence from GCaMP6s and Y-GECO-series Ca2+ indicators in Mi1 during stimulus periods when the light intensity was ramped down to give the maximum response. Using this approach, we were consistently able to identify responsive neurons when we used GCaMP6s but not when we used Y-GECO variants.
Discussion
New Y-GECO variants with a broad range of Ca2+ affinities
The Y-GECO series now contains a total of seven variants with Kd′ values ranging over 2 orders of magnitude. From highest to lowest affinity, this series includes: Y-GECO2.2s (Kd′ = 25 nM); Y-GECO2.1 s (Kd′ = 63 nM); Y-GECO2s (Kd′ = 121 nM); Y-GECO1m (Kd′ = 190 nM); Y-GECO2m (Kd′ = 204 nM); Y-GECO2f (Kd′ = 2200 nM); and Y-GECO1f (Kd′ = 2500 nM). For monitoring of neural activity with a cytosolic indicator, Ca2+ Kd′ values in the 100 to 200 nM range have been empirically found to be close to ideal, as demonstrated by the GCaMP series of highly optimized indicators9. Due to the fundamental relationship Kd = koff/kon, faster Ca2+ dissociation kinetics (described by rate constant koff) must be associated with a higher Kd, assuming no changes in Ca2+ association kinetics (described by rate constant kon). Accordingly, all genetically encoded Ca2+ indicators for neural activity imaging represent compromises between Kd′ (lower is better) and koff (higher is better). As the Y-GECO series of indicators all share very similar spectral properties, they provide researchers with the opportunity to empirically test and identify the particular indicator that is best tuned to the respond to intracellular Ca2+ dynamics under investigation. In addition, their inverse response behavior means that decreases in Ca2+ concentration associated with hyperpolarization will be reported as increasing fluorescence signals, which are generally preferred for tissue imaging. Hyperpolarization causes a decrease in Ca2+ only if voltage-gated channels (e.g., T-type Ca2+ channels) are partially active at resting potential and can be further inactivated by hyperpolarization.
Inverse response Ca2+ indicators for visualization of hyperpolarization
In vivo electrophysiological recordings17 and imaging with a genetically encoded voltage indicator19, have been used to probe depolarization of the L1 neuron of the Drosophila visual pathway, as induced by a light to dark transition16. In response to L1 depolarization, the Mi1 neuron hyperpolarizes, clearly demonstrating an inhibitory contact between L1 and Mi1. In vivo Ca2+ imaging with GCaMP6f9 has revealed that the intracellular Ca2+ concentration follows the membrane polarization (i.e., a Ca2+ decrease below resting levels during hyperpolarization), in layer M10 of stimulated Mi1 neurons19.
To further probe Ca2+ signaling in the Mi1 neuron, we used a series of Y-GECO indicators with Kd′ values ranging from 25 nM to 190 nM and expressed them in Mi neurons at the same concentration and activated the neurons identically. These experiments revealed that the Kd′ of the indicator has a substantial effect on the ΔF/F and SNR, and a lesser effect on the delay to reach the maximum signal. For an 8 s stimulation, the relationship between Kd′ and ΔF/F was clear: a lower Kd′ gave a higher ΔF/F. We speculate that, for the 8 s stimulation, free Ca2+ levels drop below Kd′ (i.e., well below 25 nM) for all variants. Variants with a greater Kd′ (i.e., 100–200 nM) have a reduced change in fluorescence because their Kd′ is closer to the resting Ca2+ concentration and so they have higher fluorescence prior to stimulation and diminished ΔF/F. Consistent with their larger values of koff (i.e., faster dissociation), the two variants with higher Kd′ (Y-GECO1m and Y-GECO2s) exhibited a decreased delay to maximum fluorescence relative to the two variants with lower Kd′ (Y-GECO2.1s and Y-GECO2.2s) with an 8 s stimulus. Lastly, the signal-to-noise ratio (SNR) depends on the amount of fluorescence signal acquired, which is necessarily dependent on both Kd′ and koff. Accordingly, neither the highest (Y-GECO2.2s) nor the lowest (Y-GECO1m) affinity variants (slowest and fastest, respectively) gave the highest SNR at stimulus durations up to 4 s. Rather, the two middle affinity variants (Y-GECO2s and Y-GECO2.1s), which must represent appropriate compromises of affinity and off-rate kinetics, tended to provide the best SNR at stimulus durations up to 4 s. Following a stimulus duration of 8 s, the slow kinetics of the high affinity Y-GECO2.2s variant are presumably no longer limiting and so this variant provides the highest SNR within the series.
While we succeeded at using Y-GECO variants to image neuronal inhibition in an ex vivo tissue preparation, we were unable to detect responses from Y-GECO with natural light stimulation in vivo. We suspect that a poor 2-photon cross section for Y-GECO variants is the primary reason for this discrepancy. For the in vivo experiments, it was challenging to discern Y-GECO expressing cells from background using 2-photon excitation. Increasing the laser power led to photobleaching which further decreased the SNR. Although it is beyond the scope of this work, we suggest that using 1-photon excitation would provide improved performance, though this would be accompanied with decreased penetration of light into the tissue. Further optimization of the Y-GECO 2-photon cross section and photostability could enhance its application for in vivo imaging.
Summary
Despite the fact that 30% of neurons in humans and Drosophila are inhibitory, there are relatively few optogenetic indicators optimized for imaging of inhibitory neuronal activity. We, and others14, propose that, for imaging of inhibitory activity, inverse response Ca2+ indicators have an inherent advantage to direct response Ca2+ indicators. Specifically, imaging of inhibitory activity with a direct response Ca2+ requires detection of a dimming response that could be readily obscured by out of focus fluorescence from adjacent bright cells. In contrast, inhibitory activity could be more easily detected with an inverse response Ca2+ indicator due to the diminished background fluorescence originating from adjacent out-of-focus cells.
Our results suggest that, for an inverse response indicator in the Mi1 neuron, a Kd′ of less than 100 nM produces greater changes in fluorescence without compromising the response time. In contrast, direct response Ca2+ indicators optimized for detection of neuronal action potentials range have been found to perform best when the Kd′ is tuned to the 100 to 200 nM range. An important caveat is that this conclusion applies only to the Mi1 neuron within the synaptic region studied. Other neurons or even regions within the same neuron could have different resting Ca2+ levels which would change the optimum Kd′. For this reason, we advocate the empirical identification of the optimal Kd′ by testing a series of variants such as the ones described in this work.
Methods
Site-directed mutagenesis for mutation introduction
Mutations were introduced by Quikchange II Site-directed Mutagenesis kit (Agilent Technologies) with primers containing desired mutations at specific positions.
Purification and in vitro characterization of Y-GECO proteins
The procedure for purification and determination of extinction coefficient, quantum yield, and Kd′ of Y-GECO variants has been previously described11. A DU-800 UV-vis spectrophotometer (Beckman) was used to measure absorption spectra, and a Safire2 plate reader (Tecan) was used to measure the excitation and emission spectra. The ratiometric response to Ca2+ of Y-GECO is defined as (Rmax − Rmin)/Rmin, where R = (I with 526 nm excitation)/(I with 416 nm excitation), and I is the fluorescence intensity at 550 nm. The intensiometric response to Ca2+ of Y-GECO is defined as (Imax − Imin)/Imin, where I is the fluorescence intensity at 550 nm with 526 nm excitation.
Kinetics of Ca2+ association and dissociation fluorescence change of Y-GECO
Stopped-flow spectroscopy was used to evaluate reaction kinetics of FP with Ca2+, using an excitation wavelength of 520 nm, a 13.95 nm bandwidth, and an emission wavelength of 540 nm with a 37 nm bandwidth. E. coli cells expressing Y-GECO variants were first suspended in TBS buffer containing 0.2 mM CaCl2 or 0.2 mM EGTA, followed by rapid mixing (1:1) with TBS buffer containing 20 mM EGTA or 20 mM CaCl2. Fluorescence signals were captured using ProData SX software.
Ca2+-dissociation kinetics of purified Y-GECO indicators were measured as described previously11. The fluorescence intensity vs. time was collected, plotted and fit to a single exponential curve, giving koff.
Construction of plasmids for mammalian cell expression
To express Y-GECO indicators in HeLa cells and dissociated hippocampal cultures for ex vivo characterizations, the Y-GECO gene in pBAD vector, used for expression in E. coli cells, was amplified by PCR with the primers FW_BamHI_Kozak_6His and RV_CaM_stop_EcoRI, followed by gel purification of the PCR products. The purified gene was then digested with the restriction enzymes BamHI and EcoRI, purified and ligated into a modified pcDNA3 plasmid vectors which have been digested with the same enzymes and purified by gel. The ligation products, the plasmids for mammalian expression of Y-GECO indicators, were transformed into electrocompetent E. coli DH10B cells which were then plated on an agar plate containing 1× ampicillin for overnight culture at 37 °C. On the following day, individual colonies were picked for 12 h liquid culture in 4 mL B/ampicillin, shaken at 250 rpm. The cultured cells were then isolated and the plasmids were purified for mammalian cell transfection and expression.
HeLa cell culture and imaging
HeLa cells were cultured and transfected as described previously11. Wide-field imaging of cells was performed on an epifluorescence inverted microscope (Eclipse Ti-E, Nikon) equipped with a digital CCD camera (QuantEM 512SC). The microscope and camera were controlled using NIS-Elements Advanced Research software. Cells were imaged with a 20× air objective lens (NA 0.8). The cells were illuminated by a 100 W mercury arc lamp and a 25% neutral density filter was used to reduce the intensity of the light. To record the long Stokes shift fluorescence, we used a filter set of 438/24 nm (excitation), 458 nm (dichroic) and 542/27 nm (emission). Exposure time was set to 700 ms. To record the short Stokes shift fluorescence, we used a filter set of 480/40 nm (excitation), 505 (dichroic) nm and 535/40 nm (emission), with exposure time 500 ms.
To image histamine induced Ca2+ dynamics, images were acquired every 4 s for ~20 min. After ~30 s of initial recording, 100 μM histamine solution was added to the dish to reach a final concentration of 10 μM. After ~10 min recording, 10 mM EGTA, 40 μM ionomycin in Ca2+-and Mg2+-free HHBSS was then added to reach a final concentration of 1 mM EGTA, 4 μM ionomycin. Then, 20 mM Ca2+, 40 μM ionomycin in Ca2+ and Mg2+ free HHBSS was then added to reach a final concentration of 2 mM Ca2+, 4 μM ionomycin.
Dissociated rat hippocampal culture preparation and imaging
Dissociated rat hippocampal cells are prepared and transfected as described previously11. Tissues were collected from animals that were being sacrificed for unrelated experiments that were approved by the University of Alberta Animal Care and Use Committee and carried out in compliance with guidelines of the Canadian Council for Animal Care and the Society for Neuroscience’s Policies on the Use of Animals and Humans in Neuroscience Research. Y-GECO indicators were imaged under conditions similar to those described above. A 60× oil objective lens was used for higher amplification and a 12.5% neutral density filter was used to reduce the excitation intensity. Only short-Stokes fluorescence was recorded by using the filter set 480/40 nm (excitation), 505 (dichroic) nm and 535/40 nm (emission). Exposure time was 69 ms and images were acquired every 1 s for 4 min.
Drosophila imaging
Y-GECO variants were codon optimized for Drosophila, cloned into 13XLexAOP2-IVS-Syn21-[insert]-p10 plasmid (gift from Barret Pfeiffer) and inserted into the genome at the su(Hw)attP8 landing site. Brains from females (Genotype: 10xUAS-Chrimson-tdTomato (attP18)/LexAOP2-Y-GECO (suHwattP8); 19F01-LexA (suHwattP5) (Mi1)/+; 27G06-GAL4 (attP2) (L1)/+) expressing Chrimson-tdTomato in L1 neurons and Y-GECO variants in Mi1 neurons were tested.
Flies were reared at 25 °C on retinal supplemented (0.2 mM) cornmeal medium that was shielded from light. All experiments were performed on female flies, 1–4 days after eclosion. Brains were dissected in a saline bath (103 mM NaCl, 3 mM KCl, 2 mM CaCl2, 4 mM MgCl2, 26 mM NaHCO3, 1 mM NaH2PO4, 8 mM trehalose, 10 mM glucose, 5 mM TES, bubbled with 95% O2/5% CO2). After dissection, the brain was positioned anterior side up on a coverslip in a Sylgard dish submerged in 3 ml saline at 20 °C.
The sample was imaged with a resonant scanning 2-photon microscope with near-infrared excitation (920 nm, Spectra-Physics, INSIGHT DS DUAL) and a 25× objective (Nikon MRD77225 25XW). The microscope was controlled by using ScanImage 2015.v3 (Vidrio Technologies)21. Images were acquired with 141 μm × 141 μm field of view at 512 × 512 pixel resolution, approximately 9 Hz frame rate after averaging 5 frames. The excitation power for Ca2+ imaging measurement was 12 mW.
For the photostimulation, the light-gated ion channel Chrimson was activated with a 660 nm LED (M660L3 Thorlabs) coupled to a digital micromirror device (Texas Instruments DLPC300 Light Crafter) and combined with the imaging path with a FF757-DiO1 dichroic (Semrock). On the emission side, the primary dichroic was Di02-R635 (Semrock), the detection arm dichroic was 565DCXR (Chroma), and the emission filters were FF03-525/50 and FF01-625/90 (Semrock). Photostimulation light was delivered in a pulse train that consisted of three 5 pulses with increasing pulse durations (1, 2, 4, 8 and 1 seconds) every 30 seconds as outlined in Supplementary Fig. 4c. The light intensity was 0.24 mW/mm2, as measured using Thorlabs S170C power sensor.
In custom python scripts, an ROI was drawn over layers M8–10 on a figure containing the standard deviation over time. Before calculating the change in fluorescence (ΔF), the offset was subtracted from the fluorescence and then baseline fluorescence was subtracted. Baseline fluorescence is the median fluorescence over a 5 s time period before stimulation started. The ΔF was then divided by baseline to normalize signal (ΔF/F). The final signal was run through a gaussian filter (sigma = 1).
The time period included in the maximum fluorescence, delay to maximum signal, and signal to noise ratio start from stimulation start to 2 s after stimulation ended. Signal to noise ratio was calculated by taking the maximum ΔF/F signal and dividing by the baseline (5 s period before simulation started) standard deviation.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Tian, L., Hires, S. A. & Looger, L. L. Imaging neuronal activity with genetically encoded calcium indicators. Cold Spring Harb. Protoc. 2012, 647–656 (2012).
Pérez Koldenkova, V. & Nagai, T. Genetically encoded Ca2+ indicators: Properties and evaluation. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1833, 1787–1797 (2013).
Nakai, J., Ohkura, M. & Imoto, K. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat. Biotechnol. 19, 137–141 (2001).
Nagai, T., Sawano, A., Park, E. S. & Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proceedings of the National Academy of Sciences 98, 3197–3202 (2001).
Zhao, Y. et al. An expanded palette of genetically encoded Ca2+ indicators. Science 333, 1888–1891 (2011).
Wu, J. et al. Improved orange and red Ca2+ indicators and photophysical considerations for optogenetic applications. ACS Chem. Neurosci. 4, 963–972 (2013).
Wu, J. et al. A long Stokes shift red fluorescent Ca2+ indicator protein for two-photon and ratiometric imaging. Nat. Commun. 5, 5262 (2014).
Tian, L. et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881 (2009).
Chen, T.-W. W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013).
Dana, H. et al. Sensitive red protein calcium indicators for imaging neural activity. Elife 5 (2016).
Zhao, Y. et al. Microfluidic cell sorter-aided directed evolution of a protein-based calcium ion indicator with an inverted fluorescent response. Integr. Biol. 6, 714–725 (2014).
Hoi, H. et al. An Engineered Monomeric Zoanthus sp. Yellow Fluorescent Protein. Chem. Biol. 20, 1296–1304 (2013).
Matz, M. V. et al. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 17, 969–973 (1999).
Hara-Kuge, S. et al. An improved inverse-type Ca2+ indicator can detect putative neuronal inhibition in Caenorhabditis elegans by increasing signal intensity upon Ca2+ decrease. PLoS One 13, e0194707 (2018).
Zhao, Y., Zhang, W., Zhao, Y., Campbell, R. E. & Harrison, D. J. A microfluidic cell sorter for multiparameter screening in directed evolution. Submitted (2017).
Joesch, M., Schnell, B., Raghu, S. V., Reiff, D. F. & Borst, A. ON and OFF pathways in Drosophila motion vision. Nature 468, 300–304 (2010).
Behnia, R., Clark, D. A., Carter, A. G., Clandinin, T. R. & Desplan, C. Processing properties of ON and OFF pathways for Drosophila motion detection. Nature 512, 427–430 (2014).
Strother, J. A., Nern, A. & Reiser, M. B. Direct observation of ON and OFF pathways in the Drosophila visual system. Curr. Biol. 24, 976–983 (2014).
Yang, H. H. et al. Subcellular Imaging of Voltage and Calcium Signals Reveals Neural Processing In Vivo. Cell 166, 245–257 (2016).
Klapoetke, N. C. et al. Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346 (2014).
Pologruto, T. A., Sabatini, B. L. & Svoboda, K. ScanImage: flexible software for operating laser scanning microscopes. Biomed. Eng. Online 2, 13 (2003).
Acknowledgements
The authors thank Wei Zhang, Ahmed Abdelfattah, Vladimir Rancic, Klaus Ballanyi, Andy Holt, and the University of Alberta Molecular Biology Services Unit, for technical support. R.E.C. was supported by grants from CIHR (MOP-123514), NSERC (RGPIN 288338-2010), Brain Canada, and NIH (U01 NS094246 and UO1 NS090565). Yufeng Zhao was a recipient of Alberta Innovates Technology Futures Scholarship.
Author information
Authors and Affiliations
Contributions
R.E.C., D.J.H. and A.M.W. conceived and designed the study. E.R.S. made the Drosophila expression constructs. D.B. collected and analyzed the Drosophila imaging data. Yufeng Z. and R.E.C. drafted the manuscript, which was revised and edited by D.B., Yongxin Z., E.R.S., D.J.H. and A.M.W. All authors read, commented, and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, Y., Bushey, D., Zhao, Y. et al. Inverse-response Ca2+ indicators for optogenetic visualization of neuronal inhibition. Sci Rep 8, 11758 (2018). https://doi.org/10.1038/s41598-018-30080-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-30080-x
This article is cited by
-
Structure- and mechanism-guided design of single fluorescent protein-based biosensors
Nature Chemical Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
