
Abstract
Hepatitis delta virus (HDV), a satellite virus of hepatitis B virus (HBV), infects an estimated 15–20 million people worldwide and confers a greater risk for accelerated progression to liver disease. However, limited HDV surveillance data are available in sub-Saharan Africa where HDV diversity is high. To determine the prevalence and diversity of HDV in Cameroon, serological and molecular characterization was performed on 1928 HBsAg positive specimens selected from retrospective viral surveillance studies conducted in Cameroon from 2010–2016. Samples were screened for HDV antibodies on the Abbott ARCHITECT instrument and for HDV RNA on the Abbott m2000 instrument by research assays. HDV positive specimens with sufficient viral load were selected for genomic sequencing. The seroprevalence of HDV in HBsAg positive samples from Cameroon was 46.73% [95% CI; 44.51–48.96%], with prevalence of active HDV infection being 34.2% [95% CI; 32.09–36.41%]. HDV genotypes 1, 6, 7 and 8 were identified amongst N = 211 sequences, including N = 145 genomes. HDV prevalence is high within the study cohort, indicating that a large portion of HBV infected individuals in Cameroon are at elevated risk for severe hepatitis and death. Collectively, these results emphasize the need for HBV vaccination and HDV testing in HBsAg positive patients in Cameroon.
Similar content being viewed by others

Introduction
Hepatitis delta virus (HDV) is the smallest virus known to infect humans. HDV infection is restricted to co-infection with hepatitis B virus (HBV) or superinfection of HBV-infected individuals due to the requirement for HBV surface antigen (HBsAg) in the HDV viral envelope. An estimated 15–20 million people are co-infected with HBV and HDV worldwide; with areas of high endemicity in the Middle East, the Amazonian region of South America, the Mediterranean, and western and central Africa1. The impact of HDV infection is substantial: co-infection results in a more severe clinical outcome than HBV alone, while superinfection of HBV infected patients with HDV results in greater probability of chronic infection, more rapid progression to cirrhosis2, and overall greater risk of morbidity3. Further, hepatitis treatment regimens which decrease HBV viral loads (e.g. nucleos(t)ide analogs) do not impact HDV and pegylated interferon alpha shows poor efficacy with relapse common4,5. Additionally, HDV inhibits HBV replication, lowering HBV DNA levels detectable in patient blood6,7,8, and thereby potentially complicating HBV patient management. The ability to detect and monitor for HDV is clearly critical to patient care and the need for novel therapeutics is imperative9,10,11,12, especially in regions of elevated HDV prevalence1,13.
The 1.7 kb RNA genome of HDV is highly variable14,15; phylogenetic analyses have described eight genotypes which differ by up to 30%. The greatest variety of genotypes and subtypes has been found in hyperendemic regions of western and central Africa, where genotypes 1, 5, 6, 7, and 8 are present and may be associated with better clinical outcomes than strains found elsewhere16,17,18,19. The influence of HDV genotype on the clinical course of infection has not been fully established, albeit there is evidence that genotype 3 viruses may cause more aggressive disease20 and respond better to treatment21. The extreme genetic diversity of HDV has impacted viral detection by molecular methods, with detection of divergent African and South American isolates proving particularly problematic22. Despite the global burden of HDV, only 399 genome sequences are available in the National Center for Biotechnology (NCBI) repository, while 10,550 complete HBV genomes are available in the database (accession date March 21st, 2018). Investigation of the genetic diversity of this highly variable virus is essential for HDV epidemiology and for the design of robust detection assays that can accommodate viral diversity regardless of the geographic region where an infection was acquired.
We recently completed a large scale surveillance study in the South region of Cameroon (Fig. 1)23. Amongst the 13,700 participants in the study, HBV prevalence was 10.45% from 2011–2015,23 although the HDV prevalence was unknown. In parallel, samples have also been collected in an ongoing HIV diversity surveillance study in urban centers of Cameroon (Fig. 1), although HBV or HDV prevalence has not previously been examined in this cohort24. Herein, we retrospectively interrogated the HDV status of 1928 HBsAg-positive plasma samples from these two surveillance studies using recently developed automated research assays for detection of HDV IgG and HDV RNA on the Abbott ARCHITECT and m2000 platforms, respectively25, and found a high prevalence of both HDV exposure (46.73% seropositive) and active infection (34.2% HDV RNA positive). Furthermore, we report sequence data from 211 patient samples (145 near complete genomes and 66 partial sequences), with genotypes 1, 6, 7, and 8 present in the study population. Overall, the high prevalence and diversity of HDV infection observed within this population highlights the importance of HDV testing in the care of HBV infected individuals in Cameroon.

Map of study locations in Cameroon. The locations of study participants from two HIV surveillance studies included in the retrospective cohort for HDV testing are indicated on a map of Cameroon. The first study included specimens from the two largest cities in Cameroon, Douala (Littoral region, red) and Yaoundé (Centre region, violet) as indicated by large black dots. The second study included specimens from 14 towns in the South region (blue), with major study sites indicated by small black dots.
Materials and Methods
Samples
The retrospective cohort included HBsAg-positive specimens from two surveillance studies conducted in Cameroon from 2010–201623,24. Both studies were approved by the Cameroon National Ethical Review Board, the Faculty of Medicine and Biomedical Science IRB, and the Ministry of Health of Cameroon23,24. In accordance with study protocols, written informed consent was provided and plasma was collected anonymously. Unique populations were sampled in the two studies included in this retrospective cohort: participants were recruited from blood bank donors, hospitals, and chest clinics in the urban centers of Douala and Yaoundé24, or patients with illness of unknown etiology, antenatal patients, and participants of door-to- door and voluntary testing campaigns were recruited from 14 villages and towns in the South region of Cameroon23. All samples were tested on-site in Cameroon for HIV according to the national algorithm using the Determine HIV1/2 test (Alere International Limited, Zug, Switzerland) and/or Murex HIV Ag/Ab Combo test (DiaSorin, Saluggia, Italy). All specimens in the combined retrospective cohort were pre-screened using the Abbott ARCHITECT HBsAg (1L80) or HBsAg Qualitative (4P53) assays (Abbott Diagnostics, Abbott Park, IL) and confirmed by secondary screening for HBsAg by a previously described research mutant screening assay23. A limited number of samples were also tested for HBV viral loads using the Abbott HBV real time assay (2G34, Abbott Molecular, Des Plaines, Illinois, USA). HBV sequencing was performed on a subset of samples as previously described23. All samples were tested for HIV using ARCHITECT assays (2P37 or 3A77, Abbott Diagnostics, Abbott Park, Illinois, USA).
HDV testing algorithm
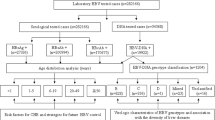
Initial HDV IgG version 1.0 testing was performed on HBsAg reactive samples collected in 2010–2015 followed by reflex screening of serology reactive samples for HDV RNA. To correct for potential deficiencies of IgG only screening, HDV seronegative samples were mini-pooled (5 per pool) and tested for HDV RNA. HDV viremic samples without evidence of HDV IgG were subjected to additional diagnostic testing for IgM or a modified version of the IgG assay using a recombinant HDV large antigen (version 1.5). The additional diagnostic testing presented deficiencies in the original HDV IgG assay using peptides and a subsequent version 2.0 assay was developed. The version 2.0 HDV IgG assay was applied to screen all specimens collected in 2016. In parallel, this set of specimens was screened for HDV RNA regardless of serology results. A flowchart detailing the complete testing algorithm can be found in Figure S1.
HDV antibody testing
HDV IgG version 1.0
The version 1.0 research use prototype assay for the detection of HDV IgG on the ARCHITECT immunoassay platform has demonstrated 100% specificity, 100% sensitivity, and 69.6% positive predictive value for HDV RNA in a seropositive sample as previously described25 (Abbott Diagnostics, Abbott Park, IL, USA). A provisional cut-off for the assay was previously established based on screening an HBsAg and HBV DNA negative population from US volunteer donors (non-endemic area) (ProMedDx, LLC, Norton, MA was set at median plus 10 standard deviations (SD) to result in 100% specificity25.
Additional serologic testing
The HDAg protein (R18420, Meridian, Memphis, Tennessee, USA) was coated onto magnetic microparticles and used in an indirect IgG assay format as described above. A signal to noise (S/N) >/= 10.0 was considered positive. HDV IgM testing was performed by modifying the HDV IgG assay using peptides by replacement of the conjugate with an acridinium conjugated mouse anti-human IgM antibody (Abbott Diagnostics, Abbott Park, Illinois, USA). A signal to noise (S/N) >/= 10.0 was considered positive.
HDV IgG version 2.0
A refined HDV IgG assay was designed to contain both HDV peptides and recombinant antigen. Specificity was established using a US negative donor population and provisional cut-off was determined.
HDV Molecular testing
HDV RNA was detected as described previously25. Quantitation was performed using the World Health Organization (WHO) HDV nucleic acid test standard (Paul Ehrlich Institute, Germany). Where sample volume was limiting, samples were diluted 1:10 or 1:100 in HBV negative normal human plasma prior to extraction; final quantitation was adjusted to account for dilution. Seronegative specimens collected in 2010–2015 were screened for HDV RNA in pools of 5 followed by dissection of positive pools. All other specimens were tested individually as indicated in Figure S1.
Sanger sequencing and phylogenetic analyses
Remaining volumes of HDV RNA eluates from specimens with viral load >4.5 log IU/ml were used as templates for sequencing amplicons covering 3 overlapping regions of the genome as previously described25. Sanger sequencing of PCR products was completed as previously described23. Resulting sequences were analyzed and classified by phylogenetic analysis as previously described25. Divergent sequences branching basal to known references were analyzed to identify recombinants using the SimPlot v3.5.1 software26.
Data availability
The complete and partial HDV genome sequences were deposited in Genbank (MG711661 - MG711805).
Statistical analysis
Confidence intervals were determined by the Wilson Score method27 in Microsoft Excel 2016.
Results
Study population
Blood samples collected from participants of two viral surveillance studies conducted from 2010–2016 were screened to identify N = 1928 HBsAg-positive specimens with sufficient volume for HDV screening23,24. Of these HBsAg positive specimens, 79.6% were obtained from participants in towns in South Cameroon through a large-scale HIV/HBV/HTLV surveillance study23; whereas 20.4% were collected from participants in the two largest cities in Cameroon, Douala and Yaoundé, through an HIV diversity surveillance study24. Specimen collection sites were blood banks, hospitals, clinics, and voluntary testing campaign locations in the Littoral, Centre, and South regions of Cameroon as shown in Fig. 1. Demographic data collected from N = 1702 participants indicated that 52% (896/1702) of were female, with ages ranging from 2 to 91 and an average age of 31 years old (Table 1). Previous serological screening for HIV indicated that 48.9% (942/1928) of the study population was HIV positive (Table 1). All N = 1928 specimens were screened for HDV IgG antibodies and N = 1850 of these specimens were screened for HDV RNA by the testing algorithm shown in Figure S1, with supplemental testing for IgM and revised versions of the HDV IgG assay for discordant specimens as indicated.
HDV prevalence
Overall, serological testing revealed a high prevalence of exposure to HDV: 901/1928, 46.73% [95% CI; 44.51–48.96%] samples were reactive for HDV IgG or IgM antibodies amongst HBsAg positive carriers from Cameroon (Table 1, Fig. 2). Molecular screening identified HDV RNA in N = 633 specimens (34.2%), with an average viral load of 4.53 (±1.9 standard deviation) log IU/ml and a maximum viral load of 9.77 log IU/ml. Within the HDV seropositive cohort; 69.25% (624/901) of samples were also positive for HDV RNA (Fig. 2). Nine HDV RNA positive seronegative samples had HDV viral loads that were less than 1.5 log IU/ml, with only 2 exceeding 100 IU/ml. These specimens were collected from 4 different cities and towns, including Yaounde, Sangmelima, Kribi, and Ebolowa, indicating that RNA positive, seronegative results were not unique to one particular site. Notably, two of the HDV RNA positive, seronegative specimens were from HIV positive donors. The overall seroprevalence of HDV in the HIV positive cohort was 41.4% (390/942), and HDV RNA was present in 26.5% (250/942) of all HIV positive specimens (Table 1). In contrast, HDV prevalence was higher in the HIV negative study cohort, with HDV seroprevalence being 51.8% (511/986) and HDV RNA prevalence being 38.8% (383/986) amongst HIV negative participants.

Prevalence of anti-HDV antibodies and HDV RNA. Pie charts summarizing results of HDV serological testing (center) and HDV RNA testing for HDV IgG or IgM seropositive samples (right) and HDV seronegative samples (left) for all HBsAg+ samples in the cohort.
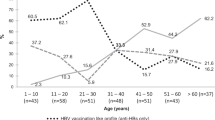
The prevalence of HIV in the study population (48.86%) was much higher than the observed HIV prevalence in the general population of Cameroon in a 2011 national survey (7.2%)28, and is likely due to the bias of the study sampling towards patients seeking healthcare. The majority of samples in the study were obtained through collection of specimens from patients with illness of unknown etiology (60.4%) and other hospital settings (e.g. Chest clinic, antenatal, hospital patient) (Fig. 3). The age range of HBsAg-positive samples in this study was 2–91 years (Table 1), with the highest number of participants being within the ages of 18–40 and a mean age of 31 (Fig. 4a). We observed a downward trend in HDV seropositive and RNA positive rates with increasing ages of participants (Fig. 4b). No appreciable gender difference in HDV prevalence was observed (Table 1).

Summary of specimen collection source types. At right is a pie chart showing representation of collection risk group categories among the 1928 samples tested within this study. The N and % per collection type is given at center. Percent HDV serology or molecular testing, as well as HIV positive status, by collection category is given in the bar graph (right) with % positive results given relative to the number of all tested samples within respective sample population. VTC, volunteer testing campaign.

Testing results by patient age. (A) Distribution of patient ages in the study population. (B) HDV seroprevalence and RNA prevalence by age group. The number of participants per age group is noted in A.
HDV phylogenetic analyses
To determine the genetic diversity of HDV in the study population, molecular characterization and phylogenetic analysis was performed for specimens with viral loads >4.5 log IU/ml. Sequences were obtained from N = 211 specimens that were RT-PCR positive in at least one of three overlapping regions targeted. In total, 145 complete or near complete genomes of at least 1 kb in length and 66 shorter sequences were aligned to reference sequences of the same length and classified according to branching patterns in neighbor joining trees. Amongst all of the HDV sequences generated, genotypes 1, 6, 7, and 8 were identified in the study population, indicating a high level of HDV diversity is present in Cameroon (Figs 5 and 6). Genotype 1 was predominant (65.4%, 138/211), with three sample clusters branching distinct from reference sequences (bootstrap values > 85) (Fig. 6). Genotype 7 was also well-represented (28.9%, 61/211), including five sequences branching basal to genotype 7 reference sequences with high bootstrap values (>85) (Fig. 6). Genotypes 6 (5.2%, 11/211) and 8 (0.5%, 1/211) were less common. Sequences branching basal to references were examined in SimPlot for potential recombinant breakpoints, though no evidence of recombination was observed.

Summary of HDV genotypes observed in the study cohort. Distribution of complete/near complete and subgenomic sequences by genotype are given.

HDV genomic phylogenetic tree of complete and near complete genomes. The phylogenetic tree for N = 145 near complete genomes and N = 43 reference strains is presented with references depicted in black and relevant nodes with bootstrap values >70 labeled with a black box. Genotypes for each major branch are labeled (GT1-8).
HBV genotype data was available for 334 samples in this cohort23; 32% of HBV genotyped samples were genotype A, 67.4% were genotype E, with 2 samples having dual infections of genotype A and E23. HDV seroprevalence was highest in the HBV genotype E cohort (Figure S2), consistent with a higher overall prevalence of genotype E. Among the 25 samples with both HBV and HDV sequence data, a clear co-circulation pattern was not observed: HDV genotype 1 (2A, 11E), HDV genotype 6 (1A, 1E), HDV genotype 7 (2A, 8E).
Discussion
We investigated HDV seroprevalence and RNA prevalence in a retrospective cohort of 1928 HBsAg-positive specimens collected from 2010–2016 in Cameroon. Within these samples, we report a high prevalence of HDV infection: the observed overall seroprevalence was 46.73%, indicating a high rate of HDV exposure. Further, HDV RNA testing revealed that 34.2% of patients were infected with actively replicating HDV at the time of sample collection. In Cameroon, the reported HDV seroprevalence ranges from 7 to 62% depending on the population tested and the assay used for screening13,16,18,29,30,31. When HDV status is stratified by age, a downward trend is observed wherein the prevalence of HDV is reduced with increasing age in this cohort (Fig. 4). This trend was evident to a lesser degree with HDV seroprevalence, likely due to sustained HDV IgG response from recovered illnesses in the older populations (Fig. 4b). Overall, our data highlight the necessity for HDV monitoring in HBV carriers of all ages within this region and support the need for HBV vaccination at an early age.
Limitations of this study include the disparate composition of the convenience population tested. We considered that the relatively high HDV prevalence observed herein could be influenced by over-representation of HIV infected individuals as 48.86% samples were HIV positive due to sample collection bias. Serological evidence of HDV infection was found in 41.4% of HIV carriers tested, which is indeed elevated compared to other studies in Cameroon32; however, higher HDV seroprevalence (51.8%) was observed in HIV negative samples, repudiating bias due to HIV-positive status (Table 1). Sample collection from patients seeking healthcare at clinical sites could also introduce bias toward elevated HDV prevalence, though we note that HDV prevalence was not higher in samples collected from clinical sites (unknown illness, chest clinic, hospital) compared to healthy donors (voluntary testing campaign, blood donors) (Fig. 3).
Another study complication was the use of prototype assays. The initial HDV IgG test (version 1.0) detected 691/1528 HBsAg positive samples with 501/691 having active HDV infection. By screening seronegative samples for HDV RNA, we identified an additional 34 samples with detectable HDV RNA. Using additional serology testing (for IgM or using recombinant antigen for IgG testing) we found evidence of HDV antibodies in the majority (26/34) of these initially seronegative samples, which included genotype 1, 6, and 7 specimens (Table S1). The supplemental testing informed revision of the anti-HDV assay (version 2.0) (Figure S1). Overall, using anti-IgG as a primary screen resulted in 9 seronegative/RNA positive and 3 IgM only/RNA positive samples, suggesting the value of IgM testing is limited in highly endemic areas. There were nine HDV RNA positive samples that were identified with no evidence of HDV seroconversion (Table S1), leaving the possibilities that these specimens were from acute infections, had other unknown underlying clinical complications, or evaded detection with the current prototype anti-HDV assays. All had HDV viral loads less than log 1.5 IU/ml and could not be sequenced (Table S1, indicated in bold). Unfortunately, no additional clinical data was available for these samples beyond the diagnostic testing we could perform. Interestingly of these 9 samples, 4 were from blood donors in the same collection site and year (Yaoundé, 2015). Overall, HDV seroprevalence was higher than RNA prevalence, with the exception of the blood donor cohort (Fig. 3). Little is known about the diagnostic profile of incident HDV infections since most HDV surveillance reports primarily rely on serology testing18,33,34,35,36. The seroconversion profile of a chimpanzee superinfected with HDV indicates that RNA is detectable in the absence of IgG or IgM for 30 days25; acute infections are likely HDV RNA positive prior to seroconversion in humans as well. Indeed, human HDV IgM and IgG responses have been reported to be late or undetected during HDV infection37,38,39. Future HDV surveillance studies may find additional RNA positive specimens if seronegative samples are screened, potentially allowing for better estimations of the prevalence of incident HDV infections. To our knowledge, HDV incidence has not previously been measured in a surveillance study, although it can be inferred from HDV prevalence rates amongst HBV incident infections as previously demonstrated in an Italian cohort40.
In addition to HDV molecular and serologic testing, we sequenced 211 HDV RNA positive samples, generating complete or near complete genomes from 145 samples and 66 sub-genomic sequences. Phylogenetic analyses of sequence data revealed genotype 1 to be dominant with additional sequences classified to genotype 7, genotype 6, and genotype 8, consistent with other analyses from central Africa17,18,33. Evidence of genotype 1 sub-clades has been noted in Africa15,18,33 and elsewhere41,42; the sequences obtained in the present study provide further evidence of rich diversity within this clade and, overall, expand greatly upon available HDV sequence data. Specifically, we find 3 branches of 13, 7, and 32 genotype 1 sequences, respectively, with bootstraps of >85 that cluster independent of reference sequences. Likewise, we find evidence of further genetic diversity in genotype 7 in our phylogenetic analysis: five Cameroon sequences are basal to reference samples. No evidence of recombination was observed in any of these sequences. Of note, no representatives of genotype 5 were observed in this cohort despite this genotype being previously identified in the region18. Further, we identified only one subgenomic sequence representative of genotype 8 within this cohort, despite evidence that this genotype is circulating within central and western Africa17,36,43.
Overall, we report high prevalence of HDV in Cameroon, including diverse viruses from genotypes 1, 6, 7, and 8; divergent sequences reported here increase the known diversity of these clades. Our data indicate that HBV patients in Cameroon are at elevated risk for severe hepatitis and death due to illness compounded by HDV infection. HDV worsens hepatitis symptoms and clinical outcomes, including a higher likelihood of progression to hepatocellular carcinoma30. Screening and diagnosis of HDV in HBV patients in Cameroon might identify individuals at increased risk for developing liver disease. Due to the high endemic seroprevalence of HDV in HBsAg carriers in Cameroon, access to serologic testing from dried blood spots could provide a means to diagnose HDV in high risk populations with limited access to venipuncture, although studies comparing assay sensitivity and specificity for dried blood spot specimens are necessary to evaluate this diagnostic approach. Importantly, HBV vaccination could help reduce the hepatitis burden in Cameroon. As HDV continues to diversify, HDV surveillance will be critical for monitoring emerging strains of HDV that may challenge diagnostic tests.
References
Manns, M. P. Hepatitis D virus in Africa: several unmet needs. The Lancet. Global health 5, e953–e954, https://doi.org/10.1016/S2214-109X(17)30345-5 (2017).
Sureau, C. & Negro, F. The hepatitis delta virus: Replication and pathogenesis. Journal of Hepatology 64, S102–S116, https://doi.org/10.1016/j.jhep.2016.02.013 (2016).
Béguelin, C. et al. Hepatitis delta-associated mortality in HIV/HBV-coinfected patients. Journal of Hepatology 66, 297–303, https://doi.org/10.1016/j.jhep.2016.10.007 (2017).
Ciancio, A. & Rizzetto, M. Chronic hepatitis D at a standstill: where do we go from here? Nat Rev Gastroenterol Hepatol 11, 68–71, https://doi.org/10.1038/nrgastro.2013.164 (2014).
Wranke, A. & Wedemeyer, H. Antiviral therapy of hepatitis delta virus infection — progress and challenges towards cure. Current opinion in virology 20, 112–118, https://doi.org/10.1016/j.coviro.2016.10.002 (2016).
Wu, J. C. et al. Production of hepatitis delta virus and suppression of helper hepatitis B virus in a human hepatoma cell line. Journal of virology 65, 1099–1104 (1991).
Sureau, C., Taylor, J., Chao, M., Eichberg, J. W. & Lanford, R. E. Cloned hepatitis delta virus cDNA is infectious in the chimpanzee. Journal of virology 63, 4292–4297 (1989).
Alfaiate, D. et al. HDV RNA replication is associated with HBV repression and interferon-stimulated genes induction in super-infected hepatocytes. Antiviral Research 136, 19–31, https://doi.org/10.1016/j.antiviral.2016.10.006 (2016).
Rizzetto, M. Investigational drugs in development for Hepatitis D. Expert opinion on investigational drugs 26, 999–1005, https://doi.org/10.1080/13543784.2017.1357695 (2017).
Bazinet, M. et al. Safety and efficacy of REP 2139 and pegylated interferon alfa-2a for treatment-naive patients with chronic hepatitis B virus and hepatitis D virus co-infection (REP 301 and REP 301-LTF): a non-randomised, open-label, phase 2 trial. The lancet. Gastroenterology & hepatology. https://doi.org/10.1016/S2468-1253(17)30288-1 (2017).
Bogomolov, P. et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J Hepatol 65, 490–498, https://doi.org/10.1016/j.jhep.2016.04.016 (2016).
Koh, C. et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: a proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. The Lancet. Infectious diseases 15, 1167–1174, https://doi.org/10.1016/S1473-3099(15)00074-2 (2015).
Stockdale, A. J. et al. Prevalence of hepatitis D virus infection in sub-Saharan Africa: a systematic review and meta-analysis. The Lancet. Global health 5, e992–e1003, https://doi.org/10.1016/S2214-109X(17)30298-X (2017).
Huang, C.-R. & Lo, S. J. Evolution and Diversity of the Human Hepatitis D Virus Genome. Advances in Bioinformatics 2010, 323654, https://doi.org/10.1155/2010/323654 (2010).
Gal, F. L. et al. Genetic diversity and worldwide distribution of the deltavirus genus: A study of 2,152 clinical strains. Hepatology, https://doi.org/10.1002/hep.29574 (2017).
Radjef, N. et al. Molecular Phylogenetic Analyses Indicate a Wide and Ancient Radiation of African Hepatitis Delta Virus, Suggesting a Deltavirus Genus of at Least Seven Major Clades. Journal of virology 78, 2537–2544, https://doi.org/10.1128/JVI.78.5.2537-2544.2004 (2004).
Le Gal, F. et al. Eighth Major Clade for Hepatitis Delta Virus. Emerging Infectious Diseases 12, 1447–1450, https://doi.org/10.3201/eid1209.060112 (2006).
Foupouapouognigni, Y., Noah, D. N., Sartre, M. T. & Njouom, R. High Prevalence and Predominance of Hepatitis Delta Virus Genotype 1 Infection in Cameroon. Journal of clinical microbiology 49, 1162–1164, https://doi.org/10.1128/JCM.01822-10 (2011).
Spaan, M. C. et al. Outcome in chronic hepatitis delta: differences between African and non-African patients. Journal of Hepatology 66, S255–256 (2017).
Casey, J. L. et al. Hepatitis B Virus (HBV)/Hepatitis D Virus (HDV) Coinfection in Outbreaks of Acute Hepatitis in the Peruvian Amazon Basin: The Roles of HDV Genotype III and HBV Genotype F. Journal of Infectious Diseases 174, 920–926, https://doi.org/10.1093/infdis/174.5.920 (1996).
Borzacov, L. M. P. et al. Treatment of hepatitis delta virus genotype 3 infection with peg-interferon and entecavir. International Journal of Infectious Diseases 46, 82–88, https://doi.org/10.1016/j.ijid.2016.03.017 (2016).
Brichler, S., Le Gal, F., Butt, A., Chevret, S. & Gordien, E. Commercial Real-Time Reverse Transcriptase PCR Assays Can Underestimate or Fail to Quantify Hepatitis Delta Virus Viremia. Clinical Gastroenterology and Hepatology 11, 734–740, https://doi.org/10.1016/j.cgh.2013.01.025 (2013).
Rodgers, M. A. et al. Identification of rare HIV-1 Group N, HBV AE, and HTLV-3 strains in rural South Cameroon. Virology 504, 141–151, https://doi.org/10.1016/j.virol.2017.01.008 (2017).
Brennan, C. A. et al. The Prevalence of Diverse HIV-1 Strains Was Stable in Cameroonian Blood Donors From 1996 to 2004. JAIDS Journal of Acquired Immune Deficiency Syndromes 49, 432–439, https://doi.org/10.1097/QAI.0b013e31818a6561 (2008).
Coller, K. E. et al. Development and performance of prototype serologic and molecular tests for hepatitis delta infection. Scientific Reports 8, 2095 (2018).
Lole, K. S. et al. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. Journal of Virology 73, 152–160 (1999).
Wilson, E. B. Probable Inference, the Law of Succession, and Statistical Inference. Journal of the American Statistical Association 22, 209–212 (1927).
Measure Demographic Health Survey. HIV Prevalence in Cameroon: Findings from the 2011 DHS-MICS. (2012).
Ndumbe, P. M. Hepatitis D in Yaounde, Cameroon. APMIS 99, 196–198, https://doi.org/10.1111/j.1699-0463.1991.tb05138.x (1991).
Amougou, M. A., Noah, D. N., Moundipa, P. F., Pineau, P. & Njouom, R. A prominent role of Hepatitis D Virus in liver cancers documented in Central Africa. BMC Infectious Diseases 16, 647, https://doi.org/10.1186/s12879-016-1992-2 (2016).
Ducancelle, A. et al. High Endemicity and Low Molecular Diversity of Hepatitis B Virus Infections in Pregnant Women in a Rural District of North Cameroon. PloS one 8, e80346, https://doi.org/10.1371/journal.pone.0080346 (2013).
Romina, S. et al. High Burden of HBV-Infection and Atypical HBV Strains among HIV-infected Cameroonians. Current HIV research 14, 165–171, https://doi.org/10.2174/1570162X13666150930114742 (2016).
Andernach, I. E. et al. Characterization of Hepatitis Delta Virus in Sub-Saharan Africa. Journal of clinical microbiology 52, 1629–1636, https://doi.org/10.1128/jcm.02297-13 (2014).
Sy, B. T. et al. High Prevalence and Significance of Hepatitis D Virus Infection among Treatment-Naïve HBsAg-Positive Patients in Northern Vietnam. PloS one 8, e78094, https://doi.org/10.1371/journal.pone.0078094 (2013).
Makuwa, M. et al. Prevalence and Molecular Diversity of Hepatitis B Virus and Hepatitis Delta Virus in Urban and Rural Populations in Northern Gabon in Central Africa. Journal of clinical microbiology 47, 2265–2268, https://doi.org/10.1128/JCM.02012-08 (2009).
François-Souquière, S., Makuwa, M., Bisvigou, U. & Kazanji, M. Epidemiological and molecular features of hepatitis B and hepatitis delta virus transmission in a remote rural community in central Africa. Infection, Genetics and Evolution 39, 12–21, https://doi.org/10.1016/j.meegid.2015.12.021 (2016).
Aragona, M. et al. Serological Response to the Hepatitis Delta Virus In Hepatitis D. The Lancet 329, 478–480, https://doi.org/10.1016/S0140-6736(87)92090-3 (1987).
Gupta, S., Govindarajan, S., Cassidy, W. M., Valinluck, B. & Redeker, A. G. Acute delta hepatitis: serological diagnosis with particular reference to hepatitis delta virus RNA. The American journal of gastroenterology 86, 1227–1231 (1991).
Opaleye, O. O. et al. Molecular epidemiology of hepatitis D virus circulating in Southwestern Nigeria. Virology Journal 13, 61, https://doi.org/10.1186/s12985-016-0514-6 (2016).
De Paschale, M. et al. Epidemiology of hepatitis D virus (HDV) infection in an urban area of Northern Italy. Infection 40, 485–491, https://doi.org/10.1007/s15010-012-0247-4 (2012).
Perveen, S., Nasir, M. I., Shahid, S. M., Azhar, A. & Khan, O. Y. Phylogenetic analysis of HDV isolates from HBsAg positive patients in Karachi, Pakistan. Virology Journal 9, 162–162, https://doi.org/10.1186/1743-422X-9-162 (2012).
Shirvani-Dastgerdi, E., Amini-Bavil-Olyaee, S., Alavian, S. M., Trautwein, C. & Tacke, F. Comprehensive analysis of mutations in the hepatitis delta virus genome based on full-length sequencing in a nationwide cohort study and evolutionary pattern during disease progression. Clinical Microbiology and Infection 21, 510.e511–510.e523, https://doi.org/10.1016/j.cmi.2014.12.008 (2015).
Makuwa, M. et al. Prevalence and Genetic Diversity of Hepatitis B and Delta Viruses in Pregnant Women in Gabon: Molecular Evidence that Hepatitis Delta Virus Clade 8 Originates from and Is Endemic in Central Africa. Journal of clinical microbiology 46, 754–756, https://doi.org/10.1128/JCM.02142-07 (2008).
Acknowledgements
We would like to acknowledge Dr Mary Kuhns for critical review of the manuscript and Jill Fuhrman for laboratory support.
Author information
Authors and Affiliations
Contributions
E.K.B. and M.A.R. contributed equally to this work. E.K.B., M.A.R., K.E.C., D.B., E.K., A.O., and M.C. collected, analyzed and interpreted the data. D.M., L.K., and N.N. provided key expertise and specimens. E.K.B., M.A.R., K.E.C., L.K., N.N. and G.C. designed the study. E.K.B., M.A.R., and K.E.C. wrote the manuscript. All authors (E.K.B., M.A.R., K.E.C., D.B., E.K., A.O., M.C., D.M., L.K., N.N., and G.C.) critically reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Butler, E.K., Rodgers, M.A., Coller, K.E. et al. High prevalence of hepatitis delta virus in Cameroon. Sci Rep 8, 11617 (2018). https://doi.org/10.1038/s41598-018-30078-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-30078-5
This article is cited by
-
Endemicity and genetic diversity of Hepatitis delta virus among Pygmies in Cameroon, Central Africa
BMC Research Notes (2022)
-
Evolutionary trends in the prevalence of anti-HDV antibodies among patients positive for HBsAg referred to a national laboratory in Cameroon from 2012 to 2017
BMC Research Notes (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
