
Abstract
Matrix metalloproteinases (MMPs) are involved in tissue remodeling. Accordingly, MMP inhibitors and related radiolabeled analogs are important tools for MMP-targeted imaging and therapy in a number of diseases. Herein, we report design, synthesis, and evaluation of a new Arginine-containing macrocyclic hydroxamate analog, RYM, its hydrazinonicotinamide conjugate, RYM1 and 99mTc-labeled analog 99mTc-RYM1 for molecular imaging. RYM exhibited potent inhibition against a panel of recombinant human (rh) MMPs in vitro. RYM1 was efficiently labeled with 99mTcO4− to give 99mTc-RYM1 in a high radiochemical yield and high radiochemical purity. RYM1 and its decayed labeling product displayed similar inhibition potencies against rhMMP-12. Furthermore, 99mTc-RYM1 exhibited specific binding with lung tissue from lung-specific interleukin-13 transgenic mice, in which MMP activity is increased in conjunction with tissue remodeling and inflammation. The results support further development of such new water-soluble Arginine-containing macrocyclic hydroxamate MMP inhibitors for targeted imaging and therapy.
Similar content being viewed by others

Introduction
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases, best known for their role in the degradation of extracellular matrix components. In part based on their enzymatic activity, these secreted or membrane-bound proteins may be classified into several classes i.e., collagenases, gelatinases, stromelysins, membrane-type MMPs, and others. As such, MMPs are involved in complex physiological and biological processes such as, tissue remodeling, wound healing, embryotic development, angiogenesis, cell adhesion, and proliferation. Tissue MMP activity is regulated by the MMP activation state and endogenous inhibitors such as tissue inhibitor of metalloproteinases (TIMPs). Many diseases, such as cancer, atherosclerosis, stroke, aortic aneurysm, cardiomyopathy, arthritis, and chronic obstructive pulmonary disease are associated with dysregulated expression and activation of MMPs1,2,3. Therefore, MMPs are attractive targets for developing new imaging and therapeutic strategies in various diseases.
Over the past decades, various MMP inhibitors including some synthetic small molecules have been developed4,5,6. MMP inhibitors labeled with radioisotopes and fluorophores have been investigated for imaging and characterization of MMP activity in vivo7,8,9,10,11,12,13,14,15. Accordingly, MMP-targeting has emerged as a promising approach for early detection, risk stratification, and therapy in a number of diseases. However, existing agents are suboptimal for in vivo use and novel MMP inhibitors and related imaging agents with improved properties and targeting activities are in high demand.
RP805, is a 99mTc-labeled macrocyclic hydroxamate MMP inhibitor (Fig. 1). Over the past decade, this tracer has been studied for MMP-targeted single-photon emission computed tomography (SPECT) imaging of cardiovascular and other pathologies in several preclinical models16,17,18,19,20,21,22. In vascular pathology, such as atherosclerosis and aneurysm, tissue uptake of RP805 correlates with vessel wall MMP activity and inflammation. Accordingly, this agent may be used to characterize atherosclerotic lesions, and evaluate aortic aneurysm progression and rupture risk. However, RP805 has a relatively prolonged blood circulation which may not be optimal for early time imaging of vascular diseases. In addition, the poor water solubility of RP805 precursor has impeded blocking studies in vivo at suitable concentrations. Therefore, we sought to pursue new macrocyclic hydroxamate analogs and related imaging agents with improved physicochemical properties for in vivo MMP-targeted imaging and therapy, with the ultimate goal of future clinical translation.

Structures of MMP-targeted SPECT imaging agent RP805 and its precursor.
Previous studies performed on anti-succinate-based hydroxamic acids have elucidated the structural requirements of this class of inhibitors for anti-MMP activity, which include a hydroxamate as a zinc-binding site, as well as α, P1′, P2′ and P3′ substituents1. The preferred conformation of this type of inhibitors displays α and P2′ substituents on the same side with close proximity spatially, which can be cyclized as shown in RP805 and its precursor. Wide variations of P3′ substituent can be tolerated. Based on this structure-activity relationship (Fig. 2), we designed a new macrocyclic hydroxamate-based analog, RYM, containing an Arginine (Arg) residue at P3′ position for improving the hydrophilicity. Furthermore, we designed an analog, RYM1, containing a hydrazinonicotinamide (HYNIC) as a precursor for 99mTc-labeling to form 99mTc-RYM1 for pilot studies. Herein, we report the related synthesis, 99mTc labeling, MMP inhibition, and other evaluations in vitro and ex vivo. In vivo evaluation of 99mTc-RYM1, including biodistribution, imaging and binding specificity in murine models of carotid and aortic aneurysm, is reported elsewhere23.

Molecular design of novel arginine-containing hydroxamate-based MMP inhibitors. RYM, RYM1, and SPECT imaging agent 99mTc-RYM1.
Results
Synthesis
As shown in Fig. 3, the synthetic strategy includes an initial synthesis of a protected anti-succinate-based macrocyclic acid analog (I-7)24,25. The synthesis was started from an anti-succinic acid derivative, i.e. (2R,3S)-3-(tert-butoxycarbonyl)-2-isobutylhex-5-enoic acid which was first converted into its benzyl ester (I-1) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and benzyl bromide in toluene. Hydroboration of I-1 by 9-borabicyclo[3.3.1]nonane (9-BBN) in tetrahydrofuran (THF), followed by oxidation with H2O2, gave the hydroxyl derivative (I-2). The conversion of I-2 into its bromide derivative (I-3) was achieved using CBr4 and triphenylphosphine (Ph3P) in dichloromethane (DCM). The benzyl ester group of I-3 was removed by catalytic hydrogenation to give its acid analog (I-4). Coupling of I-4 with L-tyrosine benzyl ester in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) and hydroxybenzotriazole (HOBT) in N,N-dimethylformamide (DMF) gave the anti-succinate-tyrosine benzyl ester (I-5). Cyclization of I-5 in the presence of Cs2CO3 in anhydrous acetonitrile gave the protected anti-succinate macrocyclic derivative (I-6). Finally, the macrocyclic acid analog (I-7) was obtained by removing the benzyl group of I-6 in the presence of 10% palladium on carbon (Pd/C) and HCOONH4 in methanol.

Synthesis of an anti-succinate-based macrocyclic acid intermediate (I-7).
As depicted in Fig. 4, we used Fmoc-Arg(Mtr)-OH as the starting material because the protecting group Mtr is stable in trifluoroacetic acid (TFA) solution compared with another commonly used protecting group Pbf in Fmoc-Arg(Pbf)-OH26. Therefore, Fmoc-Arg(Mtr)-OH was first reacted with CH3NH2 in the presence of EDCI/HOBT in DMF to give Fmoc-Arg(Mtr)-NHCH3 (I-8). De-protection of I-8 with piperidine in DCM gave H-Arg(Mtr)-NHCH3 (I-9). The reaction between I-7 and I-9 in the presence of EDCI/HOBT afforded the Arg(Mtr)-containing macrocycle derivative (I-10), followed by de-protection of I-10 with TFA to give the carboxylic acid analog (I-11). The pre-activation of I-11 in the presence of HOAT, HATU, and DIEA in DMF, followed by reaction with O-(tert-butyldimethylsilyl)hydroxylamine (TBDMS-ONH2) gave I-12. Finally, the target compound RYM was obtained by de-protection of I-12 in a solution of TFA, water, TIS, and phenol.

Synthesis of an Arg-containing anti-succinate-based macrocyclic hydroxamate, RYM.
We used a similar strategy for synthesis of the HYNIC conjugate (2). Therefore, Fmoc-Arg(Mtr)-OH was first reacted with NH2CH2CH2NH-Boc in the presence of EDCI/HOBT in DMF to give Fmoc-Arg(Mtr)- NH2CH2CH2NH-Boc (I-13) as depicted in Fig. 5. De-protection of I-13 with 5% piperidine in DCM gave H-Arg(Mtr)- NH2CH2CH2NH-Boc (I-14). The reaction between I-7 and I-14 in the presence of EDCI/HOBT afforded the Arg(Mtr)-containing macrocycle derivative (I-15), followed by de-protection of I-15 with TFA to give the carboxylic acid analog (I-16). I-16 was reacted with (Boc)2O in aqueous solution of Na2CO3 to form the Boc-protected analog I-17. The pre-activation of I-17 in the presence of HOAT, HATU, and DIEA in DMF, followed by reaction with TBDMS-ONH2 gave I-18. I-18 was de-protected in a solution of TFA, water, TIS, and phenol to give I-19. Boc-HYNIC was prepared in two steps27 and activated in the presence of HATU, HOAT, DIEA, and DIEA in DMF. The activated Boc-HYNIC-OAT was reacted with I-19 to give I-20 which was de-protected in a solution of TFA, TIS, phenol, and water to give the desired HYNIC conjugate, RYM1.

Synthesis of an Arg-containing macrocyclic hydroxamate HYNIC conjugate, RYM1.
The two crude products (RYM and RYM1) were purified by semi-preparative HPLC using aqueous acetonitrile as mobile phase to give the desired fractions as identified by LC-MS. The final products were obtained as white powder from lyophilization, which should be their corresponding TFA salts of the guanidine group. Both were further identified by analytical HPLC, high resolution mass spectroscopy (HR-MS, Supplemental Figure) and 1H/13C NMR analysis.
MMP inhibition assays
MMP inhibition assays were carried out based on the kinetic effects of an inhibitor on the MMP-mediated catalytic cleavage of a fluorogenic substrate, namely Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, as reported by others previously9,23,28. In the first set of studies, the inhibitory effect of RYM was evaluated against 5 activated recombinant human (rh)MMPs, including rhMMP-2, rhMMP-7, rhMMP-9, rhMMP-12, and rhMMP-13. GM-6001, a broad-spectrum MMP inhibitor, was also tested for comparison. As expected, GM-6001 showed potent inhibitory activities against the five rhMMPs without significant selectivity and with the Ki values close to those reported in the literature (Table 1). RYM also had potent MMP inhibition with Ki values at nM concentrations. Unlike GM-6001, RYM showed modest selectivity against MMP-12 among the 5 tested rhMMPs.
Tumor necrosis factor α converting enzyme (TACE) structurally belongs to the ADAM (short for a disintegrin and metalloproteinase) family of proteases, but its catalytic site is similar with that of MMPs25. Therefore, we evaluated RYM and GM-6001 for inhibition against TACE using rhTACE and the fluorogenic peptide substrate, Mca-Pro-Leu-Ala-Gln-Ala-Val-Dpa-Arg-Ser-Ser-Ser-Arg-NH2. RYM had strong TACE inhibition with a Ki value of 4.9 nM, which is close to the Ki value of rhMMP-2 inhibition. Interestingly, GM-6001 showed much weaker inhibition against rhTACE (Ki 113.1 nM) than rhMMPs. Next, we evaluated RYM1 for its inhibition against rhMMP-12 and TACE. This showed Ki values of 2.2 ± 0.5 nM (rhMMP-12)23 and 8.6 ± 2.2 (TACE). As such, both RYM and RYM1 had similar inhibitory potency against rhMMP-12 and TACE.
Radiochemistry
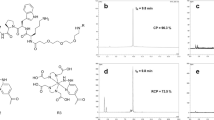
We first labeled RYM1 with 99mTc in a formulation comprised of tricine, mannitol, TPPTS, and pluronic acid in succinate buffer (pH 5.0), as described previously (Fig. 6)22,29. For optimizing radiolabeling, we tested a range of RYM1 from 2 to 10 µg (2.1~10.5 nmol, considering 2 associated TFA molecules) in 2–10 µL dimethyl sulfoxide (DMSO) mixed with 200.0 µL vehicle solution, followed by adding 50–100 µL (5.0–10.0 mCi) of 99mTcO4− solution. The mixture was heated at 95 °C for 10 min and cooled down to room temperature before subjecting it to radio-HPLC analysis. HPLC analysis was performed using aqueous acetonitrile (containing 0.16% ammonium formate) as mobile phase for gradient elution, and showed one single peak of the desired 99mTc-labelled product, i.e. 99mTc-RYM1 at retention time of 13.8 min (Fig. 7a). 99mTc-RYM1 was consistently obtained in a high radiochemical yield and radiochemical purity (>95%) with minimal formation of 99mTc-colloid or 99mTc-co-ligand product (<2%) even adding 10 mCi of 99mTcO4− solution. Based on the initial activity of 5~10 mCi (185~370 MBq) of 99mTcO4− and 2 µg (2.1 nmol) RYM1 added in 250~300 μL solution, the specific activity of 99mTc-RYM1 was calculated as 2.4~4.8 mCi/nmol (89~178 MBq/nmol). We also prepared RP805 precursor and used it for 99mTc labeling similarly. Compared with 99mTc-RYM1 at retention time of 13.8 min, RP805 had a longer retention time of 14.6 min.

99mTc-labeling of RYM1 using 3 different methods.

Radio-HPLC analysis of 99mTc-labeled compounds in saline and blood: (a) comparison of free 99mTcO4−, 99mTc-RYM1 and RP805; (b) stability of 99mTc-RYM1 in saline; (c) stability of RP805 in saline; (d) stability of 99mTc-RYM1 in blood; (e) stability of RP805 in blood; (f) stability of 99mTc-RYM1(tricine)2 in saline. A.U.: arbitrary units.
To further confirm the radiolabeling product, RYM1 was also labeled with tricine and TPPTS in the presence of SnCl2 (Fig. 6). The 99mTc-RYM1 samples obtained from both methods had identical retention time in radio-HPLC spectra. In addition, a mixture of 99mTc and the succinate solution without RYM1 was also heated at 90 °C for 10 min. Radio-HPLC analysis showed only one major peak of [99mTc]-colloid or [99mTc]-co-ligand product at 3.3 min but there was no radioactive peak at 13.8 min. Combined, these results confirmed the successful labeling to give 99mTc-RYM1.
One advantage of HYNIC ligand is to allow the use of different co-ligands such as TPPTS, tricine, and EDDA to give various99mTc labeling analogs of different hydrophilicity30,31,32. Therefore, we did 99mTc-labeling of RYM1 in the presence of SnCl2 and tricine to give a 99mTc-labeled product which was expected to be 99mTc-RYM1 analog with two tricine co-ligands, i.e. 99mTc-RYM1 (tricine)2 (Fig. 6). To this end, a solution of 5 µL/1 mM in DMSO, 99mTc, and tricine-Sn solution was heated at 90 °C for 20 min to give the 99mTc-labeling analog. Radio-HPLC showed the desired radioactive peak at 15.5 min in high radiochemical purity and yield (98%).
Stability studies
The resulting 99mTc-RYM1 was monitored by the radio-HPLC analysis for stability over 24 h post-labeling. As shown in Fig. 7, both 99mTc-RYM1 and RP805 showed good radiochemical stability in saline buffer at room temperature over 24 h. Both also exhibited good stability in murine blood samples incubated at 37 °C for 2 h (Fig. 7). However, the 99mTc-labeled analog containing two tricine co-ligands, i.e., 99mTc-RYM1(tricine)2, showed low stability in saline. The desired peak at 15.5 min was decreased to ~90% after keeping at room temperature for 8 h (Fig. 7f). A new peak at 5.6 min was increased, which might be related with free 99mTc release from 99mTc-RYM1(tricine)2, as suggested by radio-HPLC analysis of the 99mTcO4− solution under the same condition. Based on the ratio of the two radioactive peaks, 90% of 99mTc-RYM1(tricine)2 was decomposed when kept at room temperature for 24 h.
LogP measurements
Compounds RYM and RYM1 have cLogP values of −0.23 and 0.50, respectively. Accordingly, RYM1 has increased hydrophilicity compared with RP805 precursor (cLogP 3.65). The two 99mTc labeled compounds, 99mTc-RYM1 and RP805 were evaluated for their partitions between n-octanol and water or Tris buffer (pH 7.4) (Table 2)33. The negative Log values indicated that both 99mTc-labeled compounds were water soluble. Compared with RP805, the lower partition coefficient and shorter retention time in radio-HPLC indicated the improved hydrophilicity of 99mTc-RYM1, as expected.
Effects of 99mTc-labeling on MMP binding
Next, we evaluated the effect of 99mTc-labeling on RYM MMP inhibition, using rhMMP-12 as a representative enzyme. A solution of 99mTc-RYM1 prepared from 2 µg of RYM1 was decayed at −80 °C for two days, lyophilized, and re-dissolved in saline for MMP inhibition assays. The decayed solution (which contains a mixture of the unlabeled precursor (RYM1), the added co-ligands, and the decayed products 99mTc-RYM1) showed similar MMP inhibition potencies with RYM1 based on the fluorescence change kinetics, i.e. relative fluorescence units per min (RFU/min), suggesting that the labeling procedure had no significant effect on MMP binding (Fig. 8).

MMP-12 inhibitory activities of RYM1 and decayed RYM1 labeling mixture. The data represent the mean ± SD of duplicate determinations. A.U.: arbitrary unit.
Tissue binding ex vivo
We have shown that the lungs of lung-specific interleukin (IL)-13 transgenic (tg) [Club cell 10-kDa protein (CC10)-IL-13 Tg] mice, a pre-clinical model of pulmonary pathology, express elevated MMP activity in conjunction with tissue remodeling and inflammation20. Therefore, as a prelude to in vivo imaging studies23, we evaluated binding of 99mTc-RYM1 to lung tissue homogenates of CC10-IL-13Tg mice (n = 2). Consistent with the MMP-binding property of 99mTc-RYM1, there was considerable retention of this tracer, which was reduced by 87% upon co-incubation with an excess of RYM, demonstrating its binding specificity in the lung tissue (Fig. 9).

Ex vivo binding of 99mTc-RYM1 to lung tissue from lung-specific interleukin-13 transgenic mice without (control) or with (blocking) co-incubation of excess precursor, RYM. The data represent the mean ± SD of duplicate determinations. A.U.: arbitrary unit.
Discussion
We have designed, synthesized, and evaluated a new zwitterionic anti-succinate-based macrocyclic hydroxamate analog RYM, its HYNIC conjugate RYM1 and related 99mTc labeled analog 99mTc-RYM1. The compounds, which have good water solubility and nanomolar inhibition in vitro against a panel of rhMMPs and TACE, represent a new category of dual MMP/TACE inhibitors.
Hydroxamate-based MMP inhibitors such as GM-6001, batimastat (BB-94) and marimastat (BB-2516) bind to the zinc ion of the catalytic domain of activated MMPs for potent broad-spectrum inhibition2,3,34. The clinical use of these inhibitors is hampered by toxicity and side effects, which may stem from lack of targeting specificity and limited selectivity. Accordingly, other non-hydroxamate and hydroxamate MMP inhibitors with improved MMP selectivity have been developed, and are undergoing preclinical and clinical evaluation9,28,35,36,37,38. Despite the limitations of hydroxamate inhibitors as therapeutic agents, these compounds are of value in molecular imaging, which is governed by a different set of criteria. Accordingly, we have used RP805, a 99mTc-labeled hydroxamate tracer, to image MMP activation by SPECT imaging in pre-clinical models of cardiovascular and pulmonary pathology without any evidence of toxicity17,19,20,21,22,39,40,41. Prior to this work, RP805 was the only 99mTc-labeled imaging agent that has shown promise in imaging MMP activation in cardiovascular pathology. However, RP805 has a relatively long circulation time, which could be a barrier to vascular imaging. Indeed, given the physical proximity of the blood pool and vessel wall as well as the small size of the arterial wall, imaging in atherosclerosis, aneurysm and other vascular pathologies is critically dependent on the contrast between their signals. A long clearance time would require delayed imaging which can adversely affect the contrast and signal to background ratio. In addition, the limited water-solubility of RP805 precursor is a barrier for establishing the specificity of the MMP signal in blocking studies. This is a key issue, as most cardiovascular pathologies are associated with inflammation and enhanced tissue permeability that promote non-specific uptake of the tracers. Therefore, it is critical to demonstrate signal specificity for further development and translation. We hypothesized that novel hydroxamate-based MMP inhibitors with distinct physicochemical properties, favorable pharmacokinetics and better in vivo targeting could be attractive candidates for development of alternative tracers with improved imaging characteristics. Given the paucity of promising MMP-targeting imaging agents and aforementioned limitations of RP805, the development of such rationally-designed, improved MMP tracers would have a major impact on cardiovascular medicine with paradigm-shifting potential.
Water solubility or hydrophilicity is a determinant of a drug governing its formulation, pharmacokinetics and in vivo performance. Many MMP inhibitors such as GM-6001 and batimastat (BB-94) have very poor water solubility, and it is reported that glycine incorporation at P3′ is a critical structural component to achieve both good in vitro and oral activity. To develop novel water soluble MMP inhibitors, we first calculated the cLogP values GM-6001 and several related compounds using ChemDraw Professional 16.0 (Fig. 10). Compared with GM-6001 (cLogP: 0.89), the RP805 parent macrocyclic hydroxamate amide analog (cLogP: 1.42) and its glycine-containing analog (cLogP: 0.85), incorporation of an Arg side chain was expected to considerably increase the hydrophilicity of the resulting novel analogs. Indeed, both RYM (cLogP: −0.23) and RYM1 (cLogP: 0.50) had relatively low cLogP values. In addition, the positively charged guanidine of Arg and negatively charged hydroxamate groups displayed around the anti-succinate-based macrocycle scaffold show the structural characteristics of a zwitterionic molecule, a feature which may improve in vivo distribution and specific targeting42,43,44. Therefore, as a novel approach to improving water-solubility and biodistribution, we synthesized several new Arg-containing macrocyclic hydroxamate analogs, including RYM, its HYNIC conjugate (RYM1) and related 99mTc-labeled analog, 99mTc-RYM1.

Chemical structures and cLogP values of hydroxamate-based MMP inhibitors.
Macrocyclic hydroxamate MMP inhibitors can be synthesized starting from anti-succinate derivatives, as reported previously24,25. To synthesize new Arg-containing analogs, we used alternative multiple protection and de-protection strategies in 13 and 16 steps (Figs 3, 4 and 5). This novel synthetic strategy included: (1) synthesis of the orthogonally protected macrocyclic intermediate (I-7), (2) Arg introduction, (3) conversion of carboxylic acid into hydroxamic acid, and (4) HYNIC conjugation. As expected, the Arg-containing macrocyclic hydroxamate MMP inhibitors, RYM and RYM1, showed good water solubility (10 µg/µL). Furthermore, similar to RP805 precursor16,20, the compounds displayed nanomolar inhibition in vitro against a panel of rhMMPs including rhMMP-2, rhMMP-7, rhMMP-9, rhMMP-12, and rhMMP-13, as well as TACE. The results confirmed our hypothesis that the Arg at P3′ can improve the water solubility greatly but does not perturb the binding of hydroxamate group to the catalytic Zn2+ of rhMMPs. Ki values of RYM against 5 rhMMPs showed the relative inhibition potency in the following order: rhMMP-12 > rhMMP-2 > rhMMP-7~rhMMP-13 > rhMMP-9. Albeit not high, the rhMMP-12 selectivity of RYM over the other rhMMPs is remarkable compared with GM-6001, one of non-selective potent hydroxamate MMP inhibitors. We deduce that the selectivity may be ascribed to the macrocyclization between α substituent and P2′ substituent which may favor for rhMMP-12 especially45. Both RYM and RYM1 had similar Ki values against rhMMP-12, indicating the HYNIC motif and related linker might not impact the MMP binding significantly.
Because of the structural similarity between the ADAM and MMP families of proteases, we evaluated the binding of RYM to TACE (ADAM17), as a representative ADAM25. TACE is often upregulated and activated (and implicated) in inflammatory disorders, where MMP activation also plays a key role46. Accordingly, it is reasonable to speculate that by targeting both TACE and MMP activation, RYM-based tracers may prove to be more effective in tracking inflammation in vivo. Direct comparison of these tracers (and selectivity toward other ADAMs) would require dedicated sets of studies that are beyond the scope of this report.
HYNIC has been widely used as a chelator for 99mTc-labeling. It needs two co-ligands such as TPPTS and tricine or two tricine30,31,32. As described for RP805, RYM1 was conveniently labeled with 99mTc in the presence of both TPPTS and tricine in a formulation to form the stable 99mTc-labeling product, 99mTc-RYM1, in high radiochemical purity and yield. The labeling can also be achieved in the presence of SnCl2 to give 99mTc-RYM1 by an alternative method. We also confirmed two co-ligands i.e. TPPTS and tricine led to higher radiochemical stability than two tricine co-ligands. Importantly, compounds RYM1 and the decayed solution of 99mTc-RYM1 displayed a similar inhibition profile against rhMMP-12 enzymatic activity, indicating the 99mTc-labeling procedure does not have a significant effect on key macrocyclic hydroxamate motif, its MMP binding and inhibition. Furthermore, 99mTc-RYM1 exhibited specific binding with lung tissue from IL-13 transgenic mice ex vivo, which express high levels of MMP activity20.
We expected the structural differences between RP805 and 99mTc-RYM1would lead to different biodistribution, pharmacokinetics, and targeting localization. This was empirically demonstrated in murine models, where 99mTc-RYM1 showed faster blood clearance with residual blood activity reduced by more than 50% at 1 h post-injection, and significantly higher specific uptake in several organs relative to RP80523. Importantly, in these studies the MMP signal in aneurysm in vivo correlated with MMP activity, as determined by zymography ex vivo. Similar to aneurysm, MMPs play a key role in the pathogenesis of several pulmonary diseases. We confirmed tissue binding of RYM1 and its specificity in IL-13 Tg lungs, where there is considerable tissue remodeling and inflammation20. This is associated with upregulation of several members of MMP family, including MMP-12 and -13, and enhanced MMP-2 and MMP-12 activity. We have shown the feasibility of MMP-targeted imaging with 99mTc-RP85 in this model and demonstrated that the MMP signal correlates with tissue inflammation20. A future study comparing 99mTc-RYM1 and 99mTc-RP805 imaging in this model is of interest. However, it is impossible to determine to what extent each specific MMP (or ADAM) contributes to 99mTc-RYM1 (or 99mTc-RP805) binding, as this would require highly selective inhibitors that are currently unavailable. Similarly, MMP gene deletion (even if the animals were available) could alter the lung biology and confound the interpretation of the studies.
Combined, our data suggest that 99mTc-RYM1 is a suitable imaging agent for molecular imaging of MMP activation in vivo, especially in vascular pathologies, where rapid blood clearance is critical. While beyond the scope of this manuscript, based on these promising data we are currently working on securing permission for first in-human trial of this tracer. The Arg-containing macrocyclic hydroxamate MMP inhibitors can also serve as lead compounds for structural modification and further development to provide analogs with different linkers and metal chelators for various imaging and therapeutic applications.
Summary
To address the limitations of existing MMP imaging agents, we have designed, synthesized, and evaluated a new Arg-containing anti-succinate-based macrocyclic hydroxamate analog (RYM), its HYNIC conjugate (RYM1) and related 99mTc labeling analog (99mTc-RYM1). Both RYM and RYM1 displayed nanomolar potent inhibition in vitro against a panel of rhMMPs and TACE. RYM1 allows for facile efficient 99mTc-labeling to give its 99mTc-labeled analog, 99mTc-RYM1 in a high radiochemical yield. As a new generation MMP-targeted tracer, 99mTc-RYM1 exhibits specific binding with inflamed lung tissue ex vivo. These results, along with favorable in vivo imaging studies reported elsewhere23, support further development of such water soluble Arg-containing macrocyclic hydroxamate MMP inhibitors with improved physicochemical properties and favorable pharmacokinetics for molecular imaging and potentially, therapeutic applications.
Materials and Methods
All methods were carried out in accordance with relevant guidelines and regulations.
Reagents
(2R,3S)-3-(tert-butoxycarbonyl)-2-isobutylhex-5-enoic acid was purchased from Chem-Stone Co., Ltd., Guangzhou, China; Fmoc-Arg(Mtr), L-Tyrosine benzyl ester, and O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) were fromChem-Impex International, Inc., Wood Dale, Illinois, USA. Dichloromethane, ethyl acetate, methanol, and acetonitrile were from Avantor Performance Materials, Inc, Center Valley, Pennsylvania, USA. Silica gel (70–90 µm, 60 Å) was purchased from Agela Technologies (Wilmington, Delaware, USA). Other solvents and chemicals were purchased from Sigma-Aldrich, St. Louis, Missouri, USA.
Instruments
(1) HPLC Waters HPLC system 2489, UV-Vis detector, 600 controller, and Empower Pro software. (2) LC-MS: Agilent LC-MS 6120B Quadrupole, 6550 A iFunnel Q-TOF. (3) NMR: Agilent NMR DD2 400 MHz with OneProbe (Agilent Technologies, Santa Clara, CA, USA). (4) Fluorescent plate reader (BIO-TEK/Synergy HT).
Synthesis
Intermediate 1 (I-1)
To a stirred solution of (2 R,3 S)-3-(tert-butoxycarbonyl)-2-iso-butylhex-5-enoic acid (10.0 g, 95%, 35.1 mmol) and benzyl bromide (15 g, 85.9 mmol) in toluene (40 mL), were added dropwise 13.0 g (86 mmol) 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in 20 mL toluene. The resulting mixture was stirred at room temperature for 2 h and then at 60 °C for 1 h. The toluene solution was separated from the precipitated solid residue. The residue was dissolved in water (20 mL), and extracted with ethyl acetate (20 mL × 3). The combined toluene and ethyl acetate solution (100 mL) was washed with 1 N HCl (20 mL × 2), water (20 mL × 2) and brine (20 mL × 2). It was dried over anhydrous MgSO4, filtered and concentrated under vacuum. The resulting residue was purified by chromatography (silica gel, 2% ethyl acetate/hexane) to afford 9.0 g (71%) of the title compound as an oil. ES-MS: Observed [MH]+ 360.1 and [MNa]+ 383.2.
Intermediate 2 (I-2)
To a stirred solution of I-1 (9.0 g, 24.9 mmol) in 30 mL anhydrous THF, at 0 °C, was added dropwise 9-BBN in THF (200 mL, 100 mmol) over a period of 30 min and allowed to stir at room temperature overnight. 10 mL water was added dropwise after the solution was cooled in an ice bath. A solution of 9.9 g NaOAc in water (30 mL) was added, followed by the addition of 30% H2O2 (30 mL) dropwise. The mixture was stirred at room temperature for 60 min and concentrated under vacuum. The resulting aqueous solution was extracted with ethyl acetate (20 mL × 4). The combined ethyl acetate solution (80 mL) was washed with 1 N HCl (20 mL × 2), water (20 mL × 2), and brine (20 mL × 2). After it was dried over anhydrous MgSO4, the solution was filtered and concentrated under vacuum. The resulting residue was purified by silica gel chromatography (silica gel, 20% ethyl acetate/hexane) to give the title compound (7.2 g, 70%). ES-MS: observed [MH-tert-butyl]+ 323.0, [MNa]+ 401.1, and [MH-tert-butyl-H2O]+ 305.2.
Intermediate 3 (I-3)
To a stirred solution of I-2 (7.0 g, 19.0 mmol) and CBr4 (12.6 g, 38 mmol) in anhydrous DCM (30 mL), was added Ph3P (10.4 g, 40 mmol) in small portions for 30 min. The resulting mixture was stirred at room temperature for another 2 h, followed by adding 30 mL hexanes. The resulting mixture was transferred to a short column of silica gel for quick elution using DCM and hexane (1:1). The desired fractions were combined and concentrated to get a crude product which was further purified by column chromatography (silica gel, 2% ethyl acetate/hexane). 5.1 g (63%) of the title compound was obtained. ES-MS: Observed double peak [MNa]+ 463.0/465.2 and [M-tert-butyl]+ 385.0/387.0.
Intermediate 4 (I-4)
A mixture of I-3 (5.0 g, 11.3 mmol) and Pd/C (2.0 g, 10%, wet) in 30 mL methanol was stirred under hydrogen gas at room temperature for 30 min. The mixture was filtered, followed by washing the Pd/C with methanol (5 mL × 4). The combined methanol filtrate was concentrated and the product (3.3 g, 85%) obtained was used for the next step without further purification. ES-MS: Observed double peak [MNa]+ 373.0/375.0, [M-tert-butyl-H2O]+ 277.0/279.0 (100%), and [MNa-tert-butyl]+ 318.0/320.0.
Intermediate 5 (I-5)
To a stirred solution of I-4 (3.0 g, 8.5 mmol), HOBT (1.7 g, 12.7 mmol), and Tyr-OBz (3.4 g, 12.7 mmol) in 20 mL anhydrous DMF cooled in an ice bath, was added EDCI (2.4 g, 12.7 mmol) and stirred at room temperature for 2.5 h. The resulting mixture was concentrated under high vacuum, re-dissolved with ethyl acetate (100 mL), and washed with water, 1 N HCl, water, 1 N Na2CO3 solution, water, and brine. The ethyl acetate solution was dried over anhydrous MgSO4, filtered, and concentrated under vacuum. The resulting residue was purified by chromatography (silica gel, 20% ethyl acetate/hexane). 3.8 g (74%) of the title compound was obtained. ES-MS: Observed [MNa]+ double peak 626.0/628.0 and [MH]+ 604.2/606.0.
1H NMR (400 MHz, CDCl3) δ 0.8 (6 H, d, J = 8 Hz), 1.01 (1 H, m), 1.24 (2 H, m), 1.45 (9 H, s), 1.60–1.77 (4 H, m), 2.36 (2 H, m), 2.97 (1 H, m), 3.09 (1 H, m), 3.21 (1 H, m), 3.35 (1 H, m), 4.97 (1 H, m), 5.16 (2 H, m), 5.50 (1 H, s), 6.01 (1 H, m), 6.50–7.37 (9 H, aromatic H); 13C NMR (100 MHz, CDCl3) δ 21.52, 23.96, 25.85, 28.21, 29.77, 30.60, 33.28, 37.64, 40.69, 48.42, 49.17, 53.15, 67.54, 81.41, 115.72, 127.70, 128.73, 128.75, 128.79, 130.51, 135.14, 155.09, 171.58, 173.42, 173.68.
Intermediate 6 (I-6)
To a stirred solution of Cs2CO3 (3.2 g, 23.4 mmol) in 500 mL anhydrous acetonitrile at 60 °C, was added dropwise a solution of I-5 (3.0 g, 5.3 mmol) in 50 mL acetonitrile, over a period of 1 h. The resulting mixture was stirred at 60 °C for another 3 h and concentrated under vacuum. The product was re-dissolved with ethyl acetate and filtered, followed by washing the solid with ethyl acetate for 5 times (10 mL × 5). The combined ethyl acetate filtrate was washed with 1 N HCl solution, water, and brine. The ethyl acetate solution was dried over anhydrous MgSO4, filtered, and concentrated. The resulting residue was purified by silica gel chromatography using (silica gel, 20% ethyl acetate/hexane) to give the product (1.6 g, 60%). ES-MS: observed [MH]+ 524.2, [MNa]+ 546.2, [MH-tert-butyl]+ 468, and [MCs]+ 656.2; 1H NMR (400 MHz, CDCl3) δ −0.47 (1 H, m), 0.61 (1 H, m), 0.75 (6 H, m), 0.81 (1 H, m), 1.21–1.55 (4 H, m), 1.37 (9 H, s), 1.86 (1 H, m), 2.02 (1 H, m), 2.52 (1 H, m), 3.57 (1 H, m), 4.04 (1 H, m), 4.2 (1 H, m), 5.11–5.24 (3 H, m), 5.51 (1 H, m), 6.94–7.40 (9 H, aromatic H); 13C NMR (100 MHz, CDCl3) δ 21.29, 24.05, 25.61, 28.20, 29.93, 31.20, 37.85, 40.55, 49.45, 50.01, 51.61, 67.56, 73.91, 80.81, 120.52, 123.59, 128,67, 128.77, 128.83, 129,87, 131.44, 132.01, 135.21, 159.38, 171.73, 173.11, 174.01.
Intermediate 7 (I-7)
A mixture of I-6 (5.0 g, 9.5 mmol), 10% Pd/C (2.0 g), and HCOONH4 (5 g) in 15 mL CH3OH was stirred at room temperature for 2–3 h until the hydrogen gas evolved was observed. The mixture was further stirred for another 20 min and filtered, followed by washing the Pd/C with CH3OH (5 mL × 4). The methanol filtrate was concentrated and acidified with 1 N HCl solution to get a residue which was extracted with ethyl acetate. The ethyl acetate solution was washed with water (10 mL × 2) and brine (10 mL × 2), dried over anhydrous MgSO4, filtered, and concentrated. The product (3.7 g, 90%) was obtained and used for the next step without further purification. LC-MS: Observed [MH]+ 434.2 and [MNa]+ 456.2.
Intermediate 8 (I-8)
A mixture of Fmoc-Arg(Mtr)-OH (1.2 g, 1.97 mmol), HOBT (0.27 g, 2.0 mmol), CH3NH2 2 M in THF(2 mL, 4.00 mmol) was dissolved in 5 mL anhydrous DMF and cooled at 0~5 °C in an ice bath, followed by adding EDCI (600 mg, 3.1 mmol). The mixture was stirred at room temperature overnight and concentrated under high vacuum. The residue was triturated with 1 N HCl, the solid obtained was filtered, and further washed with 1 N HCl, 5 N Na2CO3, and H2O. The solid product was dried to give 1.03 g (84%) of the title compound. ES-MS: Observed [MH]+ 622.2.
Intermediate 9 (I-9)
1.0 g of I-8 obtained above was dissolved in 10 mL DCM and 3 mL piperidine. The mixture was stirred at room temperature for 30 min and concentrated under vacuum. The product was purified by flash column chromatography (2% Methanol/DCM) to give the title compound (0.54 g, 85%). ES-MS: Observed [MH]+ 400.2 and [M-H]− 398.1.
Intermediate 10 (I-10)
A mixture of I-7 (200.0 mg, 0.46 mmol), I-9 (219.7 mg, 0.55 mmol), and HOBT (115.0 mg, 0.55 mmol) were dissolved in 5 mL DMF and cooled at 0~5 °C in an ice bath, followed by adding EDCI (100 mg, 0.6 mmol). The mixture was stirred at room temperature overnight and concentrated. The residue was dissolved in 10 mL CH2Cl2, washed with 1 N HCl, H2O, and brine. The DCM solution was dried over MgSO4, filtered, and concentrated. The product was purified by flash column chromatography (2% methanol/DCM) to give the title compound (300.0 mg, 80%). ES-MS: Observed [MH]+ 815.4.
Intermediate 11 (I-11)
250 mg of I-10 was dissolved in 4 mL TFA and 1 mL DCM. After stirred at room temperature for 2 h, the solution was concentrated and dried under vacuum to get the title compound in a quantitative yield. ES-MS: Observed [MH]+ 759.3, [MH2]2+ 392.1.
Intermediate 12 (I-12)
A mixture of I-11 (200 mg, 0.26 mmol), HOAT (86.0 mg, 0.6 mmol), HATU (236.0 mg, 0.6 mmol), and DIEA (16.0 mg, 0.9 mmol) was dissolved in 4 mL anhydrous DMF. The mixture was stirred at room temperature for 20 min, followed by adding TBDMS-ONH2 (137.0 mg, 0.93 mmol). The mixture was further stirred at room temperature overnight and concentrated. The residue was triturated with 1 N HCl, the solid obtained was filtered, and washed with 1 N Na2CO3, water and brine. The product was further purified by flash column chromatography to give 100.0 mg (50%) of the title compound.
Target compound, RYM
I-12 (50 mg, 0.065 mmol) was dissolved in a mixture of 9.0 mL TFA, 0.5 mL H2O, 0.25 mL TIS, and 0.25 g phenol. The mixture was stirred at room temperature overnight and concentrated under vacuum. The residue was added into 10 mL cooled diethyl ether. The precipitated product was collected by centrifugation and purified by semi-preparative HPLC using an eluent of aqueous acetonitrile. The desired fractions were collected and lyophilized to give the title compound (10.0 mg, 22%) as confirmed by both LC-MS and analytical HPLC. ES-MS: Observed [MH]+ 562.3, [MH2]2+ 281.7, and [M-H]− 560.2; 1H NMR (400 MHz, CDCl3) δ 0.8 (3 H, dd, J = 6.6, 2.6 Hz), 0.85 (3 H, d, J = 6.5 Hz), 1.46–1.22 (4 H, m), 1.79–1.55 (2 H, m), 1.85 (1 H, dq, J = 9.8, 6.7 Hz), 2.17 (1 H, td, J = 11.1, 3.4 Hz), 2.77–2.60 (1 H, m), 3.21 (2 H, p, J = 6.7 Hz), 3.39 (1 H, dt, J = 13.6, 4.4 Hz), 4.24–4.07 (2 H, m), 4.38 (1 H, ddd, J = 13.8, 8.0, 5.7 Hz), 4.38 (1 H, ddd, J = 13.8, 8.0, 5.7 Hz), 5.03–4.89 (1 H, m), 6.91 (1 H, dt, J = 8.3, 2.7 Hz), 7.09 (1 H, dd, J = 8.3, 2.4 Hz), 7.17 (1 H, dd, J = 8.3, 2.2 Hz), 7.26 (1 H, dd, J = 8.4, 2.2 Hz), 8.30 (1 H, dd, J = 23.6, 9.7 Hz); 13C NMR (100 MHz, CDCl3) δ 20.27, 23.08, 24.97, 25.60, 29.33, 29.53, 29.92, 36.52, 40.10, 40.10, 40.60, 46.95, 47.17, 47.38, 47.59, 47.66, 47.81, 47.88, 48.02, 48.23, 52.73, 53.58, 73.15, 120.17, 122.42, 128.88, 132.38, 157.26, 158.99, 171.34, 172.10, 172.56, 174.81.
HYNIC
A mixture of 6-chloronicotinic acid (10.0 g, 63.4 mmol), hydrazine (20 mL), and water (20 mL) was refluxed at 95~100 °C for 4 h. The mixture was concentrated under vacuum and the residue was dissolved in 40 mL water. The resulting solution was acidified with 1 N HCl to reach a pH 5~5.5. The solution was kept in a refrigerator overnight. The precipitated solid was collected by filtration, washed with cold water (10 mL × 2) and ether (10 mL × 2). The solid was dried to give 8.5 g (87%) of the title compound. ES-MS: Observed [MH]+ 154.1.
Boc-HYNIC
A mixture of 4.0 g HYNIC and 8.0 g Na2CO3 in 60 mL of 1,4-dioxane and 60 mL of water was cooled in an ice bath. 8.2 g of Boc2O was added, and stirred at room temperature for 5 h. The mixture was concentrated and the residue was acidified with 1 N HCl solution, the precipitated solid was collected by filtration and dried to give the title compound.
Intermediate 13 (I-13)
A mixture of Fmoc-Arg(Mtr)-OH (1.2 g, 1.97 mmol), HOBT (0.32 g, 2.36 mmol), and NH2CH2CH2NHBoc (0.38 g, 2.36 mmol) were dissolved in 5 mL anhydrous DMF and cooled at 0~5 °C in an ice bath, followed by adding 543.0 mg EDCI (2.83 mmol). After stirred at room temperature overnight, the mixture was concentrated under high vacuum. The residue was triturated with 1 N HCl, filtered, and washed with 1 N HCl, 1 N Na2CO3, and H2O. The solid product was dried to give 1.3 g (90%) of the title compound. ES-MS: Observed [MH]+ 529.6.
Intermediate 14 (I-14)
I-13 (1.0 g, 1.33 mmol) was dissolved in 7 mL DCM and 3 ml piperidine. After stirred at room temperature for 30 min, the mixture was concentrated under high vacuum. The residue was purified by flash column chromatography using DCM and methanol to give 0.52 g (75%) of the title compound. ES-MS: Observed [MH-Boc]+ 429.2.
Intermediate 15 (I-15)
A mixture of I-14 (200.0 mg, 0.46 mmol), H-Arg(Mtr)-NHCH2CH2NH-Boc (291.0 mg, 0.55 mmol), and HOBT (74.8 mg, 0.55 mmol) was dissolved in 5 mL DMF and cooled at 0~5 °C in an ice bath, followed by adding EDCI (126.5 mg, 0.66 mmol). The mixture was stirred at room temperature overnight and concentrated. The residue was dissolved in 10 mL CH2Cl2, washed with 1 N HCl, H2O, and brine. The DCM solution was dried over MgSO4, filtered, and concentrated. The product was purified by flash column chromatography using DCM and methanol to give 260 mg (60%) of the title compound. ES-MS: Observed [MH-Boc]+ 845.8 and [MH2-tert-butyl]2+ 394.7.
Intermediate 11 (I-16)
I-15 (0.2 g, 0.21 mmol) was dissolved in 8 mL TFA and 2 mL DCM. After stirred at room temperature for 2 h, the solution was concentrated and dried under vacuum to give 0.18 g (98%) of the title compound. ES-MS: Observed [MH]+ 788.3 and [MH2]2+ 394.8.
Intermediate 12 (I-17)
I-16 (0.15 g, 0.16 mmol) was dissolved in 5 mL dioxane and 5 mL 1 N Na2CO3 solution. The mixture was stirred in an ice bath, followed by adding (Boc)2O (70.0 mg, 0.32 mmol). After stirring at room temperature for 2 h, the solution was concentrated and the residue was acidified with 1 N HCl to pH 3. The product was extracted with DCM, washed with water, and dried over MgSO4. The filtrate was concentrated to give the title compound (135.0 mg, 95%). ES-MS: Observed [MH]+ 888.3
Intermediate 13 (I-18)
A mixture of I-17 (100.0 mg, 0.11 mmol), HOAT (30.4 mg, 0.22 mmol), HATU (83.6 mg, 0.22 mmol), and DIEA (57.0 mg, 0.44 mmol) were dissolved in 5 mL anhydrous DMF. The mixture was stirred at room temperature for 20 min, followed by adding TBDMS-ONH2 (147.2 mg, 1.0 mmol). The mixture was stirred at room temperature overnight and concentrated under vacuum. The residue was triturated with 1 N HCl, filtered, and washed with water, 1 N Na2CO3, and brine. The product was further purified by column chromatography to give 30.0 mg (30%) of the title compound. ES-MS: Observed [MH]+ 903.2 and [MNa]+ 925.4.
Intermediate 14 (I-19)
I-18 (20 mg, 0.022 mmol) was dissolved in a mixture of 9.0 mL TFA, 0.5 mL H2O, 0.25 mL TIS, and 0.25 g phenol. The mixture was stirred at room temperature overnight and concentrated under vacuum. The residue was added into 10 mL cooled diethyl ether. The precipitated product was collected by centrifugation and purified by semi-preparative HPLC using an eluent of aqueous acetonitrile. The desired fractions were collected and lyophilized to give the title compound (5.4 mg, 30%). ES-MS: Observed [MH]+ 591.4, [MH2]2+ 296.3, and [M2H3]3+ 394.6.
Intermediate 20 (I-20)
A mixture of Boc-HYNIC (10.0 mg, 0.039 mmol), HOAT (5.5 mg, 0.039 mmol), and HATU (15.0 mg, 0.039 mmol) was dissolved in 1 mL anhydrous DMF, followed by adding DIEA (10.0 mg, 0.078 mmol). The mixture was stirred at room temperature for 30 min. To the resulting solution was added a solution of I-19 (16.0 mg, 0.0195 mmol) and DIEA (5 µL, 3.71 mg, 0.028 mmol) in 0.5 mL anhydrous DMF. The resulting solution was stirred at room temperature for 1 h and concentrated under vacuum. The residue was triturated with 1 N HCl, filtered, and washed with 1 N Na2CO3 and water. The solid product collected was used in the next reaction without further purification. ES-MS: Observed [MH]+ 826.2 & [MH2]2+ 413.6.
RYM1, ((6 S,7 R,10 S)-N10-((S)-5-guanidino-1-((2-(6-hydrazinylnicotinamido)ethyl) amino)-1-oxopentan-2-yl)-N6-hydroxy-7-isobutyl-8-oxo-2-oxa-9-aza-1(1,4)-benzenacyclo undecaphane-6,10-dicarboxamide)
I-20 was dissolved in a mixture of 4.5 mL TFA, 0.25 mL H2O, 0.125 mL TIS, and 0.125 g phenol. The mixture was stirred at room temperature for 30 min and concentrated under vacuum. The residue was added into 10 mL cooled diethyl ether. The precipitated product was collected by centrifugation and purified by semi-preparative HPLC using an eluent of aqueous acetonitrile. The desired fractions were collected and lyophilized to give the title compound (3.0 mg, 16%) as confirmed by both LC-MS and analytical HPLC. ES-MS: Observed [MH]+ 726.3 and [MH2]2+ 363.7.
MMP and TACE inhibition assays
The recombinant human MMPs (rhMMPs) including rhMMP-2, rhMMP-7, rhMMP-9, rhMMP-12, and rhMMP-13 were purchased from R&D Systems (Minneapolis, MN, USA). A buffer consisting of 50 mM Tris, 10 mM CaCl2, 150 mM NaCl, 0.05% Brij-35 (w/v) (pH 7.5, 1,3,5-Trichloro-2-nitrobenzene) was used for all the MMP assays. A mixture of the rhMMPs (500 ng, 9 µL in assay buffer) and p-aminophenylmercuric acetate (APMA, 1 µL, 10.0 mM in DMSO) was incubated at 37 °C (1 h/rhMMP-2, 2 h/rhMMP-7, 24 h/rhMMP-9, 4 h/rhMMP-12, and 2 h/rhMMP-13) according to the procedures described by R&D Systems. The activated MMPs were diluted to 500 µL in the assay buffer for inhibition assays. The assays were carried out in 96-well black non-binding surface microplates (Fisher Scientific). Seventy microliter (for inhibitor wells) and 80 µL (control wells) of the assay buffer were first added into each well as designed, followed by adding 10 µL (10 ng) of the activated MMP solutions into each well. The inhibitor solutions at 4 different concentrations (10 µL each well) were added into the corresponding inhibitor wells in the microplate which was then swirled at room temperature for 30 min. Ten microliter of the fluorogenic MMP substrate solutions (R&D SYSTEMS®) at 4 different concentrations (20.0, 30.0, 40.0, and 50.0 µM) in assay buffer were added into each well to make a total volume of 100 µL. The microplate fluorescence was measured at 320 nm (excitation) and 405 nm (emission) wavelengths for 30 min (1.44 min intervals, sensitivity 100, shaking intensity 4 and duration 30 s) by a fluorescent plate reader (BIO-TEK/Synergy HT) to generate the fluorescence kinetic curves and related mean velocity values (v), i.e. relative fluorescence units per min (RFU/min). Finally, the competitive inhibition constants (Ki value) were calculated from the mean velocity values, and related substrate (S) and inhibitor (I) concentrations using GraphPad Prism 6 by the following equation for competitive inhibition, where \({V}_{{\max }}\) is the maximal velocity, the \({K}_{M}\) Michaelis-Menten constant:
TACE inhibition was evaluated similarly. Recombinant Human TACE/ADAM17 and a fluorogenic peptide substrate i.e. Mca-P-L-A-Q-A-V-Dpa-R-S-S-S-R-NH2 (R&D SYSTEMS®) were purchased from R&D Systems (Minneapolis, MN). A buffer consisting of 25 mM Tris, 2.5 μM ZnCl2, 0.005% Brij35 (w/v) (pH 9.0) was used for TACE inhibition assays.
Radiolabeling with 99mTc and HPLC analysis
RYM1 was labeled with 99mTc by heating it with 99mTcO4 in a vehicle solution containing tricine (6.5 mg/ml), 3,3′,3″-phosphanetriyltris(benzenesulfonic acid) trisodium salt (TPPTS) (5.0 mg/ml), pluronic F-127 (0.1 mg/ml), succinic acid (12.7 mg/ml), sodium succinate (38.5 mg/ml) and Mannitol (40 mg/ml) in high purity and yield. Typically, 2 µg of RYM1 in 2.0 µL (1 mM) DMSO was mixed ~10 mCi/100 µL 99mTcO4, followed by adding 200 µL of the vehicle solution in a vial. The mixture was heated at 95 °C for 10 min, and cooled to room temperature to yield the 99mTc-labeled product, 99mTc-RYM1, as analyzed by radio-HPLC. Radio-HPLC analysis was performed using Waters RP-HPLC (Milford, MA) on a C-18 reverse-phase analytical column (Phenomenex, Jupiter® 4 µm Proteo 90 Å, 250 × 4.6 mm, Torrance, CA). The mobile phase consisted of Solvent A (aqueous solution containing 25 mM ammonium formate) and solvent B (90% aqueous CH3CN containing 25 mM ammonium formate). The gradient was initiated at a flow rate of 1 mL/min and from 90.0 A% for 2 min, followed by a linear gradient to 30% A over 15 min and isocratic run for another 5 min at a flow rate of 1 mL/min over 40 min. UV chromatograms were monitored at both 254 nm and 214 nm. For stability studies, the 99mTc-labeled product was similarly monitored for degradation by RP-HPLC. As analyzed by HPLC under such a condition, 99mTc-RYM1 showed a single peak at retention time of 13.8 min.
LogP measurement
An aliquot (10.0 µL/~100 µCi) of RP-805 or 99mTc-RYM1 was added into a mixture of 500.0 µL octanol and 500.0 µL water or tris solution (1 M, pH 7.4) in a 1.5 mL Eppendorf vial. The vial was mixed by vortex violently for 3 min and then centrifuged at 13,000 rpm for 5 min. The two phases i.e. octanol and aqueous layers separated were obtained carefully using a micropipette. The two solutions were centrifuged at 13,000 rpm for 5 min. The samples (10.0 µL) taken from the octanol layer and the aqueous layer were measured for radioactivity in triplicates by r-counter (PerkinElmer, Waltham, MA). The logPoctanol/water and logPoctanol/tris values were calculated by the following equation: logP = log (decay-corrected radioactivity in octanol sample/decay-corrected radioactivity in aqueous sample).
Tissue binding
Ex vivo binding of 99mTc-RYM1 to biological tissue was evaluated in lung tissue homogenate of CC10-IL-13Tg mice (n = 2) showing elevated basal MMP activity20. Lung tissue (247 ± 85 µg, in duplicates was incubated with 3 nM of 99mTc-RYM1 for 60 min at 37 °C with or without the preincubation of 20 µM of RYM. After tracer incubation, tissue homogenate was spin down and washed 3 times with PBS, before resuspension in protein lysis buffer, gamma-counting (Wizard2, PerkinElmer) and protein concentration was measured (Pierce BCA Protein Assay Kit, Thermo Fisher Scientific and Multiskan Ascent Microplate Photometer, Thermo Labsystems) in order to determine the amount of tissue used. All experimental protocols were approved by Yale University and VA Connecticut Institutional Animal Care and Use Committees.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Hu, J., V den Steen, P. E., Sang, Q. X. & Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov 6, 480–498, https://doi.org/10.1038/nrd2308 (2007).
Vandenbroucke, R. E. & Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov 13, 904–927, https://doi.org/10.1038/nrd4390; http://www.nature.com/nrd/journal/v13/n12/abs/nrd4390.html#supplementary-information (2014).
Cathcart, J., Pulkoski-Gross, A. & Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis 2, 26–34, https://doi.org/10.1016/j.gendis.2014.12.002 (2015).
Borkakoti, N. Matrix metalloprotease inhibitors: design from structure. Biochemical Society transactions 32, 17–20, doi:10.1042/ (2004).
Dorman, G. et al. Matrix metalloproteinase inhibitors: a critical appraisal of design principles and proposed therapeutic utility. Drugs 70, 949–964, https://doi.org/10.2165/11318390-000000000-00000 (2010).
Nam, D. H., Rodriguez, C., Remacle, A. G., Strongin, A. Y. & Ge, X. Active-site MMP-selective antibody inhibitors discovered from convex paratope synthetic libraries. Proc Natl Acad Sci USA 113, 14970–14975, https://doi.org/10.1073/pnas.1609375114 (2016).
Freskos, J. N. et al. Design and synthesis of MMP inhibitors with appended fluorescent tags for imaging and visualization of matrix metalloproteinase enzymes. Bioorg Med Chem Lett 23, 5566–5570, https://doi.org/10.1016/j.bmcl.2013.08.050 (2013).
Wagner, S. et al. Novel fluorinated derivatives of the broad-spectrum MMP inhibitors N-hydroxy-2(R)-[[(4-methoxyphenyl)sulfonyl](benzyl)- and (3-picolyl)-amino]-3-methyl-butanamide as potential tools for the molecular imaging of activated MMPs with PET. J Med Chem 50, 5752–5764, https://doi.org/10.1021/jm0708533 (2007).
Bordenave, T. et al. Synthesis and in Vitro and in Vivo Evaluation of MMP-12 Selective Optical Probes. Bioconjug Chem 27, 2407–2417, https://doi.org/10.1021/acs.bioconjchem.6b00377 (2016).
Hugenberg, V. et al. A new class of highly potent matrix metalloproteinase inhibitors based on triazole-substituted hydroxamates: (radio)synthesis and in vitro and first in vivo evaluation. J Med Chem 55, 4714–4727, https://doi.org/10.1021/jm300199g (2012).
Hugenberg, V. et al. Inverse 1,2,3-triazole-1-yl-ethyl substituted hydroxamates as highly potent matrix metalloproteinase inhibitors: (radio)synthesis, in vitro and first in vivo evaluation. J Med Chem 56, 6858–6870, https://doi.org/10.1021/jm4006753 (2013).
Hugenberg, V. et al. Radiolabeled Selective Matrix Metalloproteinase 13 (MMP-13) Inhibitors: (Radio)Syntheses and in Vitro and First in Vivo Evaluation. J Med Chem 60, 307–321, https://doi.org/10.1021/acs.jmedchem.6b01284 (2017).
Kalinin, D. V. et al. Novel Potent Proline-Based Metalloproteinase Inhibitors: Design, (Radio)Synthesis, and First in Vivo Evaluation as Radiotracers for Positron Emission Tomography. J Med Chem 59, 9541–9559, https://doi.org/10.1021/acs.jmedchem.6b01291 (2016).
Schrigten, D. et al. A new generation of radiofluorinated pyrimidine-2,4,6-triones as MMP-targeted radiotracers for positron emission tomography. J Med Chem 55, 223–232, https://doi.org/10.1021/jm201142w (2012).
Razavian, M. et al. Optical imaging of MMP-12 active form in inflammation and aneurysm. Sci Rep 6, 38345, https://doi.org/10.1038/srep38345 (2016).
Su, H. et al. Noninvasive targeted imaging of matrix metalloproteinase activation in a murine model of postinfarction remodeling. Circulation 112, 3157–3167 (2005).
Tavakoli, S. et al. Matrix metalloproteinase activation predicts amelioration of remodeling after dietary modification in injured arteries. Arterioscler Thromb Vasc Biol 31, 102–109 (2011).
Fujimoto, S. et al. Molecular imaging of matrix metalloproteinase in atherosclerotic lesions: resolution with dietary modification and statin therapy. J Am Coll Cardiol 52, 1847–1857 (2008).
Golestani, R. et al. Imaging vessel wall biology to predict outcome in abdominal aortic aneurysm. Circ Cardiovasc Imaging 8, e002471, https://doi.org/10.1161/CIRCIMAGING.114.002471 (2015).
Golestani, R. et al. Matrix Metalloproteinase-Targeted Imaging of Lung Inflammation and Remodeling. J Nucl Med 58, 138–143, https://doi.org/10.2967/jnumed.116.176198 (2017).
Jung, J. J. et al. Multimodality and molecular imaging of matrix metalloproteinase activation in calcific aortic valve disease. J Nucl Med 56, 933–938, https://doi.org/10.2967/jnumed.114.152355 (2015).
Razavian, M. et al. Molecular imaging of matrix metalloproteinase activation to predict murine aneurysm expansion in vivo. J Nucl Med 51, 1107–1115 (2010).
Toczek, J. et al. Preclinical Evaluation of RYM1, a Matrix Metalloproteinase-Targeted Tracer for Imaging Aneurysm. J Nucl Med 58, 1318–1323, https://doi.org/10.2967/jnumed.116.188656 (2017).
Xue, C. B. et al. Rational design, synthesis and structure-activity relationships of a cyclic succinate series of TNF-alpha converting enzyme inhibitors. Part 2: lead optimization. Bioorganic & medicinal chemistry letters 13, 4299–4304 (2003).
Xue, C. B. et al. Design, synthesis, and structure-activity relationships of macrocyclic hydroxamic acids that inhibit tumor necrosis factor alpha release in vitro and in vivo. J Med Chem 44, 2636–2660 (2001).
Fujino, M. W. M. and Kitada, C. 4-Methoxy-2,3,6-trimethylbenzenesulphonyl (Mtr):a new amino protecting group in peptide synthesis. J. Chem. Soc., Chem. Commun., 445–446 (1982).
Abrams, M. J. et al. Technetium-99m-human polyclonal IgG radiolabeled via the hydrazino nicotinamide derivative for imaging focal sites of infection in rats. J Nucl Med 31, 2022–2028 (1990).
Devel, L. et al. Development of selective inhibitors and substrate of matrix metalloproteinase-12. J Biol Chem 281, 11152–11160 (2006).
Liu, S. et al. Towards developing a non-SnCl2 formulation for RP444, a new radiopharmaceutical for thrombus imaging. J Pharm Sci 90, 114–123 (2001).
Ji, S., Zhou, Y., Shao, G. & Liu, S. Evaluation of K(HYNIC)(2) as a bifunctional chelator for (99 m)Tc-labeling of small biomolecules. Bioconjug Chem 24, 701–711, https://doi.org/10.1021/bc3006896 (2013).
Liu, S. The role of coordination chemistry in the development of target-specific radiopharmaceuticals. Chem Soc Rev 33, 445–461, https://doi.org/10.1039/b309961j (2004).
Liu, S., Edwards, D. S. & Barrett, J. A. 99mTc labeling of highly potent small peptides. Bioconjug Chem 8, 621–636 (1997).
Muftuler, F. Z. et al. Synthesis, radiolabeling and In Vivo tissue distribution of an anti-oestrogen glucuronide compound, (99m)Tc-TOR-G. Anticancer Res 30, 1243–1249 (2010).
Overall, C. M. & Kleifeld, O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer 94, 941–946 (2006).
Arkadash, V. et al. Development of High Affinity and High Specificity Inhibitors of Matrix Metalloproteinase 14 through Computational Design and Directed Evolution. J Biol Chem 292, 3481–3495, https://doi.org/10.1074/jbc.M116.756718 (2017).
Gege, C. et al. Discovery and evaluation of a non-Zn chelating, selective matrix metalloproteinase 13 (MMP-13) inhibitor for potential intra-articular treatment of osteoarthritis. J Med Chem 55, 709–716, https://doi.org/10.1021/jm201152u (2012).
Li, W. et al. A selective matrix metalloprotease 12 inhibitor for potential treatment of chronic obstructive pulmonary disease (COPD): discovery of (S)-2-(8-(methoxycarbonylamino)dibenzo[b,d]furan-3-sulfonamido)-3-methylbu tanoic acid (MMP408). J Med Chem 52, 1799–1802 (2009).
Monovich, L. G. et al. Discovery of potent, selective, and orally active carboxylic acid based inhibitors of matrix metalloproteinase-13. J Med Chem 52, 3523–3538, https://doi.org/10.1021/jm801394m (2009).
Razavian, M. et al. Atherosclerosis plaque heterogeneity and response to therapy detected by in vivo molecular imaging of matrix metalloproteinase activation. J Nucl Med 52, 1795–1802 (2011).
Razavian, M. et al. Lipid lowering and imaging protease activation in atherosclerosis. J Nucl Cardiol 21, 319–328, https://doi.org/10.1007/s12350-013-9843-7 (2014).
Jung, J. J. et al. Matrix metalloproteinase inhibitor, doxycycline and progression of calcific aortic valve disease in hyperlipidemic mice. Sci Rep 6, 32659, https://doi.org/10.1038/srep32659 (2016).
Choi, H. S. et al. Synthesis and in vivo fate of zwitterionic near-infrared fluorophores. Angew Chem Int Ed Engl 50, 6258–6263, https://doi.org/10.1002/anie.201102459 (2011).
Choi, H. S. et al. Targeted zwitterionic near-infrared fluorophores for improved optical imaging. Nat Biotechnol 31, 148–153 (2013).
Chopra, A. In Molecular Imaging and Contrast Agent Database (MICAD) (2004).
Russo, A., Aiello, C., Grieco, P. & Marasco, D. Targeting “Undruggable” Proteins: Design of Synthetic Cyclopeptides. Curr Med Chem 23, 748–762 (2016).
Gooz, M. ADAM-17: The Enzyme That Does It All. Critical reviews in biochemistry and molecular biology 45, 146–169, https://doi.org/10.3109/10409231003628015 (2010).
Acknowledgements
This work was supported by grants from NIH (R01-HL112992, R01-HL138567–01), Connecticut Department of Public Health (2016–0087) and Department of Veterans Affairs (I0-BX001750).
Author information
Authors and Affiliations
Contributions
Y.Y., J.T., K.G., H.K., J.H., M.R., R.G., J.Z. and J.J. performed the experiments. T.L.W. and M.G. helped with chemical analyses. M.M.S. designed the experiments. Y.Y., J.T., and M.M.S. wrote the first draft of the paper. All authors contributed to its revision.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ye, Y., Toczek, J., Gona, K. et al. Novel Arginine-containing Macrocyclic MMP Inhibitors: Synthesis, 99mTc-labeling, and Evaluation. Sci Rep 8, 11647 (2018). https://doi.org/10.1038/s41598-018-29941-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29941-2
This article is cited by
-
Selective Imaging of Matrix Metalloproteinase-13 to Detect Extracellular Matrix Remodeling in Atherosclerotic Lesions
Molecular Imaging and Biology (2022)
-
Evaluation of cardiac allograft vasculopathy by positron emission tomography
Journal of Nuclear Cardiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
