
Abstract
MicroRNAs are potent regulators of gene expression that have been widely implicated in reproduction and embryo development. Recent studies have demonstrated that miR-21, a microRNA extensively studied in the context of disease, is important in multiple facets of reproductive biology including folliculogenesis, ovulation, oocyte maturation and early mammalian development. Surprisingly, little is known about the mechanisms that regulate miR-21 and no studies have characterized these regulatory pathways in cumulus-oocyte complexes (COCs). We therefore investigated miR-21 in an in vitro model of bovine oocyte maturation. Levels of the primary transcript of miR-21 (pri-miR-21) and mature miR-21 increased markedly in COCs over the maturation period. Cloning of the bovine pri-miR-21 gene and promoter by 5′3′RACE (rapid amplification of cDNA ends) revealed a highly conserved region immediately upstream of the transcription start site and two alternatively-spliced variants of pri-miR-21. The promoter region contained several putative transcription factor binding sites, including two for signal transducer and activator of transcription 3 (STAT3). Mutation of these sites significantly decreased both the intrinsic activity of pri-miR-21 promoter-luciferase constructs and the response to leukemia inhibitory factor (LIF) (a STAT3 activator) in cultured MCF7 cells. In COCs, treatment with a STAT3 pathway inhibitor markedly decreased pri-miR-21 expression and prevented cumulus expansion. Pri-miR-21 expression was also inhibited by the protein synthesis inhibitor cycloheximide, suggesting that a protein ligand or signaling cofactor synthesized during maturation is necessary for transcription. Together these studies represent the first investigation of signaling pathways that directly influence miR-21 expression in bovine oocytes and cumulus cells.
Similar content being viewed by others


Introduction
Oocyte maturation consists of nuclear and cytoplasmic events that are required for the oocyte to acquire competency for fertilization, and subsequent embryo development should fertilization occur. Cumulus oophorus (typically known as cumulus) cells are an essential group of somatic cells surrounding the oocyte. They develop from granulosa cells into this specialized subtype during the antral stage of folliculogenesis1,2,3 (see review by4) and support the oocyte during maturation and fertilization. Removal of oocytes from their surrounding cumulus cells negatively impacts nuclear and cytoplasmic maturation and subsequent development to the blastocyst stage once the oocyte is fertilized5,6,7,8,9. The relationship between the oocyte and cumulus cells is multi-factorial and complex. During the final stages of maturation, cumulus cells propagate endocrine and biochemical signals to promote meiotic progression and the acquisition of oocyte competence while simultaneously responding to regulatory cues from the oocyte10,11,12 (reviewed by13). Many factors present in this microenvironment strongly influence cumulus cell gene expression, which in turn leads to the functional changes necessary for extracellular matrix synthesis, cumulus expansion, and the readiness of the cumulus oocyte complex (COC) for fertilization14,15,16.
Changes in gene expression that occur in cumulus cells during maturation include substantial modulation in the levels of microRNAs (miRNAs), an important class of endogenous small non-coding RNAs17,18,19 that have emerged as potent regulators of gene expression20,21 (see commentary by22). miRNA genes are highly conserved among eukaryotes and participate broadly in development, physiology, and pathology by influencing specific processes such as cell proliferation, differentiation, signal transduction and apoptosis23. miRNAs typically act by repressing the activity of target genes through anti-sense base-pairing with complementary sequences on the 3′ untranslated region (UTR) of messenger RNAs (mRNAs), causing translational repression or deadenylation and decay of these mRNA targets24,25 and resulting in post-transcriptional gene silencing. Comparative approaches, small RNA-sequencing and bioinformatics analysis have revealed that a substantial number of miRNA genes are expressed in bovine oocytes and their surrounding cumulus cells, a subset of which undergo dynamic changes in abundance over the course of in vitro maturation (IVM) and early embryonic development26,27,28,29. These profiles of global miRNA changes occurring in COCs have been useful for generating hypotheses regarding the potential regulatory function of miRNA during IVM, however mechanistic studies that establish particular roles for specific miRNAs are limited.
One miRNA that has been the subject of considerable research in the reproductive system and other contexts is miR-21, which plays an important, though incompletely understood role in the ovary. mir-21 is dynamically expressed throughout the estrus cycle30, and is the most abundant miRNA in bovine29 and ovine30 cumulus cells and among the three most abundant miRNAs in human cumulus cells31. miR-21 expression increases in ovine follicles throughout folliculogenesis, with a marked expression in growing follicles and a further increase observed in fully grown preovulatory follicles30. miR-21 is also abundantly expressed in periovulatory follicles in mice and furthermore, injection of miR-21 antagonists into murine ovarian bursa leads to increased apoptosis in cumulus cells and a reduction in number of recovered COCs in its corresponding oviduct, with a 50% decrease in ovulation suggesting important functional roles in ovulation and the regulation of apoptosis32. In vitro, a marked rise in miR-21 expression has been reported in cumulus cell and oocyte compartments during maturation in porcine COCs33, as well as in bovine oocytes and in vitro produced embryos, where miR-21 peaks at the 8-cell stage and is suspected to play a role in the degradation of maternal transcripts at the maternal zygotic transition (MZT)26,34.
Despite the obvious temporal associations with critical periods of oocyte and embryo development, the mechanisms directly controlling the induction of this gene in COCs are presently not well understood. miRNAs are first transcribed as long primary (pri-) transcripts that undergo sequential enzymatic cleavage steps before they are functional35. The initiation of miRNA processing is executed in the nucleus by the RNase III enzyme Drosha36, in conjunction with the RNA binding protein DiGeorge syndrome critical region 8 (DGCR8)37,38 that together form the minimal constituents of the “Microprocessor complex”38,39. In the present study we have examined pri-miR-21 and miR-21 expression levels in order to identify factors and mechanisms that may participate in miR-21 induction and processing during IVM. We identify STAT3 as a potent activator of pri-miR-21 transcription in cumulus cells. STAT3 acts as both a signal transducer and activator of transcription, and becomes activated as a DNA binding protein through tyrosine phosphorylation40. In response to cytokine signaling, STAT3 is typically phosphorylated and activated by a cascade that begins with ligand-initiated dimerization of receptor components, including the common signal transducer gp13041. These signals are propagated by the phosphorylation of Janus kinases (JAKs)42, which, in turn, activate STAT3 by phosphorylation. STAT3 is a major transcriptional activator downstream of cytokines from the gp130/interleukin-6 (IL-6) family, including IL-640 and leukemia inhibitory factor (LIF)43, both of which are expressed in cumulus cells and are capable of inducing cumulus expansion44,45. We also document that miR-21 is induced under multiple IVM culture conditions after oocyte aspiration from follicles, and suggest that miR-21 is regulated by ligands synthesized and secreted by the COC itself.
Results
Primary (pri)-miR-21 and mature miR-21 increase in bovine cumulus cells and oocytes during in vitro maturation of COCs
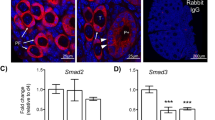
To determine whether miR-21 and its precursor transcript, pri-miR-21, undergo dynamic changes in expression in bovine COCs throughout in vitro oocyte maturation, we first performed qRT-PCR on RNA isolated from pools of oocytes and cumulus cells collected at specific time points throughout that period. We have previously analysed miR-21 over the course of in vitro oocyte maturation (IVM) and fertilization34, and found a significant rise in oocyte miR-21 levels throughout IVM. We confirmed that finding here (Fig. 1A), in order to compare the oocyte expression profile with the expression of pri-miR-21 and miR-21 in cumulus cells, which are expected to be a major contributor of miR-21 (Fig. 1B). Pri-miR-21 transcript levels rose significantly in cumulus cells over the course of maturation, with an average increase of approximately 6-fold over the first 7 hours of maturation, followed by a slight decrease at 24 hours of maturation (Fig. 1B). The rise in pri-miR-21 transcripts coincides with an overall 25–fold increase in mature miR-21 molecules from the beginning to end of IVM (Fig. 1B). In oocytes, an observable though not statistically significant change in pri-miR-21 transcripts is seen between the germinal vesicle (GV) to metaphase II (MII) stage, yet a major change in mature miR-21 levels (~80-fold) was detectable at the end of the maturation period (Fig. 1A). In order to confirm this presence of mature miR-21 in the somatic and oocyte compartments of the COC after in vitro maturation, RNA in situ hybridization was performed with Locked Nucleic Acid (LNA) oligonucleotide probes specific for miR-21 or the small nuclear RNA (snRNA) U6, which was used as control (Fig. 1C). miR-21 and snRNA U6 are both detectable in the mature oocyte and its surrounding cells, in confirmation of the qRT-PCR results. miR-21 is strongly expressed in the cumulus compartment where it localizes to the cytoplasm as expected for a mature miRNA, while snRNA U6 is detected predominantly in cumulus cell nuclei. As an additional control, hybridization was performed with a scrambled miRNA probe (Supplemental Fig. 1). The scrambled probe did not have a complete absence of fluorescent signal, suggesting that its sequence might bind partially to some RNA sequences in the COC.

Primary miR-21 transcript and miR-21 are induced in bovine COCs during maturation in vitro. qRT-PCR quantification of pri-miR-21 transcripts and mature miR-21 immediately after aspiration (0 hour) and at 7 or 24 hours of maturation in (A) oocyte (germinal vesicle (GV) to metaphase II (MII) stage) and (B) cumulus cell compartments of COCs. Gene expression is shown relative to the 0 hour or GV time point. Error bars represent SEM. N = 3 biological replicates. Different letters indicate statistically significant differences between groups. (C) RNA in situ hybridization of miR-21 or snRNA U6 in sectioned mature COCs. Nuclei counterstained with DAPI. Linear look-up tables. Scale bar represents 10 μm. (D) qRT-PCR quantification of cumulus expansion related transcripts and cytokine factors after aspiration (0 hour) and at 7 or 24 hours of maturation. Gene expression is shown relative to the 0 hour time point. Error bars represent SEM. N = 3 biological replicates. Different letters indicate statistically significant differences between groups.
The decrease observed by qRT-PCR in cumulus cell pri-miR-21 transcripts coincides with the observed marked rise in mature miR-21 at 24 hours (Fig. 1B), which is expected when transcripts are being actively processed by the Microprocessor. Similarly to cumulus cells, significant changes in mature miR-21 are not observed in oocytes until pri-miR-21 becomes detectable. Peak pri-miR-21 expression in cumulus cells occurs concurrently with an anticipated rise in the cumulus expansion gene hyaluronan synthase 2 (Has2), the EGF factor amphiregulin (Areg), and the cumulus expansion related gene TNF alpha induced protein 6 (Tnfaip6), which undergoes further induction after the 7 hour time point (Fig. 1D). The interleukin 6 (IL-6) family cytokines Il6 and Lif, as well as the downstream mediator of cytokine action Stat3 also rise over this time.
Full length primary miR-21 cloning identifies two distinct transcript variants that result from alternative splicing
To begin to elucidate whether the observed changes in pri- and mature miR-21 expression in cumulus cells might be due to changes in precursor transcript expression, we first cloned and characterized the full-length primary miR-21 transcript in Bos taurus using a rapid amplification of cDNA ends (RACE) technique. Oligonucleotides designed to prime amplification of known segments of pri-miR-21 (gene specific primers) were paired with oligonucleotides designed to prime RNA adapters ligated to the 5′ or 3′ pri-miR-21 transcript ends. RT-PCR was then used to amplify this transcript from cDNA generated from bovine testis tissue, a rich source of pri-miR-21 transcripts. Two 5′ end products and one 3′ end product were amplified, isolated and cloned into sequencing vectors. Sequence analysis of the bovine reference genome UMD 3.1.1 demonstrated that both of the amplified 5′ end products and the 3′ end product align exclusively to chromosome 19, beginning at position 11,030,051 and ending at position 11,033,585, where they flank the previously annotated MIR21 gene (a 72 nucleotide hairpin containing mature miR-21) on the forward strand. The 5′ end products reside within an intron of the protein-coding gene vacuole membrane protein 1 (VMP1) between exons 10 and 11 of that gene, and the longer of the two products contains VMP1 exon 11, intron 11–12, and exon 12. The shorter product aligns to the longer product with a 1072 nucleotide gap that represents the exclusion of VMP1 intron 11–12, and the sequence reads directly from VMP1 exon 11 to exon 12. A hexameric poly(A) signal (AAUAAA) was predicted on the 3′ end product by PolyA Signal Miner tool, at the expected location, 15 nucleotides upstream of the beginning of the sequenced poly(A) tail46. Together, these data were used to predict a 3538 nucleotide transcript and a 2466 nucleotide variant of the pri-miR-21 transcript that would result from splicing (Fig. 2A). To confirm this prediction, cloning primers were designed near the 5′ and 3′ ends of the sequenced RACE products, and these primers were used to amplify full-length pri-miR-21 from cDNA of bovine tissues and cells by RT-PCR. Both of the predicted variants are expressed in the testis, ovary, oocytes, and fetal fibroblasts (Fig. 2B). Since these transcripts can only be cloned from a cDNA template, the cloning products are slightly shorter than the actual transcripts (3458 and 2386 nucleotides, respectively) because suitable primers could not be placed at the extreme 5′ and 3′ termini. Sequencing analysis and alignment of cloned pri-miR-21 variants with the bovine reference genome UMD 3.1.1 revealed two single nucleotide differences within the region corresponding to VMP1 intron 11–12: two additional thymines are present at position 840–842, and a deletion of one thymine at position 1052 is evident, which together result in a transcript length 1 nucleotide longer than what we originally predicted based on the reference genome. The full sequence length of pri-miR-21 is 3539 nucleotides, and not 3538 nucleotides as predicted. Both the 3539 nucleotide and 2466 nucleotide transcript variant sequences were submitted to GenBank and have been assigned the accession numbers: Bos taurus microRNA pri-miR-21, transcript variant 1 (MF966934), Bos taurus microRNA pri-miR-21, transcript variant 2 (MF966935).

Bovine primary miR-21 exists as two transcript variants and contains a conserved putative upstream promoter region. (A) Predicted pri-miR-21 transcript variants based on RACE-PCR derived sequences. (B) DNA gel electrophoresis of RT-PCR amplified pri-miR-21 transcript variants in bovine tissues and cells. A very faint band representing pri-miR-21 transcript variant 1 in testis is detectable. NTC; no template control. (C) Bos taurus pri-miR-21 variant 1 plus 1 kb upstream region; alignment to Mus musculus Ch11 and Homo sapiens Ch17. Major ticks on x-axis = 1000 nucleotides, minor ticks = 500 nucleotides. Blue = coding exon, salmon = intronic region, yellow = untranslated region, red = intergenic region. Height of conservation plot at each position represents the number of nucleotides conserved out of a 100 nucleotide window centered at that position. Horizontal pink bars denote evolutionarily conserved regions (ECR) to the bovine reference genome. ECR defined as a minimum 70% identity over 100 nucleotide regions. Vertical hatched line denotes pri-miR-21 transcription start site.
The proximal promoter region and transcription start site for pri-miR-21 lie within a conserved intronic region of VMP1
Although evidence for specific factor dependent regulators of miR-21 transcription has increased recently, it has primarily been derived from studies in human cell lines and mouse models. No promoter analysis studies of the MIR21 gene have been published to date in the cow. We therefore sought to compare the bovine promoter region predicted by our analysis of the transcription start site (TSS) to previously published data on the human promoter47, and to the region of conserved sequence in the mouse. A region of the bovine reference genome UMD 3.1.1 containing full length pri-miR-21 and 1 kilobase upstream of the site TSS was used as the template to search for evolutionarily conserved regions between the cow, mouse and human genomes. As expected, the most highly conserved region among the three aligned genomes represents mature miR-21 (100% conservation) (Fig. 2C). The 72 bases representing the MIR21 hairpin is also highly conserved. The total aligned region contains intron 10–11, exon 11, intron 11–12, exon 12 and the 3′ untranslated region of the conserved VMP1 gene found in all 3 species. Across the aligned sequences, the second-most highly conserved region occurs, immediately upstream of the pri-miR-21 TSS, which lies within VMP1 intron 10–11. This region of the aligned sequences has a much higher proportion of conserved bases than protein coding exons 11 and 12 of VMP1, the 3′ untranslated region of VMP1, or intron 11–12, which is excluded by splicing in the pri-miR-21 transcript variant 2 (Fig. 2A) and shows minimal conservation between species (Fig. 2C). The highly conserved region in VMP1 intron 10–11 is described for the remainder of this text as Bos taurus miRNA proximal promoter region (miPPR)-21. Importantly, although the pri-miR-21 promoter and TSS occur within VMP1, previous studies including an investigation in granulosa cells have shown that VMP1 is expressed independently of pri-miR-21 and mature miR-2132,47. We therefore consider miPPR-21 to be the major regulatory region driving pri-miR-21 transcription.
MiRNA proximal promoter region 21 contains two conserved binding sites for signal transducer and activator of transcription 3 (STAT3)
STAT3 is a potent transcriptional regulator in gp130 cytokine signaling pathways, which is important in late follicle development and during oocyte maturation, the period during which miR-21 expression was seen to increase (Fig. 1A,B). Furthermore, the IL-6/gp130 pathway has been found to induce miR-21 via STAT3-mediated mechanisms in human myeloma cells48. We therefore postulated that STAT3 participates in pri-miR-21 transcription in the cow and examined the miPPR-21 sequence for the presence of STAT3 transcription factor binding site consensus sequences. In silico analysis revealed putative sites at 16 and 47 nucleotides upstream of the transcription start site (Fig. 3A). We next generated a promoter/reporter construct consisting of a 411 nucleotide fragment of the miPPR-21 containing both STAT3 binding sites cloned upstream of the Firefly luciferase gene (pGL3miR21-Luc). When transfected into MCF7 cells, this construct was highly active and further responsive to treatment with 15 ng/mL LIF. Elimination of one or both of the STAT3 sites by site-directed mutagenesis, significantly decreased basal reporter activity and essentially eliminated LIF-responsiveness compared to the wild-type reporter construct (Fig. 3B). This was particularly obvious with elimination of the more distal STAT3 site.

Two functional STAT3 binding sites are present on miPPR21. (A) Putative STAT3 binding sites from −47 to −38 positions and from −16 to −7 positions upstream of pri-miR-21 transcription start site (TSS) (+1) on miPPR-21. (B) Promoter reporter assay of a Firefly luciferase vector driven by bovine miPPR-21 (pGL3miR21-Luc) containing the two STAT3 binding sites shown in (A). Constructs containing mutations in both binding sites (I), a single binding site (II, III), or wild-type (IV) were co-transfected into MCF7 cells with a Renilla luciferase construct for normalization. The pGL3-Luc base construct containing no promoter element (V) was also transfected as a control. Grey rectangles indicate STAT3 binding sites, and X indicates a mutation in that binding site. MCF7 cells were treated with 15 ng/mL recombinant human LIF, and luciferase activity in untreated and LIF-treated cells is shown relative to baseline activity of wild-type construct (IV) in untreated conditions. Firefly luciferase light units normalized to Renilla luciferase light units are shown. Asterisks indicate significant differences between untreated and LIF treated cells transfected with the same construct. Constructs sharing same letters did not show significant differences in luciferase activity under the influence of LIF.
STAT3 inhibition during COC in vitro maturation reduces pri-miR-21 transcription and disrupts cumulus expansion
In order to determine whether STAT3 activation was important for pri-miR-21 expression in the context of oocyte maturation, we evaluated pri-miR-21 expression in COCs relative to STAT3 activation status. Activation by tyrosine phosphorylation at residue 705 (Y705) is required for STAT3 to induce downstream changes in gene expression in response to cytokine stimulation49. We therefore first examined the dynamics of STAT3 activation in cumulus cells during in vitro maturation of COCs and then confirmed the effect of a specific small molecule inhibitor of STAT3 phosphorylation: Stattic50. While total STAT3 protein levels did not change, the levels of phosphorylated STAT3 changed markedly in several different stages in cumulus cells over the first 8 hours of maturation. Phospho-STAT3 was detectable immediately upon COC retrieval from follicles, consistent with a previous report of STAT3 status in granulosa cells from bovine follicles dissected from ovaries37, although this decreased over the first 2 hours of culture (Fig. 4A). Re-activation of STAT3 began in culture, and phosphorylated protein first became detectable at 6 hours of maturation, with the strongest pSTAT3 signal present after 8 hours of maturation (Fig. 4A). In some samples, pSTAT3 migrates as a doublet with a faint lower band, which is expected for the commercial antibody used in this study. At all culture time points where phosphorylated STAT3 could be detected, phosphorylation was completely inhibited by Stattic. Based on the observation that STAT3 activation was robust after 6 hours of IVM, we evaluated pri-miR-21 transcript abundance in cumulus cells of COCs removed from culture at 7 hours of maturation (thus allowing sufficient time to observe the resultant transcriptional response) under standard conditions, supplemented with 10 μM Stattic or dimethyl sulfoxide (DMSO) vehicle. Under all conditions, pri-miR-21 showed increased abundance compared to the 0 hour time point, however pri-miR-21 induction was decreased by over 50% in the presence of Stattic (Fig. 4B). Importantly, after 24 hours of in vitro maturation COCs cultured in maturation media supplemented with Stattic also fail to undergo cumulus expansion compared to untreated controls (Fig. 4C). No detectable oocyte STAT3 phosphorylation was evident by Western blot (data not shown) despite abundant STAT3 protein in the oocyte, which is consistent with previous studies that suggest STAT3 is not activated in the oocyte51. We therefore focused our remaining studies on STAT3 mediated transcriptional activation on the cumulus cell compartment.

STAT3 signaling modulates the expression of pri-miR-21 in bovine cumulus cells and STAT3 inhibition disrupts cumulus expansion in vitro. (A) Representative Western blot of STAT3 phosphorylation (Y705) and inhibition by Stattic in cumulus cells from 0–8 hr IVM. Shown with total STAT3. Beta-actin is used as a loading control. Cropped images of a single blot are shown. Full-length blots are presented in Supplemental Fig. 2. Three independent experiments were performed with similar results. (B) qRT-PCR of pri-miR-21 at 0 and 7 hr IVM in cumulus cells in control media, DMSO vehicle (VEH) and 10 μM Stattic (STT). Gene expression is shown relative to the 0 hour time point. Error bars represent SEM. N = 3 biological replicates. Different letters indicate statistically significant differences between groups. (C) Light micrographs of immature (0 hr) and mature (24 hr) COCs matured in DMSO vehicle (VEH) or 10 μM Stattic (STT). Images obtained with EVOS FL Cell Imaging System using 4x objective.
miR-21 induction in COCs is independent of cumulus expansion, and is regulated by factors synthesized by the COC itself
To determine whether the observed changes in miR-21 expression occur as a result of the introduction of hormones or serum supplementation during culture in vitro, miR-21 expression was re-evaluated in the cumulus cells of COCs after 24 hours in culture under serum free conditions, without supplements, and compared to cultures supplemented with FSH alone, LIF alone, or FSH and LIF, and also with standard IVM conditions in media containing 2% FBS and supplemented with LH, FSH and estradiol. As shown in Fig. 5A, miR-21 was induced under all conditions, and increased significantly over 24 h of in vitro culture, including serum free culture without the supplementation of hormones or growth factors. This induction occurred independently from cumulus expansion, as COCs cultured without serum in the absence of supplements did not expand, and COCs cultured serum free in the presence of LIF expanded only slightly (Fig. 5B).

miR-21 is induced in bovine COCs over 24 hours in culture with or without serum and hormone supplementation but is sensitive to inhibition of protein synthesis. (A) qRT-PCR of miR-21 in cumulus cells of COCs at 0 hr or 24 hr IVM culture in serum free conditions with no growth or hormone supplementation (no supp), LIF alone, FSH alone, or FSH + LIF, as compared to standard IVM media (std IVM) which is further supplemented with LH, estradiol and FBS. Gene expression is shown relative to the 0 hour time point. Error bars represent SEM. N = 3 biological replicates. Different letters indicate statistically significant differences between groups. (B) Light micrographs of immature COCs (0 hr) and COCs matured in vitro under the same conditions described in (A). Images obtained with EVOS FL Cell Imaging System using 4x objective. (C,D) qRT-PCR quantification of pri-miR-21 transcripts and mature miR-21 in (C) oocytes and (D) cumulus cells after 24 hr culture in standard IVM media containing ethanol vehicle (VEH) or cycloheximide (CHX). Gene expression is shown relative to VEH. Error bars represent SEM. N = 3 biological replicates. Different letters indicate statistically significant differences between groups. (E) Representative Western blot of STAT3 phosphorylation (Y705) in cumulus cells at 7 hr IVM, in serum free, non-supplemented media containing ethanol vehicle (VEH), cycloheximide (CHX) or CHX + LIF. Shown with total STAT3. Beta-actin is used as a loading control. Cropped images of a single blot are shown. Full-length blots are presented in Supplemental Fig. 3. Three independent experiments were performed with similar results. (F) qRT-PCR quantification of pri-miR-21 transcripts in cumulus cells after 24 hr culture in serum free, non-supplemented media as described in (E). Gene expression is shown relative to VEH. Error bars represent SEM. N = 3 biological replicates. Different letters indicate statistically significant differences between groups.
The induction of miR-21 under basal (serum-, hormone- and growth factor-free) culture conditions in cumulus cells after COC removal from the ovary suggested that ligands synthesized by cumulus cells or the oocyte during in vitro maturation induced miR-21 expression through autocrine or paracrine signaling. To address this possibility, de novo protein synthesis was inhibited by treatment with cycloheximide, as described8,52. In oocytes treated with cycloheximide, pri-miR-21 was completely undetectable and the expression of mature miR-21 was also prevented (Fig. 5C). In the corresponding cumulus cells, cycloheximide prevented the expression of mature miR-21 while no significant effects were noted on the abundance of pri-miR-21 transcripts (Fig. 5D), consistent with a transcriptional response that does not require the synthesis of additional protein factors. Cycloheximide also inhibited the STAT3 phosphorylation that is normally observed in cumulus cells after 7 hours of IVM in the absence of serum, hormone, and growth factor supplementation (Fig. 5E) suggesting that this requires synthesis of an endogenous protein ligand, receptor, or signaling factor. The importance of the synthesis of an endogenous factor in the control of pri-miR-21 expression under these conditions is reflected in the marked reduction of pri-miR-21 expression under minimally supplemented culture conditions in contrast to the absence of suppression in the presence of serum (Fig. 5D,F).
Ligands such as LIF that activate the Janus kinase (JAK)/STAT pathway are known to be synthesized in cumulus cells during the first 6 hours of IVM in the mouse, where they are capable of activating autocrine regulatory loops45. To determine whether LIF can induce phosphorylation of STAT3 in the absence of serum, hormones, and other growth factors, recombinant LIF was added to the minimal culture condition in the presence of cycloheximide. The addition of exogenous LIF to serum and hormone free culture media supplemented with cycloheximide was sufficient to activate STAT3 phosphorylation (Fig. 5E), however LIF-driven STAT3 activation alone was not sufficient to fully restore pri-miR-21 expression in the presence of cycloheximide in the serum free conditions (Fig. 5F).
Discussion
This study characterizes the dynamics of miR-21 expression and reveals STAT3 as a transcriptional activator of miR-21 in the cumulus compartment of the bovine cumulus oocyte complex. Pri-miR-21 transcription is induced in cumulus cells over the first 7 hours of maturation while the mature form of miR-21 does not peak until 24 hours of IVM, which is consistent with active miRNA transcription and processing by the Microprocessor complex. The RNaseIII Drosha forms the enzymatic core of this complex, and has recently been shown to be expressed in cumulus cells53. Pri-miR-21 transcripts as well as mature miR-21 also accumulate in the oocyte over this time period, though mRNA transcription generally decreases in fully-grown GV oocytes and largely ceases after germinal vesicle breakdown (GVBD)54,55. The accumulation of miR-21 in the oocyte is likely the result of multiple factors. Some transcriptional activity in the oocyte, has been shown to occur for specific transcripts that may contribute to the events of oocyte maturation and early embryo development56, and transport of precursor and mature miRNA from cumulus cells to the oocyte during IVM via transzonal projections has also been suggested57. The possibility of miRNA transport from cumulus cells to the oocyte would be consistent with well-established roles for cumulus cells in providing factors to the oocyte that facilitate the acquisition of developmental competence, and demonstrates how ligands that signal through STAT3 have the potential to influence gene expression in the oocyte without activation of STAT3 protein in the oocyte itself.
Physical interactions between activated STAT3 and its cognate binding sites in the human miR-21 promoter have been demonstrated previously58. Here we show that the Bos taurus miR-21 promoter contains two functional STAT3 binding sites, and that promoter activity in response to the gp130 cytokine LIF is significantly reduced when these binding sites are mutated. Pri-miR-21 transcription is reduced in cumulus cells when COCs are cultured in the presence of an inhibitor of STAT3 phosphorylation, which markedly decreases cytokine-induced miR-21 transcription in other models59.
In our model of in vitro oocyte maturation, the increase in pri-miR-21 over the initial 7 hours of IVM coincides strongly with the induction of key cumulus expansion related genes Has2 and Tnfaip6, identified in previous bovine studies60,61 and confirmed here. Has2 and Tnfaip6 (which is induced significantly by 7 hours and undergoes a further increase throughout IVM) are involved in the formation of the hyaluronan-rich cumulus extracellular matrix14,62 and are among a subset of genes induced during cumulus expansion that are associated with inflammation. Ovulation has many characteristics typical of an inflammatory response63, and a number of genes previously considered to be limited to immune cells have been found in follicular cells64,65 (thoroughly reviewed by66). These cells are also capable of secreting and responding to inflammatory cytokines, a number of which can be detected in follicular fluid67. Evidence is mounting that cytokines play important roles at the peri-ovulatory period in vivo and during the final maturation of cumulus-oocyte complexes matured in vitro64 (see review by68. A number of genes upregulated during FSH-induced COC expansion in vitro including Prostaglandin-endoperoxide synthase 2 (Ptgs2), Has2, Tnfaip6, and Pentraxin-related protein (Ptx3) are induced during IVM by STAT3 activating cytokines45.
LIF is a member of the gp130-binding family of cytokines that also includes IL-6, IL-11 and oncostatin M (OSM) which share the common membrane receptor and signal transducer gp13069,70. A structurally similar membrane receptor to gp130 (leptin receptor, OB-R) is activated by the hormone leptin and also results in phosphorylation of STAT3 and the induction of responsive genes71. At the mRNA level, our data clearly demonstrate that Lif is strongly induced in cumulus cells during oocyte maturation in the cow, and that LIF alone can partially induce cumulus expansion under minimal culture conditions. This contrasts to some extent with findings in the mouse, where IL-6 appears to be the predominant gp130 binding cytokine. While Il6 is clearly present, Lif was strongly induced during in vitro maturation, prompting us to focus on its potential effects in this system. This is clearly relevant to species differences likely to exist between cattle and mice with respect to the predominant gp130 cytokines, since overlapping functions of these cytokines are likely to result in largely conserved outcomes in this cellular context.
The LIF/LIF-receptor complex (gp130 plus the ligand-specific receptor) is expressed in bovine cumulus cells72 and supplementation with LIF benefits oocyte maturation, cumulus expansion and embryo development in humans, mice and domestic animal species44,73,74,75. Bovine COCs matured with the addition of LIF to in vitro culture media showed a significant increase in the number of oocytes reaching metaphase II as well as enhanced cytoplasmic oocyte maturation, as indicated by a greater proportion of oocytes showing cortical granule distribution consistent with oocyte competency73. IL-6 and leptin are clearly also important in many aspects of follicular development and in vitro oocyte maturation, and may have similar effects here through their common effects on STAT3 activation. Importantly, evidence shows that LIF, IL-6, and leptin are all capable of inducing miR-2148,58,59,76 in different cell types. As miR-21 induction is the primary focus of the current study, we chose to employ this ligand as a relevant activator of STAT3 in the context of bovine cumulus and oocyte biology.
STAT3 is a critical signaling molecule and transcriptional activator in the ovary, particularly in the peri-ovulatory period and during in vitro maturation. The present study provides clear evidence of a specific role for STAT3 in the induction of miR-21 in bovine cumulus cells, however this analysis would be incomplete without a discussion of the additional signaling cascades that are likely contribute to miR-21 induction during IVM. Treatment with Stattic reduced pri-miR-21 accumulation by ~50% compared to vehicle controls, however pri-miR-21 still increased significantly from the 0 hour time point.
While we have clearly shown that STAT3 activation induces pri-miR-21 transcription during cumulus expansion and oocyte maturation, it should be emphasized here that other signaling pathways are also likely to be important in this highly conserved cellular response. Redundant and synergistic activation is likely by factors such as amphiregulin77 acting through multiple intracellular signaling cascades including STAT3, Ras/Raf/MAPK, ERK1/2 and activator protein 1 (AP-1) family transcription factors78 which can initiate pri-miR-21 transcription47. Furthermore, gp130 cytokines can also act through these other pathways to reinforce signaling responses73,79,80. Moreover, a recent report suggests that miR-21 can also be induced by oocyte-secreted factors such as Growth-differentiation factor 9 (GDF9)53. The potential for these pathways to activate pri-miR-21 transcription is strongly supported by the presence of multiple distinct cis-acting elements in the cloned promoter region (Supplemental Table 1). The presence of multiple pathways which are capable of inducing this important miRNA highlight its potential importance in oocyte and cumulus cell biology.
While the actual combination of ligands that normally induce miR-21 via JAK/STAT signaling in the cumulus oophorus remain to be determined, the data presented here and the studies discussed above demonstrate that COCs synthesize one or more ligands capable of initiating signaling cascades that increase miR-21 expression. Our data reveal that, under standard culture conditions, cycloheximide treatment abrogates miR-21 expression but does not significantly impair pri-miR-21 transcription in cumulus cells, indicating that the processing of precursor miRNAs into mature forms requires protein synthesis but the transcriptional response does not, and may be initiated by factors normally present in serum or media. When protein synthesis is inhibited under serum- and hormone-free conditions pri-miR-21 transcription is essentially absent and STAT3 phosphorylation is markedly reduced, strongly supporting the physiological relevance of the STAT3 transcriptional activation identified in our reporter studies. Importantly however, since the addition of exogenous LIF does not restore pri-miR-21 transcription in cycloheximide-treated COCs, it is likely that the synthesis of additional protein participants in the signaling pathways is necessary for pri-miR-21 induction in maturing COCs. Together with the literature discussed above, our results suggest that cumulus cell miR-21 induction is likely a product of both paracrine and autocrine signaling in the COCs for which STAT3 activation is necessary but not sufficient.
The physiologic roles that miR-21 may play in the control of cumulus cell gene expression and function are likely to be complex and lie outside the scope of this study. miR-21 has been widely implicated in apoptosis and cellular proliferation through the modulation of cellular factors such as programmed cell death 4 (PDCD4)81 and phosphatase and tensin homolog (PTEN)82, both of which are important in granulosa cell and oocyte biology33,83,84. While an increased miR-21 clearly accompanies normal oocyte maturation, some clinical studies have shown that abnormally elevated miR-21 may actually correlate with impaired fertility85. In cattle, elevated miR-21 expression correlates with large atretic follicles when compared to large healthy follicles86. Notably, follicular atresia in cattle also correlates with enhanced LIF/STAT3 signaling in granulosa cells87, which may reflect a functional association in this context. While miR-21 inhibition induces apoptosis in ovarian cells and reduces ovulation32,53, elevated miR-21 is associated with follicular atresia, poor ovarian response and inflammatory disease. It is therefore likely that an optimal level of miR-21 is necessary for a healthy periovulatory follicle, and for COCs that mature successfully in vitro. The function and overall importance of miR-21 in normal follicle development and in vitro maturation, whether expressed via STAT3 dependent or independent pathways clearly warrants further mechanistic investigation.
In summary, the data presented here have clearly shown that miR-21 expression during bovine oocyte maturation and cumulus expansion is influenced by STAT3 signaling. This study further suggests that additional pathways in the ovary are likely to contribute to this response. Identification of additional normal pathways and those stimulated by pathophysiologic processes leading to miR-21 expression in the complex follicular environment should substantially enhance our understanding of the roles it plays in normal and abnormal ovarian biology and fertility.
Methods
Cumulus oocyte complex collection and in vitro maturation
Bovine ovaries were obtained from a local abattoir (Cargill, Guelph, ON) and transported at 35 °C. Within 2 hours of ovary collection, cumulus-oocyte complexes (COC) were aspirated from follicles greater than 6 mm diameter using vacuum aspiration. Collected complexes were placed into 1 M HEPES-buffered Nutrient Mixture F-10 Ham (Sigma-Aldrich, St. Louis MO) collection media supplemented with 2% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA), Hepalene (2 IU/ml, LEO Pharma Inc, Thornhill, ON), 14.3 mM sodium bicarbonate (Sigma-Aldrich) and penicillin 50 IU/mL/streptomycin 50 IU/mL (Invitrogen, Burlington, ON).
COCs were washed twice in 1M HEPES-buffered TCM-199 maturation medium (Caisson Labs, North Logan UT) supplemented with 22 μg/mL sodium pyruvate (Sigma-Aldrich) and penicillin 50 IU/mL/streptomycin 50 IU/mL (Invitrogen). Oocytes for maturation were placed in groups of 30–40 in 400 μL maturation medium containing 0.5 μg/mL FSH, 1 μg/mL LH and 1 μg/mL estradiol (National Institutes of Health (NIH), USA) in a humidified atmosphere at 38.5 °C and 5% CO2. After 7 or 24 hours, COCs were denuded and collected. At the time of collection, COCs were washed twice in phosphate buffered saline (PBS) with 0.1% polyvinyl alcohol (PVA) (Sigma-Aldrich) and cumulus cells were separated by physical disruption in PBS/PVA. Cumulus cells were placed into 1.5 mL microcentrifuge tubes and centrifuged at 600 × g for 6 minutes, and PBS/PVA was carefully removed from pellets. After cumulus cell collection, denuded oocytes were treated with 2 mg/mL Hyaluronidase from Streptomyces hyalurolyticus (Sigma-Aldrich) to remove any remaining cumulus cells, and washed in PBS with 0.1% PVA. Upon collection, all samples were immediately flash frozen in liquid nitrogen and stored at −80 °C.
For specific experiments, IVM medium was supplemented with 25 μg/mL cycloheximide8 from microbial sources (Sigma-Aldrich) dissolved in 100% ethanol, or 10 μM Stattic (Selleckchem, Houston TX) dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich). COC collection and maturation was modified as follows for serum-free experiments: COCs were aspirated and collected in serum-free 1 M HEPES-buffered TCM-199 (Sigma-Aldrich) collection media with 2 IU/ml Hepalene (LEO Pharma), penicillin 50 IU/mL/streptomycin 50 IU/mL (Invitrogen) and 0.1% PVA, followed by in vitro culture in 1 M HEPES-buffered TCM-199 maturation medium (Sigma-Aldrich) supplemented with 22 μg/mL sodium pyruvate (Sigma-Aldrich), penicillin 50 IU/mL/streptomycin 50 IU/mL (Invitrogen) and 0.1% PVA. IVM was then supplemented with 25 ng/mL recombinant human LIF (R&D Systems, Minneapolis MN) or 0.5 μg/mL FSH (NIH). In studies where a number of treatment groups and conditions were required, it was not always possible to collect multiple samples of each condition at a single collection event. In order to appropriately analyze data under these circumstances, at least one complete set of samples from all experimental conditions were collected, and each resulting data value was divided by the average of the entire sample set, thus standardizing all trials before comparing differences between experimental conditions. In the case of protein quantification and Western blotting, all samples that appear on blots together were collected during a single experimental trial and are compared only to other experimental conditions of that same trial.
RNA isolation and quantitative PCR
Total RNA, including small RNA, was isolated using the miRNeasy Micro kit (Qiagen, Mississauga, ON) according to the manufacturer’s protocol, and with the inclusion of DNase digestion performed on-column with the RNase-free DNase Set (Qiagen). RNA was eluted in nuclease-free water and RNA quantified by Nanodrop 2000c (Thermo Fisher). RNA concentrations ranged from approximately 20–50 ng/uL. Total cellular RNA (containing pri-miRNAs) was isolated from oocytes using the PicoPure RNA isolation kit (Thermo Fisher) with on-column DNase digestion (Qiagen), as reproducible detection of these infrequent transcripts could not be achieved when RNA was isolated using the previously mentioned system. RNA was isolated from pools of 30 COCs as cumulus or oocyte fractions for use with miRNeasy kit, and from pools of 5 oocytes for use with PicoPure kit. Messenger RNA and pri-miRNA were reverse transcribed with qScript complementary DNA (cDNA) SuperMix (Quantabio, Beverly MA), and miRNA was extended by polyadenylation then reverse transcribed with qScript microRNA cDNA Synthesis Kit (Quantabio). qRT-PCR was performed with a CFX96 Touch Real-Time PCR Detection System (BioRad Laboratories, Inc., Hercules, CA) using PerfeCTa SYBR Green SuperMix (Quantabio). cDNAs encoding pri-miRNAs and mRNAs were amplified using specific forward and reverse primers (Table 1), while miRNAs were amplified with a gene-specific forward primer and PerfeCTa Universal PCR Primer (Quantabio). Three ng of cDNA template was used for each reaction. Efficiencies were calculated by standard curve for all primers designed in this study and gene expression was calculated by the efficiency-corrected ΔΔCt method88. Cumulus cell mRNA and pri-miRNA quantification were normalized to the average of reference genes Tyrosine 3-monooygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ)89 and Beta-actin (ACTB), which were determined to be the most stable using the geNorm algorithm90. Oocyte pri-miRNA quantification was normalized to YWHAZ and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which was found to be more stable in these samples. miRNA were normalized to snRNA U6, which has been shown previously to be suitable for oocytes27, and is stably expressed in cumulus cells. Expression of each gene is shown relative to the abundance of that gene in the control group for a given experiment, and so relative abundance and changes in gene expression are comparable only within an experiment and not between experiments.
miRNA in situ hybridization
All chemicals used for the in situ hybridization came from Sigma-Aldrich unless otherwise stated. Slides containing sections of mature COCs were deparaffinised in xylene and ethanol before digestion with Proteinase-K for 10 minutes at 37 °C in a humidified chamber. Sections were then fixed in 4% formaldehyde (Thermo Fisher) for 10 min at room temperature, then rinsed twice in a solution of 0.13 M 1-methylimidazole and 300 mM sodium chloride pH 8.0 and incubated in 0.16 M l-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC)/0.13 M 1-methylimidazole/NaCl for 1 hr at room temperature. Slides were then washed in 0.2% (w/v) glycine/TBS and treated with 1% hydrogen peroxide to block endogenous peroxidases, followed by dehydration in ethanol before hybridization. Double digoxigenin (DIG) LNA modified probes (Exiqon, Copenhagen, Denmark) were denatured at 90 °C for 4 min and applied to slide surface at the following concentrations: 1 nM U6 snRNA probe (5′-CACGAATTTGCGTGTCATCCTT-3′), 40 nM miR-21 probe (5′-TCAACATCAGTCTGATAAGCTA-3′), or 40 nM scramble probe (5′-GTGTAACACGTCTATACGCCCA-3′), and placed in a humidified hybridizing chamber in a 55 °C oven for 1 hr. Slides were then washed in saline-sodium citrate at hybridization temperature, and blocked in PBS with 2% sheep serum (Thermo Fisher), 1% BSA and 0.1% Tween. Anti-DIG reagent (sheep anti-DIG POD-conjugate (Roche, Indianapolis IN) was applied for 1 hr, followed by several washes in PBS before incubation with TSA-PLUS_Cy3 according to the manufacturer recommendations (TSA plus cyanine 3 system, PerkinElmer, Waltham MA) in the dark for 10 min at room temperature. After several washes in PBS, slides were counterstained with 300 nM 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) in PBS for 2 min at room temperature, rinsed multiple times in PBS and mounted with coverslips using Prolong Gold Antifade (Thermo Fisher) and cured for 24 hr before imaging.
Equipment and settings
In situ hybridized slides were imaged (16 bit) using a confocal microscope LSM 700 (Zeiss) with a 40×/0.95 objective at 555 nm for Cy3 and 405 nm for DAPI. Zen Black 2012 software (Zeiss) and ImageJ (NIH) were used to process all images. DAPI acquisitions were performed with detector gain at 520, with excitation using a 405 nm laser at 2%, and emission with a dichroic beam splitter at 582 nm with a low pass. Cy3 acquisitions were performed with detector gain at 350, with excitation using a 555 nm laser at 2% and emission with a dichroic beam splitter at 559 nm with a high pass. All images were acquired using the same setting. Live cell images of COCs were obtained with EVOS FL Cell Imaging System using a 4x objective.
5′ and 3′ RACE PCR analysis and full length pri-miR-21 cloning
The transcription start site and polyadenylation signal of the primary transcript of miR-21 were identified by 5′ and 3′ rapid amplification of cDNA ends (RACE). Total RNA was isolated from bovine testis using miRNeasy kit (Qiagen). 5′ RNA ligase mediated (RLM)-RACE and 3′RACE were performed with GeneRacer kit (Invitrogen). For 5′RACE, 5 μg RNA was dephosphorylated with calf intestinal phosphatase (CIP), followed by phenol:chloroform extraction before RNA precipitation. RNA pellets were air dried and re-solubilized in nuclease-free water for treatment with tobacco acid pyrophosphatase (TAP), then re-extracted and dried as above. RNA was 5′ ligated to adapters and reverse transcribed with random primers and Superscript III, followed by treatment with RNaseH. RT-PCR reactions were carried out using a RACE adapter primer and a 5′RACE gene specific primer (Table 2), using Platinum Taq HiFi polymerase (Invitrogen), according to manufacturer’s instructions. 5′ ends of pri-miR-21 were amplified by touchdown PCR method for a total of 35 cycles according to the following protocol: Initial denaturation at 94 °C for 2 min, followed by 6 cycles of 94 °C × 30 s and 72 °C × 2 min, followed by 6 cycles of 94 °C × 30 s and 70 °C × 2 min, followed by 23 cycles of 94 °C × 30 s, 65 °C × 30 s and 68 °C × 2 min, followed by a final extension at 68 °C for 10 min. For 3′RACE, 1 μg testis RNA was reverse transcribed with GeneRacer Oligo dT primer and Superscript III after which RT-PCR was carried out as above (see Table 2 for 3′RACE gene specific primer). cDNAs of multiple lengths were separated on 1.5% agarose gels and extracted using PureLink Quick Gel Extraction kit (Invitrogen). Sequences of cDNA ends were obtained by cloning into pGEM-T Easy vector (Promega, Madison, WI) for sequencing, according to the manufacturer’s instructions. Briefly, RACE-PCR products were ligated at 4 °C overnight into linearized vector with a single 3′-terminal thymidine overhang at each end. Circular DNA was then transformed into high-efficiency DH5α competent cells and plated onto lysogeny broth (LB) (Sigma-Aldrich) agar plates with Ampicillin/IPTG/X-Gal (Sigma-Aldrich/Thermo Fisher/Promega) for overnight culture at 37 °C. Transformants were assessed for successful cloning inserts by blue:white screening and positive colonies were selected for liquid culture in LB with Ampicillin. Plasmid DNA was purified by Miniprep (Qiagen) according to manufacturer’s instructions. The sequences and orientation of inserts were confirmed by Sanger sequencing (Laboratory Services, University of Guelph). 5′ and 3′ RACE product sequences were used to design oligonucleotides to amplify the full length primary miR-21 transcript (Table 2) from the bovine testis template used for 3′ RACE, according to the touchdown PCR protocol described above. No template control (NTC) reactions were run alongside templates to ensure that all PCR reagents were free from DNA contamination. Transcript variants were extracted from agarose gels, cloned into pGEM-T Easy and confirmed by sequencing.
Promoter analysis and transcription factor binding site prediction
Using the bovine UMD3.1 assembly as a reference genome91, the ECR Browser92 tool was employed to locate regions of pri-miR-21 and its 1 kb upstream putative regulatory region that are conserved between bovine and the human (assembly GRCh37) or mouse (assembly GRCm38) sequences. Evolutionarily conserved regions were searched for putative transcription factor binding sites in the JASPAR database93.
miR-21 proximal promoter cloning and site-directed mutagenesis
Bovine DNA was isolated from bovine testis using Zygem prepGEM tissue (Zygem, Charlottesville VA) according to manufacturer’s directions. The miR-21 proximal promoter region was amplified from bovine DNA using primers specific to a region ~400 nucleotides upstream of the putative pri-miR-21 transcription start site, using specific primers with the addition of restriction enzyme target sites for KpnI and BglII (Table 2), and cloned into pGEM-T vectors as above. To generate luciferase vectors driven by the miR-21 promoter, the most proximal 400 nucleotides upstream of the pri-miR-21 transcription start site were subcloned into pGL3 vector (Promega). Site directed mutagenesis was performed using QuikChange II site-directed mutagenesis kit (Agilent, Santa Clara CA) according to manufacturer’s instructions, by generating a GΔC, AΔG double mutation at position −41 to −40 upstream of the pri-miR-21 transcription start site, or a AΔG, AΔC double mutation at position −9 to −8 (see Table 2 for mutagenesis primers).
Promoter reporter assays
7 × 104 MCF7 human breast cancer cells (ATCC, Manassas, VA, Cat# CRL-12584, RRID:CVCL_0031) were seeded onto tissue culture-treated 24-well plates (Sigma-Aldrich) and maintained in 1 mL Roswell Park Memorial Institute (RPMI) 1640 Medium (Thermo Fisher) supplemented with 4 mM L-glutamine (Sigma-Aldrich), 10% FBS, 1 mM sodium pyruvate (both Thermo Fisher), and 1% penicillin/streptomycin (Sigma-Aldrich), and incubated at 37 °C with 5% CO2 conditioned air. Cells were cultured for 48 h after plating until they reached ~70% confluency, washed once in sterile PBS and changed to serum-free Opti-MEM media (Thermo Fisher) for serum starvation and transfection of reporter constructs. After 8 hours starvation, 450 ng Firefly-luciferase reporter construct and 50 ng pRL Renilla-luciferase control vector (Promega) were co-transfected using Lipofectamine LTX (Thermo Fisher). 1 µL Lipofectamine or 500 ng Firefly and Renilla-luciferase constructs were diluted in Opti-MEM, mixed in a 1:1 ratio and incubated for 5 min at room temperature before transfection. After 4 hours of recovery, transfected cells were cultured for 4 hours in the presence or absence of 15 ng/mL LIF, then lysed and prepared for luminescence quantification using Dual-Luciferase Reporter Assay System (Promega) according to manufacturer’s instructions. Luciferase activity was evaluated using a FLUOstar Optima microplate luminometer (BMG Labtech, Cary NC). All experiments were performed in technical duplicates and repeated a minimum of three times.
Protein isolation and Western blotting
Cumulus cells from 40 COCs were lysed in 42uL radioimmunoprecipitation assay (RIPA) buffer (50 mM pH 7.4, 1% Triton-X 100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 15 mM sodium chloride, 2 mM ethylenediaminetetraacetic acid (EDTA)), with phosphatase/protease inhibitor cocktail (Bimake, Houston TX) on ice. Samples were sonicated for 15 seconds, incubated on ice for 5 minutes and centrifuged at 12,000 × g for 10 minutes. Proteins were denatured by boiling in SDS protein loading buffer containing dithiotreitol (DTT), separated by gel electrophoresis on 8% polyacrylamide gels, and transferred to polyvinylidene fluoride membranes. Membranes were blocked for 1 hour in 5% BSA (Sigma-Aldrich) and incubated at 4 °C overnight in the following primary antibodies: phosphoSTAT3 (Y705) (Cat# 9145, RRID:AB_2491009) at a final dilution of 1:1000, native STAT3 (Cat# 4904, RRID: AB_331269) at 1:2000, Drosha (Cat# 3364, RRID: AB_2238644) at 1:1000 and β-actin (Cat# 4967, RRID: AB_330288) at 1:5000. Membranes were washed several times in Tris buffered saline pH 7.6 plus 0.1% Tween (TBS-T) (Sigma-Aldrich) before a 1 hr incubation with a horseradish peroxidase-conjugated secondary antibody (Cat# 7074, RRID: AB_2099233) at 1:5000. All antibodies used were from Cell Signaling Technology (Danvers, MA). Membranes were immersed in Clarity Western ECL Blotting Substrate (Bio-Rad) and imaged on a ChemiDoc XRS + Imaging system (Bio-Rad).
Statistical analysis
Differences among groups were analyzed by ANOVA after Tukey’s multiple comparisons test, or by one-tailed t-test when the experimental design consisted of only two groups, using GraphPad Prism 6 (GraphPad Software, La Jolla CA). Gene expression data collected from qRT-PCR were log2 transformed in order to conduct the appropriate statistical analysis. A minimum of N = 3 biological replicates were performed for all reported data, and a p-value < 0.05 was considered statistically significant. Data shown represent the mean +/− standard error of the mean (SEM). Different letters indicate statistically significant differences between groups.
References
Dong, J. et al. Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nature. 383, 531–535 (1996).
Diaz, F. J., O’Brien, M. J., Wigglesworth, K. & Eppig, J. J. The preantral granulosa cell to cumulus cell transition in the mouse ovary: development of competence to undergo expansion. Dev Biol. 299, 91–104 (2006).
Li, R., Norman, R. J., Armstrong, D. T. & Gilchrist, R. B. Oocyte-secreted factor(s) determine functional differences between bovine mural granulosa cells and cumulus cells. Biol Reprod. 63, 839–845 (2000).
Eppig, J. J. Oocyte control of ovarian follicular development and function in mammals. Reproduction. 122, 829–838 (2001).
Fukui, Y. & Sakuma, Y. Maturation of bovine oocytes cultured in vitro: relation to ovarian activity, follicular size and the presence or absence of cumulus cells. Biol Reprod. 22, 669–673 (1980).
Vanderhyden, B. C. & Armstrong, D. T. Role of cumulus cells and serum on the in vitro maturation, fertilization, and subsequent development of rat oocytes. Biol Reprod. 40, 720–728 (1989).
Dey, S. R. et al. Coculturing denuded oocytes during the in vitro maturation of bovine cumulus oocyte complexes exerts a synergistic effect on embryo development. Theriogenology. 77, 1064–1077 (2012).
Tatemoto, H., Horiuchi, T. & Terada, T. Effects of cycloheximide on chromatin condensations and germinal vesicle breakdown (GVBD) of cumulus-enclosed and denuded oocytes in cattle. Theriogenology. 42, 1141–1148 (1994).
Zhang, L., Jiang, S., Wozniak, P. J., Yang, X. & Godke, R. A. Cumulus cell function during bovine oocyte maturation, fertilization, and embryo development in vitro. Mol Reprod Dev. 40, 338–344 (1995).
Buccione, R., Vanderhyden, B. C., Caron, P. J. & Eppig, J. J. FSH-induced expansion of the mouse cumulus oophorus in vitro is dependent upon a specific factor(s) secreted by the oocyte. Dev Biol. 138, 16–25 (1990).
Hussein, T. S., Froiland, D. A., Amato, F., Thompson, J. G. & Gilchrist, R. B. Oocytes prevent cumulus cell apoptosis by maintaining a morphogenic paracrine gradient of bone morphogenetic proteins. J Cell Sci. 118, 5257–5268 (2005).
Gilchrist, R. B. et al. Molecular basis of oocyte-paracrine signalling that promotes granulosa cell proliferation. J Cell Sci. 119, 3811–3821 (2006).
Matzuk, M. M., Burns, K. H., Viveiros, M. M. & Eppig, J. J. Intercellular communication in the mammalian ovary: oocytes carry the conversation. Science. 296, 2178–2180 (2002).
Fülöp, C. et al. Impaired cumulus mucification and female sterility in tumor necrosis factor-induced protein-6 deficient mice. Development. 130, 2253–2261 (2003).
Salustri, A. et al. PTX3 plays a key role in the organization of the cumulus oophorus extracellular matrix and in in vivo fertilization. Development. 131, 1577–1586 (2004).
Dragovic, R. A. et al. Oocyte-secreted factor activation of SMAD 2/3 signaling enables initiation of mouse cumulus cell expansion. Biol Reprod. 76, 848–857 (2007).
Lee, R. C. & Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 294, 862–864 (2001).
Lagos-Quintana, M., Rauhut, R., Lendeckel, W. & Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science. 294, 853–858 (2001).
Lau, N. C., Lim, L. P., Weinstein, E. G. & Bartel, D. P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 294, 858–862 (2001).
Lee, R. C., Feinbaum, R. L. & Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 75, 843–854 (1993).
Reinhart, B. J. et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 403, 901–906 (2000).
Gurtan, A. M. & Sharp, P. A. The role of miRNAs in regulating gene expression networks. J Mol Biol. 425, 3582–3600 (2013).
Flynt, A. S. & Lai, E. C. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet. 9, 831–842 (2008).
Wu, L., Fan, J. & Belasco, J. G. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA 103, 4034–4039 (2006).
Djuranovic, S., Nahvi, A. & Green, R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. 336, 237–240 (2012).
Mondou, E., Dufort, I., Gohin, M., Fournier, E. & Sirard, M. A. Analysis of microRNAs and their precursors in bovine early embryonic development. Mol Hum Reprod. 18, 425–434 (2012).
Abd El Naby, W. S. et al. Expression analysis of regulatory microRNAs in bovine cumulus oocyte complex and preimplantation embryos. Zygote. 21, 31–51 (2013).
Miles, J. R. et al. MicroRNA expression profile in bovine cumulus-oocyte complexes: possible role of let-7 and miR-106a in the development of bovine oocytes. Anim Reprod Sci. 130, 16–26 (2012).
Uhde, K., van Tol, H., Stout, T. & Roelen, B. MicroRNA Expression in Bovine Cumulus Cells in Relation to Oocyte Quality. ncRNA. 3, 12 (2017).
McBride, D. et al. Identification of miRNAs associated with the follicular-luteal transition in the ruminant ovary. Reproduction. 144, 221–233 (2012).
Assou, S. et al. MicroRNAs: new candidates for the regulation of the human cumulus-oocyte complex. Hum Reprod. 28, 3038–3049 (2013).
Carletti, M. Z., Fiedler, S. D. & Christenson, L. K. MicroRNA 21 blocks apoptosis in mouse periovulatory granulosa cells. Biol Reprod. 83, 286–295 (2010).
Wright, E. C., Hale, B. J., Yang, C. X., Njoka, J. G. & Ross, J. W. MicroRNA-21 and PDCD4 expression during in vitro oocyte maturation in pigs. Reprod Biol Endocrinol. 14, 21 (2016).
Gilchrist, G. C., Tscherner, A., Nalpathamkalam, T., Merico, D. & LaMarre, J. MicroRNA Expression during Bovine Oocyte Maturation and Fertilization. Int J Mol Sci. 17 (2016).
Lee, Y. et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060 (2004).
Lee, Y. et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 425, 415–419 (2003).
Landthaler, M., Yalcin, A. & Tuschl, T. The human DiGeorge syndrome critical region gene 8 and Its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol. 14, 2162–2167 (2004).
Denli, A. M., Tops, B. B., Plasterk, R. H., Ketting, R. F. & Hannon, G. J. Processing of primary microRNAs by the Microprocessor complex. Nature. 432, 231–235 (2004).
Gregory, R. I. et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 432, 235–240 (2004).
Zhong, Z., Wen, Z. & Darnell, J. E. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 264, 95–98 (1994).
Hibi, M. et al. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 63, 1149–1157 (1990).
Lütticken, C. et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 263, 89–92 (1994).
Niwa, H., Burdon, T., Chambers, I. & Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 12, 2048–2060 (1998).
De Matos, D. G. et al. Leukemia inhibitory factor induces cumulus expansion in immature human and mouse oocytes and improves mouse two-cell rate and delivery rates when it is present during mouse in vitro oocyte maturation. Fertil Steril. 90, 2367–2375 (2008).
Liu, Z. et al. Interleukin-6: an autocrine regulator of the mouse cumulus cell-oocyte complex expansion process. Endocrinology. 150, 3360–3368 (2009).
Liu, H., Han, H., Li, J. & Wong, L. An in-silico method for prediction of polyadenylation signals in human sequences. Genome Inform. 14, 84–93 (2003).
Fujita, S. et al. miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J Mol Biol. 378, 492–504 (2008).
Loffler, D. et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 110, 1330–1333 (2007).
Kaptein, A., Paillard, V. & Saunders, M. Dominant Negative Stat3 Mutant Inhibits Interleukin-6-induced Jak-STAT Signal Transduction. J. Biol. Chem. 271, 5961–5964 (1996).
Schust, J., Sperl, B., Hollis, A., Mayer, T. U. & Berg, T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 13, 1235–1242 (2006).
Murphy, K., Carvajal, L., Medico, L. & Pepling, M. Expression of Stat3 in germ cells of developing and adult mouse ovaries and testes. Gene Expr Patterns. 5, 475–482 (2005).
Sirard, M. A. et al. Timing of nuclear progression and protein synthesis necessary for meiotic maturation of bovine oocytes. Biol Reprod. 40, 1257–1263 (1989).
Han, X. et al. MicroRNA-21 plays a pivotal role in the oocyte-secreted factor-induced suppression of cumulus cell apoptosis. Biol Reprod. 96, 1167–1180 (2017).
Crozet, N., Kanka, J., Motlik, J. & Fulka, J. Nucleolar Fine Structure and RNA Synthesis in Bovine Oocytes From Antral Follicles. Gamete Research. 14, 65–73 (1986).
Fair, T., Hyttel, P. & Greve, T. Bovine oocyte diameter in relation to maturational competence and transcriptional activity. Mol Reprod Dev. 42, 437–442 (1995).
Mamo, S. et al. Sequential analysis of global gene expression profiles in immature and in vitro matured bovine oocytes: potential molecular markers of oocyte maturation. BMC Genomics. 12, 151 (2011).
Macaulay, A. D. et al. The gametic synapse: RNA transfer to the bovine oocyte. Biol Reprod. 91, 90 (2014).
Iliopoulos, D., Jaeger, S. A., Hirsch, H. A., Bulyk, M. L. & Struhl, K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 39, 493–506 (2010).
Yue, X. et al. Leukemia inhibitory factor promotes EMT through STAT3-dependent miR-21 induction. Oncotarget. 7, 3777–3790 (2016).
Assidi, M. et al. Identification of potential markers of oocyte competence expressed in bovine cumulus cells matured with follicle-stimulating hormone and/or phorbol myristate acetate in vitro. Biol Reprod. 79, 209–222 (2008).
Matoba, S. et al. Predictive value of bovine follicular components as markers of oocyte developmental potential. Reprod Fertil Dev. 26, 337–345 (2014).
Fülöp, C., Salustri, A. & Hascall, V. C. Coding sequence of a hyaluronan synthase homologue expressed during expansion of the mouse cumulus-oocyte complex. Arch Biochem Biophys. 337, 261–266 (1997).
Espey, L. L. Current status of the hypothesis that mammalian ovulation is comparable to an inflammatory reaction. Biol Reprod. 50, 233–238 (1994).
Shimada, M., Hernandez-Gonzalez, I., Gonzalez-Robanya, I. & Richards, J. S. Induced expression of pattern recognition receptors in cumulus oocyte complexes: novel evidence for innate immune-like functions during ovulation. Mol Endocrinol. 20, 3228–3239 (2006).
Hernandez-Gonzalez, I. et al. Gene expression profiles of cumulus cell oocyte complexes during ovulation reveal cumulus cells express neuronal and immune-related genes: does this expand their role in the ovulation process. Mol Endocrinol. 20, 1300–1321 (2006).
Richards, J. S., Liu, Z. & Shimada, M. Immune-like mechanisms in ovulation. Trends Endocrinol Metab. 19, 191–196 (2008).
Büscher, U., Chen, F. C., Kentenich, H. & Schmiady, H. Cytokines in the follicular fluid of stimulated and non-stimulated human ovaries; is ovulation a suppressed inflammatory reaction. Hum Reprod. 14, 162–166 (1999).
Field, S. L., Dasgupta, T., Cummings, M. & Orsi, N. M. Cytokines in ovarian folliculogenesis, oocyte maturation and luteinisation. Mol Reprod Dev. 81, 284–314 (2014).
Gearing, D. P. et al. The IL-6 signal transducer, gp130: an oncostatin M receptor and affinity converter for the LIF receptor. Science. 255, 1434–1437 (1992).
Yin, T. et al. Involvement of IL-6 signal transducer gp130 in IL-11-mediated signal transduction. J Immunol. 151, 2555–2561 (1993).
Baumann, H. et al. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci USA 93, 8374–8378 (1996).
Eckert, J. & Niemann, H. mRNA expression of leukaemia inhibitory factor (LIF) and its receptor subunits glycoprotein 130 and LIF-receptor-beta in bovine embryos derived in vitro or in vivo. Mol Hum Reprod. 4, 957–965 (1998).
Mo, X. et al. Leukemia inhibitory factor enhances bovine oocyte maturation and early embryo development. Mol Reprod Dev. 81, 608–618 (2014).
Dang-Nguyen, T. Q. et al. Leukemia inhibitory factor promotes porcine oocyte maturation and is accompanied by activation of signal transducer and activator of transcription 3. Mol Reprod Dev. 81, 230–239 (2014).
Fry, R. C., Batt, P. A., Fairclough, R. J. & Parr, R. A. Human leukemia inhibitory factor improves the viability of cultured ovine embryos. Biol Reprod. 46, 470–474 (1992).
Pourhoseini, S. et al. Upregulation of miR21 and repression of Grhl3 by leptin mediates sinusoidal endothelial injury in experimental nonalcoholic steatohepatitis. PLoS One. 10, 0116780, https://doi.org/10.1371/journal.pone.0116780 (2015).
Park, J.-Y. et al. EGF-like Growth Factors as Mediators of LH Action in the Ovulatory Follicle. Science. 303, 682–684 (2004).
Sharma, S. C. & Richards, J. S. Regulation of AP1 (Jun/Fos) factor expression and activation in ovarian granulosa cells. Relation of JunD and Fra2 to terminal differentiation. J Biol Chem. 275, 33718–33728 (2000).
Craig, J., Zhu, H., Dyce, P. W., Petrik, J. & Li, J. Leptin enhances oocyte nuclear and cytoplasmic maturation via the mitogen-activated protein kinase pathway. Endocrinology. 145, 5355–5363 (2004).
van Tol, H. T., van Eerdenburg, F. J., Colenbrander, B. & Roelen, B. A. Enhancement of Bovine oocyte maturation by leptin is accompanied by an upregulation in mRNA expression of leptin receptor isoforms in cumulus cells. Mol Reprod Dev. 75, 578–587 (2008).
Frankel, L. B. et al. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 283, 1026–1033 (2008).
Meng, F. et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 133, 647–658 (2007).
Ding, L. et al. Higher PDCD4 expression is associated with obesity, insulin resistance, lipid metabolism disorders, and granulosa cell apoptosis in polycystic ovary syndrome. Fertil Steril. 105, 1330–1337.e3 (2016).
Reddy, P. et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science. 319, 611–613 (2008).
Karakaya, C. et al. Poor ovarian response in women undergoing in vitro fertilization is associated with altered microRNA expression in cumulus cells. Fertil Steril. 103, 1469–76.e1 (2015).
Sontakke, S. D., Mohammed, B. T., McNeilly, A. S. & Donadeu, F. X. Characterization of microRNAs differentially expressed during bovine follicle development. Reproduction. 148, 271–283 (2014).
Ilha, G. F. et al. Lack of FSH support enhances LIF-STAT3 signaling in granulosa cells of atretic follicles in cattle. Reproduction. 150, 395–403 (2015).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, 45, https://doi.org/10.1093/nar/29.9.e45 (2001).
Goossens, K. et al. Selection of reference genes for quantitative real-time PCR in bovine preimplantation embryos. BMC Dev Biol. 5, 27 (2005).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034, https://doi.org/10.1186/gb-2002-3-7-research0034 (2002).
Zimin, A. V. et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 10, R42, https://doi.org/10.1186/gb-2009-10-4-r42 (2009).
Ovcharenko, I., Nobrega, M. A., Loots, G. G. & Stubbs, L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 32, 280–2866, https://doi.org/10.1093/nar/gkh355 (2004).
Mathelier, A. et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42, 142–147, https://doi.org/10.1093/nar/gkt997 (2014).
Acknowledgements
The authors wish to thank Elizabeth St. John for excellent technical assistance; Monica Antenos, Stewart Russell, and Graham Gilchrist for helpful discussions. This work is supported by the Natural Sciences and Engineering Research Council of Canada (to JL) and the Ontario Ministry of Food and Rural Affairs (to JL).
Author information
Authors and Affiliations
Contributions
A.T. and J.L. designed the study and prepared the manuscript. A.T., A.B., L.S., J.K. and I.D. performed the experiments. M.S. and J.L. supervised the research and M.S. assisted in the preparation of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tscherner, A., Brown, A., Stalker, L. et al. STAT3 signaling stimulates miR-21 expression in bovine cumulus cells during in vitro oocyte maturation. Sci Rep 8, 11527 (2018). https://doi.org/10.1038/s41598-018-29874-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29874-w
This article is cited by
-
IFN-γ enhances the efficacy of mesenchymal stromal cell-derived exosomes via miR-21 in myocardial infarction rats
Stem Cell Research & Therapy (2022)
-
MiR-191-5p is upregulated in culture media of implanted human embryo on day fifth of development
Reproductive Biology and Endocrinology (2021)
-
In vitro maturation in the presence of Leukemia Inhibitory Factor modulates gene and miRNA expression in bovine oocytes and embryos
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
