
Abstract
Neurodegenerative diseases, characterised by the progressive and selective neuronal death in the central nervous system, are frequently accompanied by an activated immune system. In Huntington’s disease (HD), clinical and animal studies show evidence of immune activity, along with hyper-reactive monocyte/macrophage responses, while application of immunosuppressive regimens have imparted beneficial effects to HD mice. These findings suggest a contributory role of the immune system in HD pathology, with immune-based interventions offering a potential therapeutic strategy. Herein, we show that peripheral and CNS immune system activity increased with disease progression in HD mouse models and defined the phenotype of the immune response. Additionally, the depletion of monocytes and macrophages in vivo, via clodronate liposome treatment, revealed a major contributory role of these innate immune cells to the chronic inflammatory milieu observed during the course of the disease. This suggests that peripheral immunomodulatory strategies targeting monocytes and macrophages could be relevant for HD.
Similar content being viewed by others
Introduction
The contribution of the immune system to the pathology of neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and Huntington’s disease (HD) has gathered increasing interest and momentum with the ultimate hope of finding effective immune-based therapeutic strategies1,2,3,4,5. HD is an autosomal dominant neurodegenerative disorder characterized by progressive cognitive, psychiatric and motor impairments caused by neuronal dysfunction and cell death. The causative mutation is a CAG trinucleotide repeat expansion in exon 1 of the huntingtin (HTT) gene leading to an expanded stretch of 38 or more glutamine residues in the N-terminal region of the HTT protein6.
HTT is more highly expressed in neurons as compared to other cell types7. However, immune cells including B cells, T cells, monocytes and/or macrophages from patients show detectable levels of HTT that are positively correlated to disease burden and brain caudate atrophy8,9. While microglial cells, the major component of the central nervous system (CNS) immunity, have long been observed to be activated in presymptomatic HD10, more recent results suggest a role for the peripheral immune system in HD progression. This has taken the form of elevated levels of plasma cytokines9 and chemokines11 such as IL6 and TNFα in HD mouse models and patients, along with dysregulated monocyte and macrophage in vitro responses9,12,13. Indeed, studies utilising immunomodulatoy regimens in HD mouse models substantiate this premise. Beneficial effects have been obtained through the replacement of the HD immune system with a normal one, via bone marrow transplantation14, or by treatment with immunosuppressive agents15,16 whose effects have been specifically limited to immune cells outside of the brain. The ability of mutant HTT to promote the activation/signalling of the pro-inflammatory transcription factor NFκB may provide one mechanism for the enhanced monocyte/macrophage cytokine secretions seen in HD13.
Knock-in HD mice precisely model the genetic basis of HD. They have been generated by either inserting a highly expanded CAG repeat into the mouse Htt gene, e.g. HdhQ15017, or by replacing mouse exon 1 Htt with a mutated version of human exon1 HTT e.g. zQ17518. Incomplete splicing of the Htt gene in all knock-in models19 and HD patient brains20 results in the production of a small polyadenylated transcript that encodes the highly pathogenic exon 1 HTT protein. The R6/2 HD mouse line is transgenic for the 5′ region of the HTT gene21, expresses an exon 1 HTT protein and is a model of this incomplete splicing event. R6/2 mice with approximately (CAG)200 and knock-in mice with similar CAG repeat expansions (e.g. HdhQ150 and zQ175) develop highly comparable behavioural, molecular and neuropathological phenotypes22, but over different time scales: late-stage disease is at ~14 weeks of age for R6/2 mice and close to two years for the HdhQ150 and zQ175 knock-in lines.
Herein, we assessed HD mouse monocyte, macrophage and other immune cells from blood, brain and/or spleen during early symptomatic and late stage HD for signs of immune activity, as indicated by increased immune cell activation markers and cytokine production. We find dysregulated monocyte/macrophage activity in agreement with previous studies9,13, but also detect increased dendritic cell (DC) activation. The major role of macrophages and DCs as antigen presenting cells during T cell activation, coupled with our observation of increases in cytokines that are predominantly secreted by T cells, prompted us to additionally investigate T cell activation during HD. Our results revealed that these cells were also dysregulated and showed an increase in activity during the latter stages of disease. Subsequently, we used clodronate liposomes23,24 to selectively deplete phagocytic monocyte/macrophage and DC populations from blood and tissues, resulting in cytokine normalisation, and demonstrating that macrophages contribute to the in vivo chronic inflammatory milieu that we observed in R6/2 mice from an early symptomatic stage of disease.
Results
Dysregulation of brain and plasma cytokine levels in late stage R6/2 mice
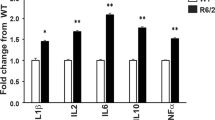
Neuroinflammation due to the activation of microglial cells in the brain of HD patients10 and mice25, as well as the possible activation of hyper-responsive monocytes and macrophages9,12, may lead to increased production of cytokines in both the periphery and the CNS of HD subjects. In order to investigate this, we measured plasma and brain cytokine levels by mesoscale discovery (MSD) multiplex ELISA and quantitative real-time PCR (qPCR) respectively. The MSD results revealed a rise in blood IL6 by approximately two-fold in late symptomatic 14 week-old R6/2 mice, while IL1β and TNFα levels were also elevated by just under 50% and IL2 and IL10 increased by over 70% (Fig. 1a).

Immune activation in late-stage HD mice as indicated by cytokine levels. (a) Cytokine upregulation in the blood of 14 week old R6/2 mice. Plasma levels for R6/2 mice compared to WT are shown ± SEM, (n = 20/group IL1β; 16/group IL2; 19/group IL6; 26/group IL10 and 28/group TNFα). (b) Increases in cytokine gene expression was limited to the striatum of late-stage R6/2 mice. Il1β, Il2, Il4, Il6, Il10, Il12, Il17 and Tnfα gene expression levels in cerebellum, striatum and cortex of R6/2 mice (n = 5/group) compared to WT (n = 5/group) ± SEM as measured by qPCR. (c) Increased cytokine gene expression in R6/2 splenic and peritoneal macrophage enriched cell populations. R6/2 mouse splenic (n = 10/group) and peritoneal macrophage (n = 7/group) gene expression for cytokines relative to WT ± SEM quantified by qPCR. *p < 0.05 and **p < 0.01 vs WT by Student’s t test. WT = wild type, pM0 = peritoneal macrophages.
In the brain, IL6 and TNFα have been documented to have detrimental neurotoxic effects26,27,28. Quantification of cytokine mRNA levels in striatum, cortex and cerebellum showed increased production of Tnfα only in the striatum by more than twofold, but there was no increase in Il6 expression in any of the brain regions tested (Fig. 1b). Furthermore, an approximately threefold increase in Il12 expression was evident in the striatum of R6/2 mice while the reverse pattern was seen in the cortex, with decreases occurring in Il12 and Il1β by approximately 60% and 40% respectively.
In the periphery, R6/2 splenocyte Il10, Il12 and Il17 expression levels were at least 50% higher than WT, whereas Il1β, Il4, Il6 and Tnfα levels remained unchanged. In contrast, cell samples from the peritoneal cavity enriched for macrophages, had approximately four times more Il4, and almost ten-fold higher Il6 than in WT, with more modest increases for Il10 and Il12 of approximately 1.8 and 1.3 fold respectively (Fig. 1c). As observed for splenocytes, Tnfα and Il1β levels were not affected. The results may suggest that blood IL6 upregulation in late stage HD is primarily due to increased macrophage secretion, as increases were only observed in the macrophage enriched samples, and not in splenocytes, where macrophages make up only a small percentage of the cell population. Conversely, the increase in splenocyte Il12 and Il10 levels could suggest that cells additional to macrophages, such as helper T cells, are contributing to the increases in the plasma levels of these cytokines.
The cytokine gene expression results for macrophages in the periphery and microglial cells in the brain indicate phenotypically different populations of activated macrophages/microglia in these two compartments, with cells at the periphery exhibiting a more M2-like phenotype, while microglia in the brain favour an M1 phenotype29.
Increased frequencies of activated immune cells in late-stage R6/2 mice
Although the gene expression results for the macrophage enriched peritoneal cells identify myeloid cells as a major source of the upregulated blood cytokines during late stage HD, the possible contribution of other cytokine secreting immune cells such as T lymphocytes and dendritic cells (DC) cannot be completely dismissed. More importantly, some cytokines that were elevated in the periphery in R6/2 mice, such as IL2 in blood and splenic Il17 mRNA, are not typically secreted by macrophages. This finding prompted us to identify the other cells responsible for the increased peripheral cytokine levels.
Following activation, DC and macrophage/microglia upregulate their expression of several activation markers including OX40 ligand (L) and CD40, while, similarly, activated T cells can be identified via the increased expression of cell surface markers such as OX40, CD40L and CD2530,31,32,33,34. Gene expression levels for Ox40L and Cd40 in the spleen, peritoneal macrophages and brain regions were measured in order to detect DC and macrophage/microglia activation, and Ox40 in the spleen to detect T cell activity. In the spleen, which contains T cells and a small percentage of DCs and macrophages, expression of Ox40L and Cd40 were increased in R6/2 mice by approximately four-fold and two-fold respectively (Fig. 2a). Similarly, Cd40 and Ox40L levels were elevated in the peritoneal macrophage enriched cell population (Fig. 2b). Therefore, DC and/or macrophage cell activation can be detected in both spleen and peritoneal macrophages during late-stage HD. Surprisingly, Ox40 expression did not increase in the spleen (data not shown), suggesting a lack of splenic T cell activity. In the striatum, an increase in Cd40 expression was also evident (Fig. 2c), which could not be observed in the cortex or cerebellum (data not shown), consistent with the observation of increased cytokine production being confined to the striatum (Fig. 1b). We failed to detect any Ox40L expression in any of the brain regions assessed in either WT or R6/2 mice (data not shown).

Increased activity of immune cells in late-stage R6/2 mice. (a) Increased splenic Ox40L and Cd40 gene expression in late-stage R6/2 compared to WT. Splenic Ox40L (n = 8/group) and Cd40 (n = 5/group) in R6/2 mice relative to WT ± SEM as measured by qPCR. (b) Increased Cd40 (n = 5/group) and Ox40L gene (n = 5/group) expression in peritoneal macrophages enriched cell populations in late-stage R6/2 compared to WT ± SEM as measured by qPCR. (c) Higher Cd40 expression (n = 5/group) in striatum of 14 week old R6/2 mice vs. WT ± SEM as analysed by qPCR. (d) Increased frequencies of activated CD40hi peritoneal macrophages in late-stage R6/2 mice. Percentage of peritoneal macrophages (gated as; CD11b+, CD49b−, CD8−, CD11c−, Ly6G−) highly expressing cell surface CD40 in 14 week old R6/2 mice and WT as assessed by FACS. The Figure shows a typical example of FACS dot plots obtained for each group analysed. (e) Increased frequencies of activated OX40L high and CD40 high splenic DC in late-stage R6/2 mice. Mean percentage ± SEM of DC (gated as; CD11b+, CD8−, CD11c+, Ly6G−) highly expressing cell surface OX40L and CD40 in 14 week old WT (n = 4/group) and R6/2 mice (n = 6/group) as assessed by FACS. (f) Increased frequencies of activated OX40 high CD4 and CD8 splenic T cells in late-stage R6/2 mice. Mean percentage ± SEM of T cells (gated as; CD3+, CD4+ or CD8+) highly expressing cell surface OX40 in 14 week old WT (n = 5/group) and R6/2 mice (n = 6/group) as assessed by FACS. *p < 0.05 and **p < 0.01 vs WT by Student’s t test. WT = wild type, pM0 = peritoneal macrophages, DC = dendritic cells.
In order to verify the qPCR results, FACS analysis of OX40L and CD40 expression was performed for (CD11b+, CD11c−, CD3−, Ly6G−) splenic and peritoneal macrophages and (CD11b+, CD11c+, CD3−, Ly6G−) splenic DCs. Additionally, CD3 and CD4 or CD8 expressing splenic T cells were assessed for OX40 cell surface expression. Figure 2d depicts a representative FACS analysis (n = 5/group) of purified peritoneal macrophages from 14 week old R6/2 mice showing that 52% of the cells are active based on their high expression of CD40 as compared to 10% in WT mice. These results indicate a probable role for macrophages in elevating the plasma cytokine levels during HD. Further assessment of OX40L high expressing activated macrophages showed an increase in their frequency in the peritoneal cavity during late-stage HD (Figure S1).
Assessment of splenic DCs (CD11b+ CD11c+ CD8− Ly6G−) for high expression of OX40L and CD40 by FACS also revealed increases in the numbers of OX40L high and CD40 high cells in late-stage R6/2 mice compared to WT (Fig. 2e). R6/2 mice at 14 weeks of age had almost two fold more activated DCs that were OX40L high, and over three times more activated DCs that were CD40 high than WT.
The FACS analysis of CD3+, CD4+ helper or CD3+, CD8+ cytotoxic T cells for high expression of OX40, as a marker for activated T cells, revealed significantly higher percentages of OX40 high expressing CD4 and CD8 T cells in the 14 week old R6/2 mice versus WT. Both cell populations showed approximately twice as many activated cells in their total helper (~7% ± 1 for WT and ~14% ± 3 for R6/2) and cytotoxic (~12% ± 4 for WT and ~30% ± 5 for R6/2) T cell pools in the R6/2 mice (Fig. 2f). These FACS results are contrary to the level of Ox40 expression, which was not increased in R6/2 spleens as compared to WT.
Peripheral immune activation precedes immune activity in the brain of R6/2 mice
We have previously observed that the activation of the peripheral immune system, as evidenced by significant increases in a plethora of cytokines in blood, occurs in both HD patients and mouse models from early stages of disease, with macrophages and microglial cells prone to hyperactivity following their in vitro stimulation9,12. We therefore sought to determine whether immune activity in the R6/2 mouse brain could also be detected at an earlier stage. Therefore, we assessed gene expression and/or protein cytokine levels, and immune activation markers in 8-week-old early symptomatic R6/2 mouse brains, blood and lymphoid tissues/cells. We focused on the cytokines and activation markers that we had found to be upregulated in late stage R6/2 mice. Gene expression analysis showed little change in striatal cytokine expression in R6/2 as compared to WT (data not shown). However, increases in the expression of cytokines and activation markers were identified in R6/2 splenocytes and/or peritoneal macrophages by 8 weeks of age. Il6 was increased in the peritoneal macrophage enriched cell sample by over two fold (Fig. 3a), splenic Il12 was increased by over 70% (Fig. 3b), Cd40 was increased in the spleen by over 90% (Fig. 3c) and in peritoneal macrophages by approximately 25% (Fig. 3d).

Immune activity in the periphery of early stage R6/2 mice. Upregulated cytokine and activated macrophage cell marker gene expression levels in 8-week-old R6/2 mice. Gene expression of (a) Il6 in peritoneal macrophages (n = 10/group), (b) Il12 (n = 5/group) and (c) Cd40 in splenic cells (n = 5/group), (d) Cd40 in peritoneal macrophages (n = 10/group) in R6/2 mice compared to WT. (e) Increased frequencies of activated macrophages and DC in early-symptomatic stage R6/2 mice. Mean % ± SEM of peritoneal macrophages (gated as; CD11b+, CD8−, CD11c−, Ly6G−) and splenic DC (gated as; CD11b+, CD8−, CD11c+, Ly6G−) highly expressing cell surface CD40 in 8 week old WT (n = 5/group) and R6/2 mice (n = 6/group) as assessed by FACS. (f) Increased frequencies of activated T cells in early-symptomatic stage R6/2 mice. Mean percentage ± SEM of splenic T cells (gated as; CD3+, CD4+ or CD3+, CD8+) highly expressing cell surface OX40 in 8 week old WT (n = 5/group) and R6/2 mice (n = 8/group) as assessed by FACS. (g) Presence of exon 1 mutant huntingtin (mHTT) in splenic mononuclear cells and peritoneal macrophages in 8 week old R6/2 (n = 6) but not in WT (n = 5) mice as assessed by qPCR, mean CT values ± SEM are shown for both mHTT and the geometric mean of ATP5B and B2M housekeeping genes (HK). Agarose gel electrophoresis of the corresponding mHTT qPCR products are shown below each graph. #p = 0.06, *p < 0.05 vs WT by Student’s t test. WT = wild type, pM0 = peritoneal macrophages.
Quantification of protein levels in blood from 8-week-old R6/2 mice showed no increase in IL6, IL10, IL1β and TNFα cytokines levels (data not shown), despite the apparent increased gene expression for some of these cytokines within peripheral cell populations. This finding suggests that the immune system has not been activated for long enough to allow cytokines to accumulate. In contrast, the cell surface expression of CD40 on macrophages and DCs mirrors the gene expression findings: with a 20% increase in the percentage of activated macrophages and approximately 30% increase in the percentage of activated DCs, indicating that these cells may have become recently activated (Fig. 3e).
The percentages of OX40 high expressing cells were increased for the CD4 T cells (p = 0.06) but not for the CD8 T cell population, indicating that helper T cells are on the cusp of activation at this stage of disease (Fig. 3f). Interestingly, unlike for Cd40 (Fig. 3c,d), Ox40L gene expression, and cell surface protein expression on macrophages and DCs remained similar to WT levels (data not shown). This further supports the notion that we have analyzed these cell populations at the early stages of immune activity, since OX40L upregulation on antigen presenting cells (APC) usually occurs at a later stage of activation, after CD40 surface expression increases33,34. Furthermore, FACS assessment of peritoneal macrophages from 8-week-old mice also revealed similar results with increases in the percentages of CD40 highly expressing cells in the R6/2 mice versus WT controls (Fig. 3e) but with no differences in OX40L highly expressing peritoneal macrophage frequencies in the R6/2 and WT groups (Figure S2).
Assessment of exon 1 mutant HTT gene expression within R6/2 peritoneal macrophages and splenic mononuclear cells, containing lymphocytes, macrophages and DC, confirmed that the R6/2 transgene was expressed in these cells and as expected was absent from their WT counterparts (Fig. 3g).
Late stage zQ175 mice recapitulate the immune activation characteristics observed in 14-week-old R6/2 mice
In order to confirm the relevance of phenotypes identified in R6/2 mice, we ascertained whether these also develop in a knock-in HD mouse model, which more closely recapitulates the genetic basis of HD. The expression levels of cytokines in splenocytes from late-stage zQ175 mice at 22 months of age showed similar increases to those observed in late-stage R6/2 mice: Il10 by over 50%, Il12 by almost three-fold, and Il17 by approximately 50%. In contrast to R6/2 mice, Tnfα was also increased (Fig. 4a). In the peritoneal macrophage enriched cell populations, Il6 gene expression was almost three-fold higher, Il10 over six-fold higher and Il12 around five-fold higher in the zQ175 mice versus WT (Fig. 4a). These results indicate that the M1 activated macrophage phenotype is more evident in zQ175 than in R6/2 mice, as indicated by a greater increase in Il12, as well as the upregulation of Tnfα expression in spleen. This suggests that the zQ175 mice have a less stringent M1 or M2 macrophage response. The frequencies of activated T cells, macrophages and DCs in the spleen or peritoneal cavity were also upregulated in late-stage zQ175 mice as compared to WT, according to the cell surface expression of activation markers: OX40 (Fig. 4b) and OX40L (Fig. 4c) respectively, which were increased. Analysis of cytokine gene expression in brain regions of zQ175 mice at 12 months of age revealed that only Tnfα expression was increased in the striatum, as had been the case for R6/2 mice. However, unlike R6/2 mice, Il12 was not upregulated in the striatum, and nor was it decreased in the cortex of zQ175 mice (Fig. 4d). We were unable to observe any increase in striatal cytokine levels in late stage 21 month old zQ175 mice (Fig. 4e), which may be due to the effect of immune exhaustion in these old zQ175 mice as a result of chronic immune activation of self-renewing HD microglial cells35.

Immune activation in zQ175 mice. (a) Increased gene expression in late-stage zQ175 mice for several cytokines in splenic (n = 5–6/group) and peritoneal (n = 5–6/group) macrophage populations. 22-month-old zQ175 mice ± SEM splenic and peritoneal macrophage gene expression for cytokines relative to WT as quantified by qPCR. (b) Increased activated T cell numbers in 22-month-old Q175 mice. Mean % ± SEM of splenic T cells (gated as; CD3+, CD4+ or CD8+) expressing OX40 highly in 22 month old WT (n = 5/group) and zQ175 mice (n = 6/group) as assessed by FACS. (c) Increased activated peritoneal macrophage and DC frequencies in 22-month-old zQ175 mice. Mean percentage ± SEM of peritoneal macrophage and splenic DC expressing OX40L highly in 22 month old WT (n = 5/group) and zQ175 mice (n = 5/group) as assessed by FACS. (d) Increases in cytokine gene expression was limited to the striatum of 12 month old zQ175 mice. Mean Il1β, Il6, Il10, Il12 and Tnfα gene expression levels in cerebellum, striatum and cortex of zQ175 mice (n = 5/group) compared to WT (n = 5/group) ± SEM as measured by qPCR. (e) There were no increases in cytokine gene expression in 21 month old zQ175 mice. Mean Il1β, Il6, Il10, Il12 and Tnfα gene expression levels in cerebellum, striatum and cortex of zQ175 mice (n = 5/group) compared to WT (n = 5/group) ± SEM as measured by qPCR. *p < 0.05 and **p < 0.01 vs WT by Student’s t test. WT = wild type, pM0 = peritoneal macrophages, DC = dendritic cells.
In vivo depletion of macrophages and DC via clodronate liposome infusion normalizes cytokine levels during late stage HD
Our current and previous results13 strongly implicate macrophages as the major contributors to the immune dysregulation that occurs in HD. In order to ascertain the role of these cells, we utilized the toxic clodronate immunosuppressive drug, commonly used for the treatment of osteoporosis36. In order to specifically deplete monocyte and macrophage cells in vivo, clodronate packaged in liposomes were administered via IV or IP injection into R6/2 mice at 13–14 weeks of age37,38,39. IV injection resulted in the depletion of (CD11b+ CD8− CD11c− Ly6G−) blood monocytes and splenic macrophages, and (CD11b+ CD8− CD11c+ Ly6G−) splenic DCs, in both the blood and spleen of R6/2 mice (Fig. 5a) and WT (data not shown), but not T cells or neutrophils (data not shown). PBS liposome treatment had no impact on the frequencies of blood monocytes, and splenic macrophages and DCs (Fig. 5a). Depletion of splenic DCs and splenic macrophages was achieved one and two days post clodronate injection respectively, with maximal depletion of around 75% for macrophages and almost 90% of DCs occurring on the third day, this being maintained until day four post injection. Blood monocyte depletion was observed earlier, one day after clodronate injection. The recovery of blood monocytes two days post injection may indicate a response by the bone marrow to replete monocyte numbers, while its subsequent gradual reduction from day two may be due to the recruitment of monocytes into tissues, in order to replenish the diminished macrophage numbers post clodronate liposome depletion24,39.

Clodronate liposome depletion of splenic macrophage and DC results in reduced IL6 levels in R6/2 mice. (a) IV clodronate liposome treatment of late-stage R6/2 mice depletes splenic macrophages (gated as; CD11b+, CD8−, CD11c−, Ly6G−) and DC (gated as; CD11b+, CD8−, CD11c+, Ly6G−), and monocytes (gated as; CD11b+, CD8−, CD11c−, Ly6G−). Mean percentage ± SEM of splenic macrophages and DC, and monocytes in the total leukocyte population following clodronate liposome or PBS liposome injection into late-stage R6/2 mice as analyzed by FACS. (b) Clodronate liposome treatment only normalises IL6 to WT plasma levels in late-stage R6/2 mice. Mean concentration of TNFα, IL1β, IL2, IL6 and IL10 ± SEM following clodronate liposome or PBS liposome injection as assessed by MSD. *p < 0.05 decrease, #p < 0.05 increase as compared to R6/2 day 0, (n = 5/group). WT = wild type, IV = intravenous, DC = dendritic cells, M0 = macrophages. MNC = monocytes, Clod/lip = clodronate liposome, PBS/lip = PBS liposome.
Analysis of plasma cytokine levels following clodronate liposome treatment revealed that the drug initially promoted a gradual increase in all of the cytokines assessed (Fig. 5b). This peaked two days post injection, rising from approximately 20 pg/mL to over 80 pg/mL for IL10, from 2 pg/mL to 13 pg/mL for IL1β, by over 60% for IL2, by approximately 130 pg/mL for TNFα and by over fifty-fold for IL6 (Fig. 5b). No significant changes were evident in the PBS liposome treated control mice at day 2 and day 4 post injection (Fig. 5b). This observation can be explained by the activation of the immune system by dead or dying monocytes and macrophages, increasing cytokine production by immune cells, which includes any remaining monocytes and macrophages40. Such initial, dramatic cytokine spikes could potentially have detrimental effects, however no adverse effects were observed up to ten days post drug injection. At day 3 post injection, when maximum splenic macrophage and DC depletion was observed, most of the cytokines returned to basal levels, while at day 4 post injection IL6 levels had significantly decreased from that observed prior to injection. It is worth noting, that of the cytokines assessed here, only Il6, and Il10 levels were elevated in the peritoneal macrophage enriched cell population of late stage R6/2 mice compared to WT (Fig. 1c). Therefore, the greater likelihood of a reduction in IL6 and IL10, but not TNFα, IL1β or IL2 post macrophage depletion, was perhaps to be expected. However, the lack of a reduction in IL10 levels suggests that other cells, such as T cells and B cells, may upregulate this cytokine in order to compensate for the loss of macrophage IL10 production following the depletion of these cells41,42. Our inability to detect any differences in plasma basal levels in R6/2 compared to WT mice for the other upregulated cytokines (Fig. 1a) is likely due to the low sample size for this experiment. The detection of differences for only plasma IL6 and IL10 (Fig. 5b), the cytokines that show the greatest disparity between R6/2 and WT at 14 weeks of age would support this conclusion.
In order to resolve the limitation of a small sample size, we repeated the clodronate liposome infusion study twice and pooled the experimental results. However, the splenic macrophage and DC depletion dynamics were somewhat variable between experiments. Maximal splenic macrophage (decreasing to 0.8–0.9% CD11b+ CD8− CD11c− Ly6G− cells from 3.9–4.5% prior injection) and DC depletion (decreasing to 0.1–0.2% CD11b+ CD8− CD11c+ Ly6G− cells from 0.6–0.8% prior injection) was achieved in all experiments, but occurred at different time-points, between one to three days post injection (data not shown). This difference may be due to batch-to-batch drug variations. Despite the difference in depletion timing, macrophage frequencies and cytokine levels did not differ significantly at the start of the experiment, or at the time when highest cell depletion was seen between individual experiments.
Therefore, we decided to look specifically at the cytokine levels prior to treatment, for all three experiments, on the day when maximal macrophage depletion was achieved and on the day following maximal macrophage depletion. Our combined results not only confirmed the significant reductions in IL6 levels at the day following maximal macrophage depletion, but additionally, revealed decreases in IL1β and IL2, not only at this time-point, but also on the day of maximal splenic macrophage depletion (Fig. 6a). The reduction of IL1β and IL2 one day ahead of IL6, may be explained by their shorter half-life when compared with IL643,44,45, thus resulting in their more rapid loss post significant macrophage depletion.

Clodronate liposome normalisation of cytokine levels in late-stage R6/2 mice. (a) Clodronate liposome treatment reduced IL1β, IL2 and IL6 levels in late-stage R6/2 mice post IV treatment (n = 8–12/group). Mean concentration of IL10, IL1β, IL2, IL6 and TNFα ± SEM following clodronate liposome IV injection, at the day of greatest splenic M0 depletion and one day later as assessed by MSD. (b) Clodronate liposome treatment normalised TNFα, IL2 and IL6 levels in late-stage R6/2 mice post IP treatment (n = 9–12/group). Mean concentration of TNFα, IL1β, IL2, IL6 and IL10 ± SEM following clodronate liposome IP injection, at day of highest splenic M0 depletion and one day later as assessed by MSD. All comparisons to R6/2 levels at day 0, ANOVA with Bonferroni correction. *p < 0.05, #p = 0.06. WT = wild type, IV = intravenous, IP = intraperitoneal, M0 = splenic macrophages.
Next, we assessed whether clodronate injection via the IP route produced similar results to those of IV injected mice. An IP injection will result in macrophages and DCs within tissues being depleted without affecting blood monocytes, and will reveal whether this is sufficient to account for the effect of clodronate liposomes on R6/2 cytokine levels46. The dynamics of cell depletion in the spleen post IP injection were variable, similar to those seen by IV. The maximal depletion of macrophages (from 4% ± 0.3% to 1.1% ± 0.4%) and DCs (0.4% ± 0.09% to 0.2 ± 0.04%) were at comparable levels to those for IV, but occurred later, on days three and four post injection, as expected, there was no effect on blood monocyte numbers (data not shown). Plasma cytokine levels were measured by MSD multiplex analysis on the day when maximal cell depletion was achieved, as well as the following day. IL6 levels were decreased one day after maximum splenic macrophage depletion, but there was no effect on IL1β as had been observed for the IV injected mice. This may be due to IL1β being predominantly secreted by blood monocytes, which remain intact post IP clodronate treatment or, to the greater macrophage depletion within spleens via the IV route compared to the IP route (data not shown). However, TNFα levels were also reduced following cell depletion, which had not occurred after IV injection (Fig. 6b). Taking the IP and IV results together, these findings suggest that reductions in specific cytokines post clodronate treatment only occur if these cytokines are at higher levels in R6/2 plasma as compared to WT at treatment initiation. The results indicate that depletion of macrophages and DCs within the spleen, and probably other tissues, is sufficient to prevent the increased plasma cytokine levels in R6/2 mice, suggesting that macrophages and/or DCs are central to the observed increases in IL1β, IL2, IL6, and TNFα levels.
Discussion
Herein we have shown that the cells that promote immune hyperactivation during disease progression in HD mouse models are not only macrophages and monocytes, as inferred by previous studies, but also T cells and DCs. We additionally characterised the macrophage response in HD according to the M1 and M2 phenotypic classification, and found that this differed depending on the tissue in which these cells reside. In R6/2 mice, within the brain, microglial cells took up a predominantly M1 cytotoxic profile, whilst in the periphery, macrophages favour an M2 humoral/repair phenotype29. Thus, while microglial cells produced elevated Il12 and Tnfα but lacked Il10 and Il6 increases, their peripheral counterparts increased their production of Il6 and Il10 as well as the M2 promoting cytokine Il4, but lacked Tnfα upregulation. This indicated a phenotypic discordance in the activated macrophage cells populating these two distinct regions. Although the M1/M2 paradigm has been traditionally used to identify seemingly two phenotypically distinct macrophage populations generated following their activation, such differences have been shown to be less clear cut more recently47,48. In keeping with this, in the zQ175 mice the M1/M2 paradigm was less evident, but nevertheless some remnants of a peripheral M2 peritoneal macrophage and an M1 like microglial response could still be seen.
The absence of Il10 upregulation within the brain despite, its increases in the periphery, not only support the idea that microglial cells are predominantly M1 in phenotype but may also indicate that these cells are not trying to counter the inflammation within the CNS, at least via the secretion of anti-inflammatory IL10. Interestingly, the study by Bjorkvist et al. in three different HD mouse models, not only saw similar increases in the levels of blood M2 type cytokines, IL6 and IL10 in the periphery but also found no significant increases in the M1 promoting cytokine IFNγ and, unlike in our study, TNFα as well, thus further supporting the notion that activated peripheral monocytes/macrophages show an M2 dominant phenotype9.
Inflammation and immune activation has been reported to be a feature of HD and our analysis of cytokine levels in the brain and plasma of late-stage R6/2 and zQ175 mice further confirmed this finding. Of particular interest, this included increases in IL6 and TNFα, which are known to have neurotoxic properties26,28 and have the capacity to cross the blood brain barrier49,50. Interestingly, TNFα expression was increased in the brain and elevated levels of this cytokine were found in the blood, despite no increase in Tnfα gene expression in the peripheral immune compartments assessed. Therefore, TNFα may have migrated from the brain to the blood. However, other peripheral TNFα sources such as lymph nodes cannot be discounted as the possible source for the raised blood TNFα levels.
IL17 has been found to be secreted by astrocytes within the brain51, however we failed to detect Il17 gene expression in either WT or R6/2 mice in our qPCR analysis which might suggest a minimal role of these glial cell population to the observed cytokine upregulation in the HD brain, since they are also coincidently capable of secreting cytokines such as TNFα52. Clearly, determining the mechanisms for the observed site-specific differences in HD brain cytokine production requires further elucidation.
Subsequent analyses to quantify the frequencies of activated immune cells, responsible for the upregulated cytokines, revealed not only increases in the levels of active macrophages, as predicted, but also DCs and T cells. These results indicate a possible contribution of these cells to the macrophage-induced inflammation seen during HD. Previously, we failed to detect any increases in activated macrophage frequencies based on their MHCII cell surface expression13. This result is particularly interesting given that unlike M1 macrophages, M2 macrophages do not notably upregulate MHCII on their surface post-activation. This not only provides an explanation for the previous results but also strengthens our current findings, suggesting that peripheral macrophages predominantly adopt an M2 phenotype in R6/2 mice29,53. Late-stage zQ175 mice revealed similar increases in cytokine production, and activated immune cells, to their 14-week-old R6/2 counterparts. However, these mice were found to have a more balanced M1 and M2 macrophage response, which may be due to age differences or minor differences in disease presentation between the two models. We previously reported that the NFκB transcription factor is dysregulated in HD patient monocytes/macrophages13. It would be interesting to assess whether any of the other HD murine cell populations we have found to have enhanced activation levels also utilise NFκB for their activity.
The analysis of immune activity during specific stages of disease progression in R6/2 mice provided an insight into the initiation of immune activation. Initial signs of immune activity in macrophage and DC populations occurred at an early symptomatic stage, at which there was no immune activity in the striatum, a brain region that shows immune dysregulation during late-stage disease. Our finding that immune activation in the periphery precedes that in the brain could suggest that the peripheral immune system may, at least in part, initiate or enhance the activation of brain microglia, as previously proposed by our group and others9,11, possibly through the secretion and provision of IL6 and other blood brain barrier penetrating cytokines. These cytokines may stimulate microglia directly, or indirectly, to become activated e.g. through neuronal cell killing, as could be the case for IL654.
Clodronate liposome depletion of macrophages and DCs in vivo, resulted in the normalisation of cytokine levels in late-stage R6/2 mice, and provided the first in vivo evidence to confirm that these APCs upregulate their cytokine levels. Our results provide a valuable insight into the impact of the dysregulated macrophage and DC responses on the HD immune system, identifying these cells as major accomplices in promoting the chronic in vivo inflammation that manifests as excessive blood cytokine content in HD mice. Follow up studies would be required to decipher the effect of peripheral cytokine normalisation on HD associated neuroinflammation and ultimately, HD disease pathology. From our experience as well as from previous immunomodulatory HD studies, this would require consistent, long term and continuous blood cytokine reduction. Thus multiple, scheduled clodronate treatments would be required to prevent the observed recovery of monocytes/macrophages and DC numbers in order to investigate the impact of prolonged, sustained drug induced macrophage and DC depletion on disease- related phenotypes. Several immunomodulatory regimens, which have achieved long-term immunosuppression in HD mice, have however conferred beneficial effects in improving symptoms and/or survival. We propose that such effects are, at least in part, due to their impact on macrophages and/or DCs, be it upstream, as in the case of bone marrow transplantation, where HD myeloid cells are replaced by wild type cells14, or downstream, where specific macrophage-upregulated cytokines are inhibited, such as peripheral IL6 neutralization and TNFα signalling inhibition15,16. Although we show that clodronate liposome therapy significantly dampens mutant huntingtin induced macrophage hyperactivity, and elevated cytokine production, the long-term use of such a potent drug may be deleterious, considering the important role of macrophages in other biological functions such as red blood cell recycling55. Thus, more defined therapeutic strategies targeting the production of specific cytokines, or biasing their M1/M2 activation phenotype, are likely to have more desirable outcomes. Nevertheless, herein, we have shown that macrophages and DCs play a protagonist role in the immune hyper-activation and chronic inflammation associated with HD in vivo, providing a case for immunomodulatory regimens targeting these particular cells as potential therapeutic strategies for this disease.
Methods
Mice
Hemizygous R6/2 mice were generated by backcrossing R6/2 males to (CBA x C57BL/6) F1 females (B6CBAF1/OlaHsd, Harlan Olac, Bicester, UK)21. Heterozygous zQ175 knock-in mice18 were supplied from CHDI colonies (Jackson Laboratory, Bar Harbor, ME, USA) and maintained by backcrossing to C57BL/6J (Charles River). All animals had unlimited access to food and water, and were kept in sterile housing conditions in individually ventilated cages with environmental enrichment as previously described56. Mice were subject to a 12-h light/dark cycle. All experimental procedures were performed in accordance with Home Office regulations and approved by the King’s College London and University College London Ethical Review Committees.
R6/2 and zQ175 mice were genotyped and the CAG repeat was measured as previously described57. The mean repeat size (±SD) for all mice used in the study was 180 ± 10 for zQ175 mice and 205 ± 7 for R6/2 mice. Mice were euthanized via CO2 asphyxiation or cervical dislocation as required. Equal numbers of male and female R6/2 and zQ175 mice, and their WT littermate controls were used unless stated otherwise.
Blood, plasma, tissue and cell sample collection
Blood was obtained via tail vein puncture followed by decapitation post cervical dislocation and collected into EDTA tubes. Plasma was obtained by spinning blood at 2000 × g for 5 min obtaining the top plasma layer, while the bottom layer, containing blood cells, was used for subsequent FACS analysis. Following euthanasia, whole brains, striatum, cortex, cerebellum and whole spleens tissues were dissected. Brain samples were snap frozen in liquid nitrogen immediately after dissection and stored at −80 °C until required. Single cell suspensions from spleen samples were obtained as described previously58, in some cases mononucleated cells from isolated splenocytes were purified by density gradient centrifugation using Histopaque (Sigma). Peritoneal macrophages (pM0) were isolated as described previously12. All cell samples were treated with red blood cell lysis solution (R&D systems) in order to remove red blood cells prior to subsequent processing.
Plasma analyses
Cytokine (IL1β, IL2, IL6, IL10, IL12 and TNFα) levels in plasma were quantified using Meso Scale Discovery (MSD) assays as per the manufacturer’s protocol and analysed on a SECTOR 2400 instrument (MSD).
RNA extraction and quantitative real time gene expression analysis
Total RNA from cell sample was extracted with the mini-RNA kit according to the manufacturer’s instruction (Qiagen). Reverse transcription (RT) was performed using the MMLV Superscript reverse transcriptase (Invitrogen) and random hexamers (Operon) as described previously59. All Taqman quantitative real-time PCR (qPCR) reactions were performed using the Chromo4 real-time PCR detector (Biorad) and quantified via the 2−ΔΔCT method60. Gene expression for the gene of interest was normalised to the geometric mean of endogenous housekeeping genes (beta 2 microglobulin and Atp5b for peripheral tissue/cells or Ubc and Atp5b for brain tissue) for each sample. Primer and probe sets for genes of interest (Il1β, Il2, Il4, Il6, Il10, Il12, Il17, Tnfα, Cd40, Ox40 and Ox40L) and housekeeping genes were purchased from Biorad. For mutant huntingtin gene expression analysis: forward primer sequence = 5′gctgcacctaccgtgagt3′, reverse primer sequence = 5′cgcaggctgcagggttac3′, and FAM labelled probe sequence = 5′agctccctgtccgggcgg3′ were used.
Flow cytometry (FACS)
For detecting macrophage/monocytes, DCs, neutrophils61 and CD8 T cells62, 0.5 × 106 cells were stained with anti-mouse CD8-FITC, anti-mouse CD49b-FITC, anti-mouse CD11b-phycoerythrim (PE), anti-mouse CD11c-APC-Cy7 (for DC exclusion) or CD11c-Per-CP (for activated DC population analysis) and anti-mouse Ly6G-APC-Cy7. To detect activated macrophage and DC populations, anti-mouse CD40-APC or OX40L-APC antibodies were also added (all from eBioscence). To detect activated T cells, anti-mouse CD3-PE, anti-mouse CD4-PerCP, anti-mouse CD8-FITC and anti-mouse OX40-APC or anti-mouse CD25-APC antibody (all from eBioscience) staining was performed for 30 min at 4 °C after blocking with 10% FCS in PBS with 10 mM EDTA, then fixed with 2% paraformaldehyde in PBS with 10 mM EDTA. Cells were analysed using FACS Canto (Becton Dickinson) acquiring 20,000 leukocytes (gated according to forward and side scatters with doublets excluded according to FCS-A/FCS-H dot plots). FACS results were subsequently analysed with FACS Diva (Becton Dickinson). Corresponding unstained cell samples for each antibody were acquired to establish gating areas for positively staining cells.
In vivo clodronate liposome treatment
13 to 14 week old female mice were injected intravenously (IV) via the tail vein or intraperitoneally (IP) with 200 µl or 400 µl clodronate liposome (Liposoma) at 5 mg/mL respectively or with control liposomes containing PBS. Preliminary dosing studies were performed to determine the optimal dose required to deplete target cells over ten days. Body temperature was monitored post injection, and no adverse effects were seen. Mice were sacrificed by cervical dislocation at days 1, 2, 3 and 4 post treatment for IV injected mice or 3, 4, 5 and 6 post treatment for IP injected mice as well as day 0 (prior injection). Blood, spleen and liver samples were obtained as described above for plasma cytokine and/or cell (monocyte/macrophage, DC, CD8 T cells or neutrophil) content quantification by MSD or FACS respectively as detailed above.
Statistical Analysis
Differences between specified groups were detected using one-way ANOVA with Bonferroni correction (SPSS software) or Student’s t-test (Microsoft Excel software) as stated in the Figure legends. P-values of 0.05 or less were considered significant.
References
Shamim, D. & Laskowski, M. Inhibition of Inflammation Mediated Through the Tumor Necrosis Factor alpha Biochemical Pathway Can Lead to Favorable Outcomes in Alzheimer Disease. Journal of central nervous system disease 9, 1179573517722512, https://doi.org/10.1177/1179573517722512 (2017).
Domingues, C., da Cruz, E. S. O. A. B. & Henriques, A. G. Impact of Cytokines and Chemokines on Alzheimer’s Disease Neuropathological Hallmarks. Current Alzheimer research 14, 870–882, https://doi.org/10.2174/1567205014666170317113606 (2017).
Tiwari, P. C. & Pal, R. The potential role of neuroinflammation and transcription factors in Parkinson disease. Dialogues in clinical neuroscience 19, 71–80 (2017).
Manocha, G. D. et al. Defining the contribution of neuroinflammation to Parkinson’s disease in humanized immune system mice. Mol Neurodegener 12, 17, https://doi.org/10.1186/s13024-017-0158-z (2017).
Alcolea, D. et al. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology 85, 626–633, https://doi.org/10.1212/WNL.0000000000001859 (2015).
Huntinton’s Disease Colaborative Research Group. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntingtons-Disease Chromosomes. Cell 72, 971–983, https://doi.org/10.1016/0092-8674(93)90585-E (1993).
Landwehrmeyer, G. B. et al. Huntington’s disease gene: regional and cellular expression in brain of normal and affected individuals. Annals of neurology 37, 218–230, https://doi.org/10.1002/ana.410370213 (1995).
Weiss, A. et al. Mutant huntingtin fragmentation in immune cells tracks Huntington’s disease progression. J Clin Invest 122, 3731–3736, https://doi.org/10.1172/JCI64565 (2012).
Bjorkqvist, M. et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med 205, 1869–1877, https://doi.org/10.1084/jem.20080178 (2008).
Tai, Y. F. et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 130, 1759–1766, https://doi.org/10.1093/brain/awm044 (2007).
Wild, E. et al. Abnormal peripheral chemokine profile in Huntington’s disease. PLoS currents 3, RRN1231, https://doi.org/10.1371/currents.RRN1231 (2011).
Trager, U. et al. Characterisation of immune cell function in fragment and full-length Huntington’s disease mouse models. Neurobiology of disease 73, 388–398, https://doi.org/10.1016/j.nbd.2014.10.012 (2015).
Trager, U. et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFkappaB pathway dysregulation. Brain 137, 819–833, https://doi.org/10.1093/brain/awt355 (2014).
Kwan, W. et al. Bone Marrow Transplantation Confers Modest Benefits in Mouse Models of Huntington’s Disease. Journal of Neuroscience 32, 133–142, https://doi.org/10.1523/Jneurosci.4846-11.2012 (2012).
Hsiao, H. Y. et al. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Human molecular genetics 23, 4328–4344, https://doi.org/10.1093/hmg/ddu151 (2014).
Bouchard, J. et al. Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of Huntington’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience 32, 18259–18268, https://doi.org/10.1523/JNEUROSCI.4008-12.2012 (2012).
Menalled, L. B., Sison, J. D., Dragatsis, I., Zeitlin, S. & Chesselet, M. F. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. The Journal of comparative neurology 465, 11–26, https://doi.org/10.1002/cne.10776 (2003).
Menalled, L. B. et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PloS one 7, e49838, https://doi.org/10.1371/journal.pone.0049838 (2012).
Sathasivam, K. et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. P Natl Acad Sci USA 110, 2366–2370 (2013).
Neueder, A. et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Scientific reports 7, 1307, https://doi.org/10.1038/s41598-017-01510-z (2017).
Mangiarini, L. et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 (1996).
Woodman, B. et al. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain research bulletin 72, 83–97, https://doi.org/10.1016/j.brainresbull.2006.11.004 (2007).
Weisser, S. B., van Rooijen, N. & Sly, L. M. Depletion and reconstitution of macrophages in mice. Journal of visualized experiments: JoVE, 4105, https://doi.org/10.3791/4105 (2012).
van Rooijen, N., Kors, N. & Kraal, G. Macrophage subset repopulation in the spleen: differential kinetics after liposome-mediated elimination. Journal of leukocyte biology 45, 97–104 (1989).
Simmons, D. A. et al. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 55, 1074–1084, https://doi.org/10.1002/glia.20526 (2007).
Conroy, S. M. et al. Interleukin-6 produces neuronal loss in developing cerebellar granule neuron cultures. J Neuroimmunol 155, 43–54, https://doi.org/10.1016/j.jneuroim.2004.06.014 (2004).
Qiu, Z. H. & Gruol, D. L. Interleukin-6, beta-amyloid peptide and NMDA interactions in rat cortical neurons. J Neuroimmunol 139, 51–57, https://doi.org/10.1016/S0165-5728(03)00158-9 (2003).
Talley, A. K. et al. Tumor-Necrosis-Factor Alpha-Induced Apoptosis in Human Neuronal Cells - Protection by the Antioxidant N-Acetylcysteine and the Genes Bcl-2 and Crma. Mol Cell Biol 15, 2359–2366 (1995).
Davis, M. J. et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. mBio 4, e00264–00213, https://doi.org/10.1128/mBio.00264-13 (2013).
Gramaglia, I., Weinberg, A. D., Lemon, M. & Croft, M. Ox-40 ligand: A potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol 161, 6510–6517 (1998).
Jaiswal, A. I. & Croft, M. CD40 ligand induction on T cell subsets by peptide-presenting B cells: implications for development of the primary T and B cell response. J Immunol 159, 2282–2291 (1997).
Hosono, M. et al. Increased expression of T cell activation markers (CD25, CD26, CD40L and CD69) in atherectomy specimens of patients with unstable angina and acute myocardial infarction. Atherosclerosis 168, 73–80 (2003).
Ohshima, Y. et al. Expression and function of OX40 ligand on human dendritic cells. J Immunol 159, 3838–3848 (1997).
Murata, K. et al. Impairment of antigen-presenting cell function in mice lacking expression of OX40 ligand. Journal of Experimental Medicine 191, 365–374, https://doi.org/10.1084/jem.191.2.365 (2000).
Bruttger, J. et al. Microglia are unique tissue phagocytes with high self-renewing capacity. Vol. 275 (2014).
Muratore, M., Quarta, E., Grimaldi, A., Calcagnile, F. & Quarta, L. Clinical utility of clodronate in the prevention and management of osteoporosis in patients intolerant of oral bisphosphonates. Drug design, development and therapy 5, 445–454, https://doi.org/10.2147/DDDT.S12139 (2011).
Frediani, B. et al. Improved efficacy of intramuscular weekly administration of clodronate 200 mg (100 mg twice weekly) compared with 100 mg (once weekly) for increasing bone mineral density in postmenopausal osteoporosis. Clinical drug investigation 33, 193–198, https://doi.org/10.1007/s40261-013-0062-4 (2013).
Everse, L. A., Anderson, L. D. Jr., van Rooijen, N. & Mullen, C. A. Bone marrow transplant conditioning intensified with liposomal clodronate to eliminate residual host antigen presenting cells fails to ameliorate GVHD and increases PERI-BMT mortality. Transplantation 71, 611–618 (2001).
Van Rooijen, N., Kors, N., vd Ende, M. & Dijkstra, C. D. Depletion and repopulation of macrophages in spleen and liver of rat after intravenous treatment with liposome-encapsulated dichloromethylene diphosphonate. Cell and tissue research 260, 215–222 (1990).
Kono, H. & Rock, K. L. How dying cells alert the immune system to danger. Nat Rev Immunol 8, 279–289, https://doi.org/10.1038/nri2215 (2008).
Kalampokis, I., Yoshizaki, A. & Tedder, T. F. IL-10-producing regulatory B cells (B10 cells) in autoimmune disease. Arthritis Res Ther 15, https://doi.org/10.1186/ar3907 (2013).
Ng, T. H. S. et al. Regulation of adaptive immunity; the role of interleukin-10. Front Immunol 4, https://doi.org/10.3389/Fimmu.2013.00129 (2013).
Lotze, M. T. et al. Invivo Administration of Purified Human Interleukin-2 .2. Half-Life, Immunological Effects, and Expansion of Peripheral Lymphoid-Cells Invivo with Recombinant Il-2. J Immunol 135, 2865–2875 (1985).
Peters, M. et al. The function of the soluble interleukin 6 (IL-6) receptor in vivo: Sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. Journal of Experimental Medicine 183, 1399–1406, https://doi.org/10.1084/jem.183.4.1399 (1996).
Kudo, S., Mizuno, K., Hirai, Y. & Shimizu, T. Clearance and Tissue Distribution of Recombinant Human Interleukin-1-Beta in Rats. Cancer research 50, 5751–5755 (1990).
Biewenga, J. et al. Macrophage Depletion in the Rat after Intraperitoneal Administration of Liposome-Encapsulated Clodronate - Depletion Kinetics and Accelerated Repopulation of Peritoneal and Omental Macrophages by Administration of Freunds-Adjuvant. Cell and tissue research 280, 189–196 (1995).
Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20, https://doi.org/10.1016/j.immuni.2014.06.008 (2014).
Martinez, F. O. & Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports 6, 13, https://doi.org/10.12703/P6-13 (2014).
Banks, W. A., Kastin, A. J. & Gutierrez, E. G. Penetration of interleukin-6 across the murine blood-brain barrier. Neuroscience letters 179, 53–56 (1994).
Pan, W. & Kastin, A. J. TNFalpha transport across the blood-brain barrier is abolished in receptor knockout mice. Experimental neurology 174, 193–200, https://doi.org/10.1006/exnr.2002.7871 (2002).
Hu, M. H. et al. Neuroprotection effect of interleukin (IL)-17 secreted by reactive astrocytes is emerged from a high-level IL-17-containing environment during acute neuroinflammation. Clinical and experimental immunology 175, 268–284, https://doi.org/10.1111/cei.12219 (2014).
Choi, S. S., Lee, H. J., Lim, I., Satoh, J. & Kim, S. U. Human astrocytes: secretome profiles of cytokines and chemokines. PloS one 9, e92325, https://doi.org/10.1371/journal.pone.0092325 (2014).
Arnold, C. E., Gordon, P., Barker, R. N. & Wilson, H. M. The activation status of human macrophages presenting antigen determines the efficiency of Th17 responses. Immunobiology 220, 10–19, https://doi.org/10.1016/j.imbio.2014.09.022 (2015).
Fu, R. Y., Shen, Q. Y., Xu, P. F., Luo, J. J. & Tang, Y. M. Phagocytosis of Microglia in the Central Nervous System Diseases. Mol Neurobiol 49, 1422–1434, https://doi.org/10.1007/s12035-013-8620-6 (2014).
Delaby, C. et al. A physiological model to study iron recycling in macrophages. Exp Cell Res 310, 43–53, https://doi.org/10.1016/j.yexcr.2005.07.002 (2005).
Hockly, E., Woodman, B., Mahal, A., Lewis, C. M. & Bates, G. Standardization and statistical approaches to therapeutic trials in the R6/2 mouse. Brain research bulletin 61, 469–479 (2003).
Sathasivam, K. et al. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington's disease. Human molecular genetics 19, 65–78, https://doi.org/10.1093/hmg/ddp467 (2010).
Girdlestone, J. et al. Enhancement of the immunoregulatory potency of mesenchymal stromal cells by treatment with immunosuppressive drugs. Cytotherapy 17, 1188–1199, https://doi.org/10.1016/j.jcyt.2015.05.009 (2015).
Mielcarek, M. et al. HDAC4 Does Not Act as a Protein Deacetylase in the Postnatal Murine Brain In Vivo. PloS one 8, https://doi.org/10.1371/journal.pone.0080849 (2013).
Pido-Lopez, J. et al. The effect of allogeneic in vitro stimulation and in vivo immunization on memory CD4(+) T-cell APOBEC3G expression and HIV-1 infectivity. European journal of immunology 39, 1956–1965, https://doi.org/10.1002/eji.200939228 (2009).
Rose, S., Misharin, A. & Perlman, H. A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytom Part A 81A, 343–350, https://doi.org/10.1002/cyto.a.22012 (2012).
Mcfarland, H. I., Nahill, S. R., Maciaszek, J. W. & Welsh, R. M. Cd11b (Mac-1) - a Marker for Cd8+ Cytotoxic T-Cell Activation and Memory in Virus-Infection. J Immunol 149, 1326–1333 (1992).
Acknowledgements
We thank members of the Bates Lab for assistance with generation of mice, genotyping and CAG repeat sizing and assistance with clodronate administration. This work was supported by funding from the MRC (MR/L02053X/1) and the CHDI Foundation.
Author information
Authors and Affiliations
Contributions
J.P.-L., R.A., S.J.T. and G.P.B. designed the study. J.P.-L., R.A., N.A., A.C.B., S.F. performed the experiments. J.P.-L. and R.A. analysed the data. J.P.-L. prepared the figures. J.P.-L. and G.P.B. wrote the manuscript. All authors discussed and interpreted the data and revised the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pido-Lopez, J., Andre, R., Benjamin, A.C. et al. In vivo neutralization of the protagonist role of macrophages during the chronic inflammatory stage of Huntington’s disease. Sci Rep 8, 11447 (2018). https://doi.org/10.1038/s41598-018-29792-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29792-x
This article is cited by
-
Immunological aspects of central neurodegeneration
Cell Discovery (2024)
-
Ginsenoside Rg1 exerts neuroprotective effects in 3-nitropronpionic acid-induced mouse model of Huntington’s disease via suppressing MAPKs and NF-κB pathways in the striatum
Acta Pharmacologica Sinica (2021)
-
Ablation of kynurenine 3-monooxygenase rescues plasma inflammatory cytokine levels in the R6/2 mouse model of Huntington’s disease
Scientific Reports (2021)
-
Inhibition of tumour necrosis factor alpha in the R6/2 mouse model of Huntington’s disease by etanercept treatment
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
