
Abstract
A simple protocol to overcome the problematic trifluoromethylation of carbonyl compounds by the potent greenhouse gas “HFC-23, fluoroform” with a potassium base is described. Simply the use of glymes as a solvent or an additive dramatically improves the yields of this transformation. Experimental results and DFT calculations suggest that the beneficial effect deals with glyme coordination to the K+ to produce [K(polyether)n]+ whose diminished Lewis acidity renders the reactive anionoid CF3 counterion species more ‘naked’, thereby slowing down its undesirable decomposition to CF2 and F− and simultaneously increasing its reactivity towards the organic substrate.
Similar content being viewed by others

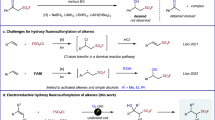
Introduction
There has been remarkable progress recently in the synthetic incorporation of a trifluoromethyl (CF3) moiety into potential bioactive molecules, prompting the discovery of new pharmaceuticals and agrochemicals1,2,3,4,5. Fluoroform (HFC-23, HCF3, trifluoromethane) is a potent greenhouse gas that is formed as a by-product in huge amounts during the synthesis of poly-tetrafluoroethylene (PTFE) and polyvinylidene difluoride (PVDF) from chlorodifluoromethane (ClCHF2). Fluoroform has a 11,700-fold higher GWP than carbon dioxide with an atmospheric lifetime of 264 years and is used to a very limited extent as a refrigerant or as a raw material6,7,8,9,10. At present, fluoroform abatement techniques involve thermal oxidation, catalytic hydrolysis and plasma destruction, so there are operation and economical limits to transform fluoroform to useful refrigerants or fire extinguishers11,12,13,14,15,16,17. HFC-23 is an easily handled, stable and non-toxic trifluoromethyl (CF3) source18,19,20,21,22,23,24. Thus the synthetic use of HCF3 serving as feedstock for various trifluoromethylations is highly desirable. However, chemoselective and efficient activation of HCF3 for nucleophilic trifluoromethylation processes, is a long-standing, challenging and intriguing issue in organic chemistry. One of the primary problems in the extensive usage of HCF3 for trifluoromethylations is the facile decomposition of the CF3− anion to difluorocarbene (:CF2) and fluoride (F−)18. This decomposition is probably induced by the strong repulsion between the lone electron pairs on the carbon and fluorine atoms of CF3− (Fig. 1a). In the presence of alkali (M+) and other metal cations, the decomposition to difluorocarbene is particularly favored due to the formation of highly stable fluoride salts, such as MF.

(a) Decomposition of CF3 anion to difluorocarbene (:CF2) and fluoride (F−). (b) Stabilization of CF3 anion by DMF. (c) Direct trifluoromethylation of carbonyl compounds by using sterically demanding P4-tBu base. (d) Encapsulation of K+ with glymes for trifluoromethylation (this work).
Several strategies have emerged to use HCF3 for trifluoromethylation via deprotonation with strong organic or inorganic bases18,19,20,21,22,23,24. In 1991, Shono and co-workers for the first time reported the trifluoromethylation of carbonyl compounds with fluoroform by electrogenerated bases as well as common bases such as NaH and tBuOK in DMF19. Subsequently, Barhdadi, Troupel and Perichon reported the trifluoromethylation of aldehydes with fluoroform by a strong base generated via cathodic reduction of iodobenzene20. Then Nomant and Roques demonstrated use of MeSOCH2K21 and KHMDS23 in DMF for trifluoromethylation of carbonyl compounds. It should be pointed out that, in all of these original developments, N,N-dimethylformamide (DMF, Me2NCHO) was used as the solvent. The crucial role of DMF was to stabilize the CF3− generated on deprotonation of HCF3 in the form of the hemiaminaloate [Me2NCH(O)CF3]−, which served as a CF3− “reservoir” in the reaction (Fig. 1b). In 2011, Grushin and co-workers reported the direct cupration of fluoroform with the dialkoxycuprate produced from CuCl and tBuOK in a 1:2 ratio to prepare CuCF325,26,27, which since then has been successfully applied to a wide variety of trifluoromethylations28,29,30,31,32,33,34,35. The cupration of fluoroform is governed by a concerted ambiphilic metal-ligand activation (AMLA) mechanism rather than simple deprotonation to give CF3− and/or difluorocarbene intermediates27. The important dual effect of the alkali-metal counterion, which would slowly decompose CuCF3 via α-fluoride elimination but also provides electrophilic assistance for the CF3H cupration, was demonstrated by adding stoichiometric amounts of 18-crown-6 or [2.2.2]crypt and (crypt-222) before and after the cupration, in order to diminish the electrophilicity of alkali-metal cation25,27. While the CuCF3 is stable, its direct synthesis from HCF3 requires an amide solvent, such as DMF, DMA, and NMP.
The first DMF-free trifluoromethylation with HCF3 was reported by Langlois and co-workers in 200024. Although a catalytic amount of DMF was still needed for trifluoromethylation of carbonyl compounds with HCF3/N(TMS)3/[Bu4N]+ [Ph3SiF2]− or Me4NF/DMF, the trifluoromethylation of dioctyl disulfide was successfully carried out in pure THF (66% yield). In 2012, Prakash et al. also reported nucleophilic trifluoromethylations of Si, B, S, and C centers by HCF3 using potassium hexamethyldisilazide (KHMDS) in the absence of DMF36. The formation of a KCF3 intermediate followed by CF3 transfer to the organic substrate was proposed in a DFT study37. Simultaneously, we reported that a sterically demanding Schwesinger base, phosphazene P4-tBu, is effective for pushing inert HCF3 to nucleophilic trifluoromethylation of carbonyl compounds, disulfides, and arylsulfonyl fluorides in the absence of DMF and any metals38,39. Being metal-free, our HCF3/P4-tBu system efficiently suppresses the decomposition of CF3− to difluorocarbene and fluoride, as explained above (Fig. 1c). Very recently, Szymczak and co-workers reported a new type of Lewis acid-CF3 adducts formed from an alkali metal hydride, HCF3 and boron-based Lewis acids40,41. Although these are important developments, simple, cost-efficient, and environmentally benign methods are needed to perform trifluoromethylation reactions with HCF3 on a large scale. We now report a simple protocol for one-step trifluoromethylation of carbonyl compounds with HCF3 in the presence of tBuOK or KHMDS. While being fundamentally similar to the previously reported methods based on deprotonation of HCF324,36, our new protocol features a dramatic improvement from performing the reaction in the presence of a suitable amount of polyethers such as glymes (Fig. 1d). A wide variety of ketones, chalcones and aldehydes are nicely converted to the trifluoromethylated carbinols by HCF3 under the optimized glyme conditions. Cyclic polyethers such as 18-crown-6 and crypt-222 are even more effective. The encapsulation of the K+ by acyclic or cyclic polyethers is the key for this transformation, which makes the reactive anionoid CF3 species more “naked”.
Results
Towards an economical and practical method, we intended to use glymes for tuning the Lewis acidity, hardness and steric bulk of the potassium-based counter-cation to CF3− 42. Glymes, saturated non-cyclic polyethers, are usually less volatile, miscible with water, and less toxic than many other organic solvents43. We initiated our investigation with the reaction of benzophenone (1a) and HCF3 (excess) with tBuOK (2.0 equiv) in THF or 1,2-dimethoxyethane (DME or monoglyme, 0.4 M) at room temperature (rt) for 6 h (runs 1 and 2, Table 1 and Fig. 2). While a desired 2,2,2-trifluoro-1,1-diphenylethan-1-ol (2a) was obtained in 52% yield in THF (run 1), a much higher yield of 88% was observed in monoglyme (run 2). This yield (88%) in monoglyme is noticeably higher than in the reported DMF-free reaction employing much more costly KHMDS (71%)36 and comparable with our tBu-P4 method (92%)38. Encouraged by the initial result, we explored the possibility of using diglyme, triglyme and tetraglyme in this reaction. The yields of 2a appeared to increase with the size of the glyme (runs 3–5). In triglyme and tetraglyme, the desired product was produced quantitatively (>99%, runs 4 and 5). By using 1.0 equiv of tBuOK in triglyme or tetraglyme, the yields were lower, 64% and 74%, respectively (runs 6 and 7). HMDS bases were also examined in triglyme, and only potassium base was effective (runs 8–10). In order to gain more insight into the importance of coordination of triglyme to the K+, control experiments were conducted (runs 11–16; see Table S1 for more details). In the absence of triglyme, a 32% yield of 2a was obtained in toluene with tBuOK. Interestingly, a steady increase in the yield (from 54% to >99%) was observed as the amount of triglyme in the system was increased from 1.0 to 4.0 equiv. These results suggested 2:1 coordination of triglyme to K+, furnishing the complex cation [K(triglyme)2]+. While the use of triglyme (4.0 equiv) in toluene was clearly a good choice for the conditions (run 15), for simplicity we selected monoglyme and triglyme as solvents rather than additives (runs 2 and 4). The 2:1 coordination was also confirmed for tetraglyme/K+, [K(tetraglyme)2]+, in a series of similar experiments. The comparison of the amount of HCF3 was finally examined (runs 4, 17–20). In principle, one equiv of HCF3 was enough for nearly quantitative transformation (runs 4 vs 17), and the slightly lower yield (99% vs 90%) was probably due to the technical issues. Thus, we concluded that one equiv of HCF3 is suitable for this transformation. More details of the optimization of the reaction conditions are shown in Table S1.

Optimization of reaction conditions of 1a to 2a by HCF3.
The substrate generality of this process in monoglyme or triglyme was next investigated using a variety of ketones, chalcones and aldehydes (Fig. 3). While one equiv of HCF3 is enough for the almost quantitative transformation (run 17, Table 1), we carried out the reaction mainly by using HCF3 in excess for simplicity. A series of diaryl ketones 1a–h with a variety of substituents on the aromatic rings, such as methyl, methoxyl, chloro, bromo and trifluoromethyl groups, were smoothly converted to corresponding trifluoromethyl carbinols 2a–h in good to excellent yield (72–93%) in monoglyme (0.4 M) and in nearly quantitative yield (up to 99%) in triglyme (0.4 M) at rt. For cyclic diaryl ketones 1o and 1p, a noticeable increase in the yield was observed in triglyme. Slightly better yields were also detected for bulky aliphatic-substituted ketones 1q and 1r. As for the nitro-substituted ketone 1i and heteroaryl substrates 1j–n, the transformation was less efficient, possibly due to coordination with potassium to the NO2 group and to the heteroatoms of the substrate. After further brief screenings of the reaction conditions (see Tables S2 and S3), the desired trifluoromethylated products 2i–n were obtained in high yields (77–91%) under modified reaction conditions employing KHMDS (2.0 equiv) as the base at −40 °C for 12 hours. Subsequently, several chalcones 1s–w with electron-donating and electron-withdrawing substituents on the aryl ring were also converted to the corresponding products 2s–w in 54–88% yields under such conditions. Aromatic aldehydes were found to be compatible with the reaction conditions using triglyme and tBuOK to produce the corresponding products 2x–2ee in 44–80% yields. The diminished yield in some cases might be due to side processes, such as the Cannizaro reaction. Unfortunately, only 6% of product 2ff was obtained in the reaction of 1ff bearing an enolizable α-proton. To demonstrate the scalability of the method, trifluoromethyl carbinol 2a was synthesized from benzophenone 1a (1.822 g, 10.0 mmol) in 93% isolated yield under the standard triglyme reaction conditions.

Substrate scope of trifluoromethylation of ketones, chalcones and aldehydes by HCF3 in the presence of tBuOK or KHMDS in monoglyme or triglyme. *The reaction of 1 (0.2 mmol) with HCF3 (excess) was carried out in the presence of tBuOK (2.0 equiv) in monoglyme (0.4 M) at rt; †The reaction of 1 (0.2 mmol) with HCF3 (excess) was carried out in the presence of tBuOK (2.0 equiv) in triglyme (0.4 M) at rt; ‡The reaction of 1 (0.2 mmol) with HCF3 (1.0 equiv) was carried out in the presence of tBuOK (2.0 equiv) in triglyme (0.4 M) at rt; §The reaction of 1 (0.2 mmol) with HCF3 (excess) was carried out in the presence of KHMDS (2.0 equiv) in triglyme (0.2 M) at −40 °C; ||The reaction of 1 (0.2 mmol) with HCF3 (excess) was carried out in the presence of tBuOK (2.0 equiv) in triglyme (0.2 M) at −40 °C; ¶19F NMR yield.
The trifluoromethylation of enolizable ketones such as 1ff could be improved by reducing the Lewis acidity of the counter cation K+ with more powerful ligands. Tetraglyme and cyclic ethers were further considered. After additional optimization of the reaction conditions (Table S4), 1ff was converted to the desired trifluoromethylated product 2ff in moderate to good yields, up to 96% depending on ligand used (triglyme, tetraglyme, 18-crown-6, crypt-222; Fig. 4). The yield of 2ff clearly increased with stronger ligation of the K+ prompting a weakening in its Lewis acidity in the order: [K(triglyme)2]+ > [K(tetraglyme)2]+ > [K(18-crown-6)]+ > [K(crypt-222)]+. Substrate generality of enolizable ketones 1 is shown in Fig. 4. These reactions were performed using 18-crown-6/tBuOK (3.0 equiv)/HCF3 in THF at rt. Using THF as the solvent is important (see Table S3) and is discussed below. The strategy of tuning the Lewis acidity of the potassium-based counter-cations enabled the effective trifluoromethylation of enolizable ketones with fluoroform, although the need to use stoichiometric amounts of rather costly 18-crown-6 may limit the applicability of the method on a larger scale.

Trifluoromethylation of enolizable ketones 1ff–kk by HCF3 with 18-crown-6. *19F NMR yield; †The reaction of 1ff with HCF3 (excess) was carried out in the presence of tBuOK (3.0 equiv) in glymes (0.1 M, triglyme or tetraglyme) at rt; ‡Crypt-222 was used.
Discussion
The reactive anionoid CF3 species in the mismatched Lewis acid-base adducts [K(polyethers)n][CF3] with diminished Lewis acidity of the K+ is rather stable, which is good agreement with the experimental observation in our previous report38. Namely, the sterically demanding and poorly electrophilic protonated tBuP4 base, [HtBuP4]+, improves the reactivity and stability of the CF3− for nucleophilic trifluoromethylation. This observation is in good agreement with the report by Prakash and co-workers that the anionoid CF3 species derived from iPr3SiCF3 in the presence of [K(18-crown-6)]+ is stable enough to be observed by NMR at −78 °C44. In spite of the apparent high degree of iconicity, the bonding between the coordinatively unsaturated and Lewis acidic K+ in [K(18-crown-6)]+ and the CF3 moiety certainly has a covalent component. Grushin and co-workers have reported the existence of the free or naked (uncoordinated) CF3− anion with the [K(crypt-222)]+ counter-cation, in which the K+ is caged inside the 3-dimensional host45. This ionic complex has been characterized by a combination of methods, including X-ray diffraction, solution NMR, and reactivity toward electrophiles data, as well as labeling, acid-base, and DFT studies45,46,47.
As [K(polyether)n]CF3 intermediates are expected to be much more stable than KCF3 (see above), a reaction mechanism in glymes (triglyme or tetraglyme) is proposed as shown in Fig. 5a. First, two molecules of glymes coordinate to tBuOK to form, reversibly, a 2:1 complex of [K(glyme)2][tBuO], followed by deprotonation of HCF3 with the tBuO− to furnish [K(glyme)2][CF3]. In this complex, the K+ is ligated by the glyme molecules, which reduces its Lewis acidity and, consequently, ability to decompose the CF3−. Similarly, if 18-crown-6 is used in place of the glyme, [K(18-crown-6)(tBuO)] is first formed, which deprotonates HCF3 to give [K(18-crown-6)(CF3)].

(a) Reaction mechanism for trifluoromethylation of 1 with HCF3/tBuOK in triglyme and tetraglyme. The optimized structures of (b) [η1-K(triglyme)2][CF3], (c) [η1-K(triglyme)2][CF3] disordered isomer, (d) [η1-K(tetraglyme)2][CF3], (e) [η2-K(tetraglyme)2][CF3] disordered isomer, (f) [K(18-crown-6)][CF3], (g) [K(18-crown-6)/THF][CF3] and [K][CF3] complexes obtained by B3LYP/6-311G** level DFT calculations.
The structures of [K(triglyme)2][CF3] and [K(tetraglyme)2][CF3] were studied by DFT calculations48,49 using reported X-ray structural data for [K(triglyme)2]+50 and [K(tetraglyme)2]+ 51. The four selected minima identified (Fig. 5; see also the Supplementary Information) display coordination of the CF3 to the glyme-ligated K+ through the C or F atoms. This is also the case with the computed structures of KCF3 and [K(18-crown-6)(CF3)], in which K-F contacts were found. A deviation from the tetrahedral geometry is observed in all of the computed structures, featuring longer C-F bonds and distorted F-C-F angles. Without glyme ligands, optimized KCF3 displayed coordination via two of the three F atoms and an overall tighter bonding, as follows from the bond distances presented in Fig. 5. Naturally, the less Lewis acidic K+ interacts with CF3− more weakly, which not only inhibits the undesired formation of KF and CF2, but also enhances the nucleophilicity of the anionoid CF3 species toward the organic substrate.
With regard to the beneficial effect of THF in the trifluoromethylation of enolizable ketones with HCF3 in the presence of 18-crown-6 (Fig. 4), we optimized the structure of [K(18-crown-6)(CF3)] in THF, using the X-data for [K(18-crown-6)(tBuO)]48,49,52,53. This structure [η2-K(18-crown-6)(CF3)] (Fig. 5f) also showed the coordination of the CF3 to the K center via two of the three fluorine atoms. Also, using the X-ray data for [K(18-crown-6)(THF)]+ 54, the structure of [K(18-crown-6)(THF)(CF3)] was computed (Fig. 5g)48,49,53. The binding energies (Ebind) for [K(18-crown-6)(tBuO)] and [K(18-crown-6)(CF3)] in THF were computed at −26.7 and −24.1 kcal/mol, respectively49,53. For [K(18-crown-6)(THF)(tBuO)] and [K(18-crown-6)(THF)(CF3)], also in THF, the K-O and K-F interactions were weaker, according to the computed Ebind values of −19.3 and −20.1 kcal/mol, respectively49,53. We therefore conclude that THF is also capable of serving as a ligand to the potassium in the reaction, thereby additionally diminishing the Lewis acidity of the cation and consequently enhancing both the reactivity of the anionoid CF3 intermediate and its stability toward fluoride elimination
In summary, we have developed an advantageous, simple and high-yielding method to trifluoromethylation of ketones, chalcones and aldehydes to the corresponding trifluoromethyl carbinols with HCF3 and a potassium base in the presence of glymes and/or 18-crown-6. The beneficial event of the polyethers deals with their coordination to the K+, rendering it less prone to fluoride abstraction from the reactive anionoid CF3 intermediate. Our data provide complementary evidence for enhanced stability of CF3− toward fluoride elimination and formation of difluorocarbene (:CF2), which is strongly induced by metal-fluorine interactions.
Methods
Trifluoromethylation of acyclic diaryl ketones 1a–1h, cyclic diaryl ketones 1o, 1p and bulky aliphatic-substituted ketones 1q, 1r by using monoglyme or triglyme as solvent in Table 1b (see Supplementary Information, the general synthetic procedure A and B)
The solution of tBuOK (45 mg, 0.4 mmol) in dry monoglyme or triglyme (0.5 mL), was cooled in liquid nitrogen followed by adding carbonyl compounds (diaryl ketones 1a–1h, cyclic diaryl ketones 1o, 1p and bulky aliphatic-substituted ketones 1q, 1r, 0.2 mmol) under argon atmosphere. After being charged with HCF3 (1.0 equiv or excess) by cooling at the same temperature under vacuum, the resulting mixture was allowed to warm to room temperature. Then the reaction mixture was stirred at rt for 6 h monitored by TLC, quenched by addition of sat. NH4Cl aq., extracted with Et2O, dried over with Na2SO4 and then concentrated in vacuo. The residue was purified by column chromatography on silica gel (n-hexane/ethyl acetate) to give corresponding α-trifluoromethyl alcohols 2a–2h, 2o–2r in good to high yields.
Trifluoromethylation of nitro group substituted diaryl ketones 1i, heteroaryl groups substituted ketones 1j–1n and chalcones 1s–w, aryl aldehydes 1x–1z and 1aa-1ee by using triglyme as solvent in Table 1b (see Supplementary Information, the general synthetic procedure C and D)
The solution of tBuOK (45 mg, 0.4 mmol) or KHMDS (80 mg, 0.4 mmol) in dry triglyme (0.5 mL) was charged with fluoroform by cooling in liquid nitrogen under vacuum. After being warmed to −40 °C, a solution of carbonyl compounds (0.20 mol) in triglyme (0.5 mL) was added slowly (over 5 min) by syringe. Then the reaction mixture was stirred at the same temperature for 12 h, quenched by addition of sat. NH4Cl aq., extracted with Et2O, dried over with Na2SO4 and then concentrated in vacuo. The residue was purified by column chromatography on silica gel (n-hexane/ethyl acetate) to give corresponding α-trifluoromethyl alcohols 2i–n and 2s–2z and 2aa–2ee in good yields.
Trifluoromethylation of enolizable ketones 1ff-1kk in the presence of 18-crown-6 in Fig. 4 (see Supplementary Information, the general synthetic procedure E)
The solution of tBuOK (67 mg, 0.6 mmol), 18-crown-6 (159 mg, 0.6 mmol) in THF (2.0 mL) was cooled in liquid nitrogen followed by adding carbonyl compounds (enolizable ketones 1ff–1kk, 0.2 mmol) under argon atmosphere. Then the resulting mixture was charged with HCF3 by cooling at the same temperature under vacuum. Then the solution was allowed to warm to room temperature. After being stirred for 6–12 h monitoring by TLC upon the completion of the reaction, the resulting mixture was quenched with sat. NH4Cl aq. extracted with Et2O, dried over with Na2SO4 and then concentrated in vacuo. The residue was purified by column chromatography on silica gel (n-hexane/ethyl acetate) to give corresponding α-trifluoromethyl alcohols 2ff–2kk in good yields.
References
Alonso, C., Martínez de Marigorta, E., Rubiales, G. & Palacios, F. Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes. Chem. Rev. 115, 1847–1935 (2015).
Charpentier, J., Früh, N. & Togni, A. Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev. 115, 650–682 (2015).
Liu, X., Xu, C., Wang, M. & Liu, Q. Trifluoromethyltrimethylsilane: Nucleophilic Trifluoromethylation and Beyond. Chem. Rev. 115, 683–730 (2015).
Yang, X., Wu, T., Phipps, R. J. & Toste, F. D. Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 115, 826–870 (2015).
Ni, C. & Hu, J. The unique fluorine effects in organic reactions: recent facts and insights into fluoroalkylations. Chem. Soc. Rev. 45, 5441–5454 (2016).
Houghton, J. T. et al. (eds) Climate Change 1995: the science of climate change. (Cambridge University Press, Cambridge, 1996).
Oram, D. E., Sturges, W. T., Penkett, S. A., McCulloch, A. & Fraser, P. J. Growth of fluoroform (CHF3, HFC-23) in the background atmosphere. Geophys. Res. Lett. 25, 35–38 (1998).
McCulloch, A. & Lindley, A. A. Global emissions of HFC-23 estimated to year 2015. Atmos. Environ. 41, 1560–1566 (2007).
Han, W., Li, Y., Tang, H. & Liu, H. Treatment of the potent greenhouse gas, CHF3—An overview. J. Fluorine Chem. 140, 7–16 (2012).
Grushin, V. V. Fluoroform as a feedstock for high-value fluorochemicals: novel trends and recent developments. Chim. Oggi 32, 81–90 (2014).
AM0001. Incineration of HFC23 Waste Streams, UNFCCC, https://cdm.unfccc.int/methodologies/DB/0MKGF12PM6TSNFNJZUESTSKG581HN6/view.html (2006).
Feaver, W. B. & Rossin, J. A. The catalytic decomposition of CHF3 over ZrO2-SO4. Catal. Today 54, 13–22 (1999).
Jeon, J. Y., Xu, X.-F., Choi, M. H., Kim, H. Y. & Park, Y.-K. Hydrolytic decomposition of PFCs over AlPO4-Al2O3 catalyst. Chem. Commun., 1244–1245 (2003).
Mukhopadhyay, S., Bell, A. T., Srinivas, R. V. & Smith, G. S. Synthesis of Trifluoromethanesulfonic Acid from CHF3. Org. Process Res. Dev. 8, 660–662 (2004).
Nagasaki, N., Morikuni, Y., Kawada, K. & Arai, S. Study on a novel catalytic reaction and its mechanism for CF3I synthesis. Catal. Today 88, 121–126 (2004).
Zhao, J. et al. New catalysts for dichlorodifluoromethane hydrolysis: Mesostructured titanium and aluminum phosphates. J. Mol. Catal. A: Chem. 242, 218–223 (2005).
Onoda, H., Ohta, T., Tamaki, J. & Kojima, K. Decomposition of trifluoromethane over nickel pyrophosphate catalysts containing metal cation. Appl. Catal., A 288, 98–103 (2005).
Langlois, B. R. & Billard, T. Some Recent Results in Nucleophilic Trifluoromethylation and Introduction of Fluorinated Moieties. Synthesis, 0185–0194 (2003).
Shono, T., Ishifune, M., Okada, T. & Kashimura, S. Electroorganic chemistry. 130. A novel trifluoromethylation of aldehydes and ketones promoted by an electrogenerated base. J. Org. Chem. 56, 2–4 (1991).
Barhdadi, R., Troupel, M. & Perichon, J. Coupling of fluoroform with aldehydes using an electrogenerated base. Chem. Commun., 1251–1252 (1998).
Folléas, B., Marek, I., Normant, J.-F. & Jalmes, L. S. Fluoroform: an efficient precursor for the trifluoromethylation of aldehydes. Tetrahedron Lett. 39, 2973–2976 (1998).
Folléas, B. T., Marek, I., Normant, J.-F. & Saint-Jalmes, L. Fluoroform: an Efficient Precursor for the Trifluoromethylation of Aldehydes. Tetrahedron 56, 275–283 (2000).
Russell, J. & Roques, N. Effective nucleophilic trifluoromethylation with fluoroform and common base. Tetrahedron 54, 13771–13782 (1998).
Large, S., Roques, N. & Langlois, B. R. Nucleophilic Trifluoromethylation of Carbonyl Compounds and Disulfides with Trifluoromethane and Silicon-Containing Bases. J. Org. Chem. 65, 8848–8856 (2000).
Zanardi, A., Novikov, M. A., Martin, E., Benet-Buchholz, J. & Grushin, V. V. Direct Cupration of Fluoroform. J. Am. Chem. Soc. 133, 20901–20913 (2011).
Mazloomi, Z. et al. Continuous Process for Production of CuCF3 via Direct Cupration of Fluoroform. Org. Process Res. Dev. 18, 1020–1026 (2014).
Konovalov, A. I., Benet-Buchholz, J., Martin, E. & Grushin, V. V. The critical effect of the countercation in the direct cupration of fluoroform with [Cu(OR)2]−. Angew. Chem. Int. Ed. 52, 11637–11641 (2013).
Lishchynskyi, A. et al. Trifluoromethylation of Aryl and Heteroaryl Halides with Fluoroform-Derived CuCF3: Scope, Limitations, and Mechanistic Features. J. Org. Chem. 78, 11126–11146 (2013).
Novák, P., Lishchynskyi, A. & Grushin, V. V. Fluoroform-Derived CuCF3 for Low-Cost, Simple, Efficient, and Safe Trifluoromethylation of Aryl Boronic Acids in Air. Angew. Chem. Int. Ed. 51, 7767–7770 (2012).
Novák, P., Lishchynskyi, A. & Grushin, V. V. Trifluoromethylation of α-Haloketones. J. Am. Chem. Soc. 134, 16167–16170 (2012).
Potash, S. & Rozen, S. A new synthesis of trifluoromethyl sulfides utilizing thiocyanates and fluoroform. J. Fluorine Chem. 168, 173–176 (2014).
Potash, S. & Rozen, S. General Synthesis of Trifluoromethyl Selenides Utilizing Selenocyanates and Fluoroform. J. Org. Chem. 79, 11205–11208 (2014).
Yang, X., He, L. & Tsui, G. C. Hydroxytrifluoromethylation of Alkenes Using Fluoroform-Derived CuCF3. Org. Lett. 19, 2446–2449 (2017).
He, L., Yang, X. & Tsui, G. C. Domino Hydroboration/Trifluoromethylation of Alkynes Using Fluoroform-Derived [CuCF3]. J. Org. Chem. 82, 6192–6201 (2017).
Lishchynskyi, A., Berthon, G. & Grushin, V. V. Trifluoromethylation of arenediazonium salts with fluoroform-derived CuCF3 in aqueous media. Chem. Commun. 50, 10237–10240 (2014).
Prakash, G. K. S., Jog, P. V., Batamack, P. T. D. & Olah, G. A. Taming of Fluoroform: Direct Nucleophilic Trifluoromethylation of Si, B, S, and C Centers. Science 338, 1324–1327 (2012).
Luo, G., Luo, Y. & Qu, J. Direct nucleophilic trifluoromethylation using fluoroform: a theoretical mechanistic investigation and insight into the effect of alkali metal cations. New J. Chem. 37, 3274–3280 (2013).
Kawai, H., Yuan, Z., Tokunaga, E. & Shibata, N. A sterically demanding organo-superbase avoids decomposition of a naked trifluoromethyl carbanion directly generated from fluoroform. Org. Biomol. Chem. 11, 1446–1450 (2013).
Okusu, S., Hirano, K., Tokunaga, E. & Shibata, N. Organocatalyzed Trifluoromethylation of Ketones and Sulfonyl Fluorides by Fluoroform under a Superbase System. ChemistryOpen 4, 581–585 (2015).
Geri, J. B. & Szymczak, N. K. Recyclable Trifluoromethylation Reagents from Fluoroform. J. Am. Chem. Soc. 139, 9811–9814 (2017).
Geri, J. B., Wade Wolfe, M. M. & Szymczak, N. K. Borazine-CF3- Adducts for Rapid, Room Temperature, and Broad Scope Trifluoromethylation. Angew. Chem. Int. Ed. 57, 1381–1385 (2018).
Tsuzuki, S. et al. Effect of the cation on the stability of cation-glyme complexes and their interactions with the [TFSA]- anion. Phys. Chem. Chem. Phys. 19, 18262–18272 (2017).
Tang, S. & Zhao, H. Glymes as versatile solvents for chemical reactions and processes: from the laboratory to industry. RSC Adv. 4, 11251–11287 (2014).
Prakash, G. K. S. et al. Long-Lived Trifluoromethanide Anion: A Key Intermediate in Nucleophilic Trifluoromethylations. Angew. Chem. Int. Ed. 53, 11575–11578 (2014).
Lishchynskyi, A. et al. The Trifluoromethyl Anion. Angew. Chem. Int. Ed. 54, 15289–15293 (2015).
Miloserdov, F. M. et al. The Trifluoromethyl Anion: Evidence for [K(crypt-222)]+ CF3−. Helv. Chim. Acta 100, e 1700032 (2017).
Harlow, R. L. et al. On the Structure of [K(crypt-222)]+ CF3−. Helv. Chim. Acta 101, e 1800015 (2018).
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988). The geometries of complexes were optimized at the B3LYP/6-311G** level. The intermolecular interaction energies for complexes (Eint) were calculated at the B3LYP/6-311G** level by the supermolecule method. The basis set superposition error (BSSE) was corrected for all the interaction energy calculations using the counterpoise method. The stabilization energy for forming a complex from isolated species (Eform) was calculated as the sum of the Eint and the deformation energy (Edef), which is the sum of the increases in energy of molecules due to the deformation during formation of the complex. Here, the Edef was calculated at the B3LYP/6-311G** level. Details and related references are shown in supplementary.
Seidel, W. W., Summerscales, O. T., Patrick, B. O. & Fryzuk, M. D. Activation of white phosphorus by reduction in the presence of a zirconium diamidodiphosphine macrocycle: formation of a bridging square-planar cyclo-P4 unit. Angew. Chem. Int. Ed. 48, 115–117 (2009).
Mandai, T. et al. Effect of Ionic Size on Solvate Stability of Glyme-Based Solvate Ionic Liquids. J. Phys. Chem. B 119, 1523–1534 (2015).
Kleeberg, C. Synthesis and characterisation of [K(18-crown-6)(OtBu)]: An usuful reagent in nonpolar solvents. Z. Anorg. Allg. Chem. 637, 1790–1794 (2011).
Tomasi, J., Mennucci, B. & Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 105, 2999–3094. The geometry optimization were carried out in tetrahydrofuran solution using Polarizable Continuum Model (2005).
Hohloch, S. et al. Benzoquinonoid-bridged dinuclear actinide complexes. Dalton Trans. 46, 11615–11625 (2017).
Acknowledgements
This work was supported by the Asahi Glass Foundation, ACT-C from the JST (JPMJCR12Z7) and Tosoh Finechem Corporation. We thank Dr Toshihiko Mandai (Iwate University) to give us the cif file of [K(tetraglyme)2]+ reported in the reference 51 and Dr. Timothy F. Day (National Institute for Basic Biology) for editing the manuscript.
Author information
Authors and Affiliations
Contributions
T.S. and J.W. contributed equally. T.S., J.W., E.T. conducted and analysed the experiments and compounds. S.T. conducted the DFT calculations. N.S. designed, directed the project, and wrote the manuscript with contributions from T.S., J.W., E.T. and S.T. All authors contributed to discussions.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saito, T., Wang, J., Tokunaga, E. et al. Direct nucleophilic trifluoromethylation of carbonyl compounds by potent greenhouse gas, fluoroform: Improving the reactivity of anionoid trifluoromethyl species in glymes. Sci Rep 8, 11501 (2018). https://doi.org/10.1038/s41598-018-29748-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29748-1
This article is cited by
-
Ex-situ generation and synthetic utilization of bare trifluoromethyl anion in flow via rapid biphasic mixing
Nature Communications (2023)
-
Transition-metal-free silylboronate-mediated cross-couplings of organic fluorides with amines
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
