
Abstract
Cigarette smoking alters the oral microbiome; however, the effect of alternative tobacco products remains unclear. Middle Eastern tobacco products like dokha and shisha, are becoming globally widespread. We tested for the first time in a Middle Eastern population the hypothesis that different tobacco products impact the oral microbiome. The oral microbiome of 330 subjects from the United Arab Emirates Healthy Future Study was assessed by amplifying the bacterial 16S rRNA gene from mouthwash samples. Tobacco consumption was assessed using a structured questionnaire and further validated by urine cotinine levels. Oral microbiome overall structure and specific taxon abundances were compared, using PERMANOVA and DESeq analyses respectively. Our results show that overall microbial composition differs between smokers and nonsmokers (p = 0.0001). Use of cigarettes (p = 0.001) and dokha (p = 0.042) were associated with overall microbiome structure, while shisha use was not (p = 0.62). The abundance of multiple genera were significantly altered (enriched/depleted) in cigarette smokers; however, only Actinobacillus, Porphyromonas, Lautropia and Bifidobacterium abundances were significantly changed in dokha users whereas no genera were significantly altered in shisha smokers. For the first time, we show that smoking dokha is associated to oral microbiome dysbiosis, suggesting that it could have similar effects as smoking cigarettes on oral health.
Similar content being viewed by others

Introduction
The human oral microbiome (OM) is the second most diverse and densely populated microbiome of the human body1. It plays key roles in human digestion, protection against pathogen colonization and nitrate reduction2 and may have a role in human health including cardiovascular disease and cancer3,4. We recently reported that cigarette smoking5 and alcohol6 are important determinants of the oral microbiome. In particular, cigarette smoking is a cause of oral dysbiosis, affecting microbial diversity and its functional potential5,7.
Despite a global decline in tobacco consumption, tobacco use is still rising in African and Eastern Mediterranean countries, which is a significant public health concern8. Although cigarettes account for much of this increase, part of the increase is related to the popularization of alternative tobacco products common in Middle Eastern countries, such as dokha9 and shisha10. Dokha is a blend of tobacco leaves, barks, herbs, dried fruits and/or flowers and spices, which is smoked using a specialized pipe (midwakh) and is known to contain a much higher nicotine content than cigarettes11. Shisha is a fruit flavored tobacco comprised of shredded tobacco leaves, glycerol and other additives, which is smoked using a waterpipe12. Alternative tobacco products such as dokha and shisha, are, like cigarettes, a source of nicotine and other toxic products; however, the effect of these different types of tobacco on the oral microbiome remains unclear.
We tested for the first time the hypothesis that the oral microbiome is differentially impacted by specific tobacco products commonly used in Middle Eastern countries. We compared the effects of cigarette, dokha and shisha use on community composition of the oral microbiome by high-throughput sequencing of the bacterial 16S Ribosomal RNA (16S rRNA) gene in 330 participants from the “UAE Healthy Future” (UAEHF) pilot study13.
Results
We studied 330 subjects, including 105 (31.8%) smokers and 225 nonsmokers (68.2%) (Fig 1, Table 1). Smokers were more likely to be men (96.2%), but other health related factors such as age, BMI and diabetic status (ascertained by HbA1c levels in blood), were not different between smokers and nonsmokers (p = 0.81, p = 0.11 and p = 0.13, respectively) with the exception of systolic blood pressure (p = 0.05). Among the 105 smokers, 39% smoked more than one tobacco product, with cigarettes most commonly used (67.6%), followed by dokha (42%) and shisha (34.3%). Participants that exclusively used cigarettes smoked an average of 9.2 cigarettes per day and exclusive dokha users smoked an average of 10.7 midwakh pipes per day. On the other hand, only 50% of the participants that used shisha exclusively smoked it on a daily basis, while 13.3% smoked it on a weekly (2–3 times per week) and 36.6% smoked it on a monthly basis.

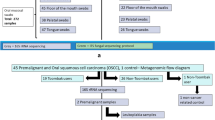
Flow chart depicting the classification of participants from the UAEHF pilot study. UAEHF pilot study participants were Emirati nationals aged 18 and above. Study participants completed a self-administered questionnaire including information on smoking habits. During the physical exam, participants provided blood, urine and mouthwash samples. From 517 consented study participants, 363 subjects completed the smoking section of the baseline questioners and provided mouthwash samples. A Cotinine test in urine was used to ascertain smoke exposure. These results were further used to validate the non-smoking self-reported data. We further excluded 33 subjects (11 had no cotinine data and 22 had self-reported as non-smokers but tested positive for cotinine). All individuals participating in the study read and signed an informed consent.
After data filtering, there were 16,132,922 high quality 16S rRNA sequence reads ready for analysis for these study subjects (mean per subject: 48887.42; SD: 13,408.67). After low count filtering, the final data set was comprised of 13 phyla, 20 classes, 26 orders, 41 families, 57 genera, 26 species and 1,080 OTUs. We observed that amongst the 13 phyla, Firmicutes (50.0%), Bacteroidetes (21.7%), Proteobacteria (15.8%), Actinobacteria (6.7%) and Fusobacteria (4.7%) were the most abundant (Supplementary Table S1) and were present in all samples. Phyla such as Tenericutes, SR1 and Synergistetes, although in very low relative abundance, were present in more than 85% of the samples.
Overall oral microbiome community
We found that microbial diversity (Shannon entropy) was marginally greater in all smokers compared with nonsmokers (p = 0.04, Fig. 2A); however, this was not observed when comparing single tobacco type users to nonsmokers (Fig. 2B). Based on Unifrac distance matrices, controlling for age, sex and batch effects, we found that the oral microbiome overall structure significantly differed between smokers and nonsmokers (p = 0.001, Fig. 3A). This finding was confirmed in the comparison of cotinine positive and cotinine negative participants (p = 0.001, Supplementary Figure S1), which was independent of their self-reported status. When considering single tobacco products independently, the oral microbiome structure of exclusively cigarette smokers (p = 0.001, Fig. 3B) and exclusively dokha users (p = 0.042, Fig. 3C) were significantly different from that of nonsmokers. However, exclusive shisha smokers’ oral microbiome was not significantly different from that of nonsmokers (p = 0.62, Fig. 3D). In addition, no significant differences were observed when comparing the overall oral microbiome structure amongst those who exclusively used one type of tobacco (cigarette, dokha and shisha smokers, p = 0.2; Supplementary Figure S2).

Characterization of the α-diversity of the Emirati oral microbiome. Diversity comparisons between (A) smokers (n = 105) and nonsmokers (n = 225) and (B) between tobacco types, cigarettes (n = 33), dokha (n = 16) and shisha (n = 15) versus nonsmokers (n = 225). Diversity was significantly greater in smokers than nonsmokers, but not when comparing single tobacco type use to nonsmokers. Only significant p values from linear regression are shown.

Principal Coordinate Analysis (PCoA) of the bacterial communities according to smoking use and tobacco types derived from Unifrac weighted distances. Significant differences between (A) smokers (n = 105) and nonsmokers (n = 225) were observed (p = 0.001), (B) cigarette (n = 33, p = 0.001) and (C) dokha smokers (n = 16, p = 0.042). However, no significant differences were identified between (D) shisha smokers (n = 15, p = 0.620) and nonsmokers. All nonsmoker participants were colored orange and all smokers independently of tobacco use or type in blue.
Bacterial taxa abundance according to tobacco consumption and type
To determine the association of different tobacco products with oral bacterial taxa, we performed further detailed analyses.
Exclusively cigarette smokers (CS) vs. nonsmokers
Contrasts between CS (n = 33) and nonsmokers (n = 225) (Table 2, Supplementary Table S2, Fig. 4) showed that CS were depleted of the phylum Proteobacteria and in particular of its genera Neisseria, Eikenella, Aggregatibacter, Actinobacillus, Haemophilus and Lautropia, and the phylum Fusobacteria, represented at the genus level by Fusobacterium and Leptotrichia. Also, significantly depleted and not previously reported were the less abundant phyla SR1, GN02 and Cyanobacteria. In contrast, CS presented higher abundances at the phylum level of Spirochaetes, Synergistetes and Tenericutes being represented at the genus level by Treponema, TG5 and Mycoplasma, respectively. Furthermore, Firmicutes, Bacteroidetes and Actinobacteria were enriched at all lower taxonomical levels in CS, being characterized at the genus level among others by Megasphaera and Dialister (Firmicutes), Paludibacter, Porphyromonas and Prevotella (Bacteroidetes), and Atopobium (Actinobacteria).

Log2 fold change of genera abundances in the oral microbiome relative to tobacco use. Heatmap of the genera that were in significantly different relative abundances when comparing nonsmokers (n = 225) to cigarette (n = 33), dokha (n = 16), shisha (n = 15) and multiple (n = 41) tobacco type smokers independently. All genera with q < 0.1 (indicated by stars) in at least one of the comparisons are shown. Heatmap displays log2 fold change when compared to nonsmokers.
Exclusively dokha smokers (DS) vs. nonsmokers
Consistent with the patterns observed in CS, taxa dynamics in DS (n = 16) differed significantly from nonsmokers with the depletion of the phylum Cyanobacteria observed in CS, and in the genera Actinobacillus, Lautropia (Proteobacteria), and Porphyromonas (Bacteroidetes), which were also depleted in CS (Table 2, Supplementary Table S2, Fig. 4). In contrast, DS were exclusively enriched in the genus Bifidobacterium (Actinobacteria).
Exclusively shisha smokers (SS) vs. nonsmokers
Consistent with overall microbial composition comparisons, only four taxa were identified as having a significantly different relative abundance between SS (n = 15) and nonsmokers (Table 2, Supplementary Table S2, Fig. 4). The phyla Cyanobacteria and SR1 and the classes Chloroplast (Cyanobacteria) and BD1-5 (GN02) were all significantly depleted in SS when compared to nonsmokers.
Multiple tobacco type smokers (MS) vs. nonsmokers
We also performed the contrast between MS (n = 41) against nonsmokers (Table 2, Supplementary Table S2, Fig. 4). Depletion and enrichment patterns of taxa relative abundances were for the most part mirroring those observed in the contrast between CS and nonsmokers with some exceptions, in particular, the significant enrichment in MS of the genera Campylobacter and Enhydrobacter (Proteobacteria), and the depletion of the genus Vagococcus (Firmicutes); the two latter genera were not observed in other contrasts.
Contrasts between tobacco types
Comparisons between CS and DS revealed no significant differences at any taxonomical level in the taxa relative abundances of their oral microbiome (Supplementary Table S3). Comparisons between CS and SS however showed similar results to those observed in the contrast between CS and nonsmokers, with patterns for SS similar to that of nonsmokers. Only the depletion of the genus Actinobacillus was consistently observed in cigarette vs. shisha users and dokha vs. shisha smokers.
Discussion
This study encompasses the first characterization of the oral microbiome of the Emirati population and describes, for the first time, the specific effects on oral bacterial community structure of two regional products, dokha and shisha, with the latter experiencing increased worldwide usage in recent years14. We found that the Emirati population exhibited a diverse oral microbiome and that overall microbial diversity and composition were associated with use of tobacco products (Fig. 4). In particular, smoking in general, exclusive use of cigarettes and exclusive use of dokha were associated with significant alterations of oral microbiome structure and relative taxa abundances. Exclusive use of shisha was not associated with alterations in overall microbiome structure; however, depletion was noted in phyla Cyanobacteria and SR1, and classes Chloroplast and BD1-5.
The oral microbiome of the Emirati population presented a composition similar to that of other populations in the United States5,15, Japan and Korea16,17, China18, and among Amazonian Amerindians19,20, characterized by community dominance of phyla Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria and Fusobacteria and genera Streptococcus, Prevotella, Haemophilus, Veillonella and Neisseria. It would appear that the oral microbiome tends to have a generally similar community structure globally, despite there being wide differences in lifestyle and oral hygiene practices between populations. Of course, at a further detailed level of analysis between populations, there may be traits that tend to be more population-specific. For example, SR1 was reported as part of the core microbiome only in a Saudi Arabian population21. In the Emirati population SR1 was observed in 87% of the samples. This high carriage could be a characteristic of Middle Eastern populations, requiring further exploration. Further detailed analyses involving for example transcriptomics, metabolomics or proteomics could potentially reveal further population-specific biomarkers.
We found that tobacco use in general, was marginally associated (p = 0.04), with greater diversity of the oral microbiome. Similar associations have been reported in other studies16,22,23. However, this is not consistently observed with some reporting no change in diversity24,25,26. Exposure to tobacco smoke results in functional and structural changes in saliva and the oral environment27,28,29, which may impact on reduced immune fitness and decreased ability of the autochthonous bacteria to compete with transient taxa for nutrients; this may result in the increased diversity of the OM7,30 observed in smokers. Perhaps of clinical importance, periodontal disease and gingivitis, which are known to be associated with tobacco use, are also characterized by a higher diversity of the OM7,22,31,32,33, potentially indicating an early pathway to oral disease in smokers.
Consistent with previous studies, we found that smokers were significantly depleted of Proteobacteria such as Neisseria, Haemophilus and Lautropia)5,23,30, and enriched of Bifidobacterium5 and TG534. In addition, we reported for the first time the depletion in all smokers of the less abundant phylum SR1 (Supplementary Table S4). Members of the SR1 phylum are predominantly uncultivated, non-respiring oral bacteria that possess an altered genetic code, where the usual UGA stop codon is reassigned to a glycine)35,36,37. This change in the genetic code of SR1 may limit synergistic relationships within the oral bacterial community36, which is potentially related to the depleted relative abundances we observed.
Exclusive cigarette use was also associated with differentials in specific oral taxa, including a wider range of taxa than that found for all smokers combined. CS were depleted when compared to nonsmokers in the genera Aggregatibacter (Proteobacteria), Capnocytophaga and Porphyromonas (Bacteroidetes) and in particular of the phylum Fusobacteria represented by significantly depleted Fusobacterium and Leptotrichia. We previously reported5 similar differentials and related the bacterial genes involved to xenobiotic metabolism of toluene, in agreement with Peralbo-Molina, et al.38 who observed that exhaled breath condensate of cigarette smokers contained lower levels of p-cresol, a toluene metabolite. We also found that CS were significantly enriched in Atopobium and Bifidobacterium (Actinobacteria), TG5 (Synergistetes), Treponema (Synergistetes), Campylobacter and Eikenella (Proteobacteria) and Megasphaera (Firmicutes) consistent overall with previous results5,30,34,39,40.
We report for the first time on the impact of dokha and shisha on the oral microbiome. Dokha was associated with similar patterns of OM dysbiosis as found for cigarette use, although significant associations were found for fewer taxa, among which, the depletion of the phylum Cyanobacteria, the genera Actinobacillus, Lautropia (Proteobacteria) and Porphyromonas (Bacteroidetes) and the enrichment of the genus Bifidobacterium (Actinobacteria). The lower number of significant associations of taxa differentials with the use of dokha is probably due to the low number of participants (n = 16) that exclusively smoked dokha. Dokha is commonly consumed in the UAE using the traditional midwakh pipe. Midwakh users in the UAE consume dokha on average 12 times per day, being equivalent to smoking 6 grams of dokha per day9, which is reflected on its effects on the oral microbiome. Dokha toxicants and health effects have received limited study, although notably Shaikh et al. reported increased systolic blood pressure, heart rate and respiratory rate in users41, indicating that these exposures constitute a potentially significant, yet understudied threat to health in the Middle Eastern region.
Although we found that shisha use was not related to overall oral microbiome structural changes, taxa relative abundance analysis identified the phyla Cyanobacteria and SR1, and the classes Chloroplast and BD1-5 as significantly depleted in shisha smokers when compared to nonsmokers, The lack of significance observed for the majority of the taxa is likely due to both the low n number available (n = 15) for exclusive shisha users as well as to the infrequency of shisha use, rather than the absence of toxicants in this product12,42. Shisha is usually associated with social gatherings, and consumption is often on a weekly to monthly basis43. As the OM is resilient44 and smoking related changes may not be permanent if cessation occurs5, the frequency with which participants smoke shisha could partially explain the patterns observed. Shisha smoking has been associated with esophageal squamous cell carcinoma45, low birth weight of infants from smoking mothers46 and cardiovascular effects47, and hence warrants for further study.
Users of multiple tobacco types in our study tended to show similar depletion/enrichment patterns of taxa relative abundances as cigarette smokers, largely because cigarette use was the most common tobacco use type in this group. Potentially of note, increased abundance of Enhydrobacter was related to joint use of cigarettes and dokha. This bacterium grows in the presence of ammonia48 which is believed to be common in both these products. In the case of cigarettes, ammonia is added to facilitate freeing of nicotine molecules by raising pH49.
This investigation is the largest study of the oral microbiome of an Arabic population. In contrast to most studies that rely exclusively on self-reported questionnaire, we validated nonsmoking status by urinary cotinine measurement. Although associations were identified for the tobacco types commonly used in this region, larger studies, which would provide stronger statistical power, with more detailed information on tobacco use patterns and frequency will be needed to further delineate differentials in tobacco products and the oral microbiome. We are currently recruiting participants to the UAEHFS to address this and other health-related issues for the UAE population. Although amplicon pyrosequencing has major advantages for human microbiome studies, it has also some limitations, such as the possible overestimation of OTU richness due to homopolymer errors (repeated nucleotides), inaccuracies of taxonomic identification due to the short length of the sequences and the introduction of primer and sequencing related biases50,51,52. While our research using the 16 S rRNA sequencing approach was appropriate for identifying taxonomies, future studies should investigate functional capacity of the microbiome, using full shotgun metagenomics sequencing and other methodologies.
In summary, we characterized the oral microbiome in the Emirati population and found that tobacco use had an important impact on the oral microbiome, particularly with regard to cigarette and dokha use. The abundance of multiple taxa and in particular that of 15 genera was significantly altered (enriched or depleted) in cigarette smokers; however, at the genus level, only the abundance of Actinobacillus, Lautropia, Porphyromonas and Bifidobacterium were significantly altered in users of dokha, and none were observed in shisha smokers. Our results suggest that cigarettes and other local tobacco products alter the oral microbiome structure and specific taxa abundance in the Emirati population.
Methods
Study Population
UAEHF pilot study participants were recruited in a 5-month period between December 2014 and April 2015 at Zayed Military Primary Health Clinic (ZMH PHCC) and Abu Dhabi Blood Bank (ADBB), both of which are licensed for clinical research by the Health Authority of Abu Dhabi. Eligible Emirati nationals (aged 18 and above) completed a self-administered questionnaire including information on socio-demographic factors, lifestyle and medical history. Study participants completed physical and clinical exams, including measurements of anthropometry, body composition, and blood pressure13. During the physical exam, participants also provided blood, urine and mouthwash samples. From 517 consented study participants, 363 subjects completed baseline questionnaires and provided mouthwash samples. We further excluded 33 subjects who had inconsistent smoking data (see smoking definition below). Therefore, our analytic dataset was comprised of 330 subjects (Fig. 1). This study was approved by the Institutional Review Boards (IRB) of Sheikh Khalifa Medical City (SKMC), Zayed Military Hospital (ZMH), New York University Abu Dhabi (NYUAD) and NYU Langone Medical Center, New York. All individuals participating in the study read and signed an informed consent. All experiments were performed in accordance with relevant guidelines and regulations.
Measurements
Definition of smoking
Detailed information on cigarette smoking, including smoking status, tobacco type used and smoking history, was ascertained by questionnaire. We also measured cotinine in urine by COT rapid test cassette (International Biomedical Supplies), with a cut off concentration of 200 ng/ml for tobacco smoke exposure. We further excluded from analysis 22 subjects with positive cotinine test who had self-reported as nonsmokers and 11 subjects with missing cotinine data. Tobacco type groups are defined as follows: smokers (all participants that self-reported as smoker independently of cotinine results), exclusively cigarette smokers (those that only smoke cigarettes), exclusively dokha smokers (those that only smoke dokha, typically in the traditional midwakh pipe), exclusively shisha smokers (those that only smoke shisha), multiple smokers (those that smoke more than one type of tobacco product), nonsmokers (those that self-reported as nonsmokers, and were further validated by a cotinine negative result).
Mouthwash sample collection
Participants were given a 10 ml sample of pharmaceutical grade normal saline (0.9%) solution and asked to vigorously swish for 30 seconds and spit it out onto a new sterile tube. Samples were stored initially at 4 °C for no more than 48 h. Samples were then vortexed for 20 seconds, pipetted up and down 10 times, aliquoted into 1 ml cryotubes and stored at −80 °C until further processing. To confirm that the saline solution used for collection of mouthwash samples contained no detectable levels of DNA, identical DNA extraction methods to those used in the study (see below), were applied to two separate saline solutions samples alongside two mouthwash samples. Neither of the two saline solution samples yielded any DNA. Extracted DNA was viewed by gel electrophoresis and concentrations were quantified using the high sensitivity Qubit assay. Only mouthwash samples yielded measurable amounts of DNA (Supplementary Table S5).
Microbiome assay
Two 1 ml aliquots per sample were pooled for DNA extraction. Thawed samples were centrifuged at 6000 g for 3 min and then at 10000 g for 10 min in order to collect the cell pellet. DNA was extracted using the Mo BioPowerSoil PowerLyzer kit following manufacturer’s instructions (Mo Bio Laboratory Inc, California, USA). Genomic DNA was visualized on a gel and quantified using the Qubit HS kit (Thermo Fisher Scientific). Amplification of DNA from the V4 region of 16S rDNA gene (515 F-5′GTGCCAGCMGCCGCGGTAA3′ - 806 R - 5′GGACTACHVGGGTWTCTAAT3′) was performed using specifically designed primers with Roche 454 FLK adaptor sequences and a 12 bp index (reverse primer only) added for posterior multiplexing. Amplification was carried out using the FastStart enzyme (Roche, IN). PCR products were visualized in an agarose gel, purified using Agencourt AMPure beads (Beckman Coulter Life Sciences, IN) and quantified using the Qubit BR kit (Thermo Fisher Scientific). Samples were then pooled for sequencing on an Illumina Miseq platform.
Quality control
Samples were sequenced in two batches. In addition to study samples, each batch contained three quality control samples, each in triplicate (shown in Supplementary Table S6) and a negative control (blank sample for DNA extraction and PCR amplification). Quality control samples showed good reliability, with the coefficient of variability ranging from 1.65–2.32% for the Shannon entropy and 1.02–7.21% for specific phyla relative abundances.
Statistical analysis
Sequence data processing and taxonomic assignment
Sequences were de-multiplexed and trimmed using the split_libraries_fastq.py QIIME script with default parameters53. Only sequences that passed quality control filters (average base score quality per read .20, reads longer than 200 bp), were further processed. Taxonomical assignment was achieved using the pick_de_novo_otus.py workflow as implemented in QIIME53. Sequences were clustered into operational taxonomical units (OTU) using a 97% pairwise-identity cutoff, executing the UCLUST algorithm54. PyNAST55 and the Greengenes database were used for taxonomical assignment, followed by removal of chimeric sequences using ChimeraSlayer as implemented in the QIIME workflow51. Low count OTUs were filtered from the analyses if they were singletons and absent in more than 10% of the participants.
Estimating α-diversity
β-diversity and taxa relative abundances. Oral microbiome richness and diversity were estimated from a rarefied dataset (16738 sequence reads per sample), in order to eliminate possible biases introduced by differences of sampling effort. Estimation of richness (observed and Chao) and diversity (Shannon entropy and Simpson diversity index) were calculated using the vegan library in R56 (Supplementary Figure S3). To compare α-diversity between cases and controls we modeled richness and Shannon entropy in linear regression, adjusting for age sex and batch effects. Because linear regression assumes a normal distribution of the outcome, Shannon entropy was previously log transformed. We conducted permutational multivariate analysis of variance (PERMANOVA) of weighted (taxa relative abundance) and unweighted (absence/presence) Unifrac distance matrices to compare overall oral microbial composition between tobacco users and nonusers and by tobacco type56. Matrices were calculated implementing the Unifrac function in the Phyloseq library in R57,58. We then generated PCoA plots to visualize sample ordination using the first two principal coordinates. All PERMANOVA analyses were adjusted for age, sex, and batch effects and were performed using the Adonis function in the vegan R library56. We used DESeq259 to explore for differential taxa abundances between smokers and nonsmokers for all tobacco categories as well as for the cotinine data. All statistical tests were two-sided. A p-value < 0.05 was considered of nominal statistical significance. In order to limit false detection of significance due to multiple comparisons, we adjusted for the False Discovery Rate (FDR)60. We determined a q-value < 0.10 as significant after adjustment. All analyses were conducted using R version 3.3.261.
Data availability
The datasets analyzed in this study are available in the Qiita database study ID - 11838.
References
Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214, https://doi.org/10.1038/nature11234 (2012).
Wade, W. G. The oral microbiome in health and disease. Pharmacol Res 69, 137–143, https://doi.org/10.1016/j.phrs.2012.11.006 (2013).
Koren, O. et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci USA 108(Suppl 1), 4592–4598, https://doi.org/10.1073/pnas.1011383107 (2011).
Ahn, J., Chen, C. Y. & Hayes, R. B. Oral microbiome and oral and gastrointestinal cancer risk. Cancer Causes Control 23, 399–404, https://doi.org/10.1007/s10552-011-9892-7 (2012).
Wu, J. et al. Cigarette smoking and the oral microbiome in a large study of American adults. ISME J 10, 2435–2446, https://doi.org/10.1038/ismej.2016.37 (2016).
Fan, X. et al. Drinking alcohol is associated with variation in the human oral microbiome in a large study of American adults. Microbiome 6, 59, https://doi.org/10.1186/s40168-018-0448-x (2018).
Camelo-Castillo, A. J. et al. Subgingival microbiota in health compared to periodontitis and the influence of smoking. Front Microbiol 6, 119, https://doi.org/10.3389/fmicb.2015.00119 (2015).
WHO. WHO report on the Global Tobacco Epidemic. (World Health Organization, 2015).
Al-Houqani, M., Ali, R. & Hajat, C. Tobacco smoking using Midwakh is an emerging health problem–evidence from a large cross-sectional survey in the United Arab Emirates. PLoS One 7, e39189, https://doi.org/10.1371/journal.pone.0039189 (2012).
Waziry, R., Jawad, M., Ballout, R. A., Al Akel, M. & Akl, E. A. The effects of waterpipe tobacco smoking on health outcomes: an updated systematic review and meta-analysis. Int J Epidemiol 46, 32–43, https://doi.org/10.1093/ije/dyw021 (2017).
Vupputuri, S. et al. Midwakh/dokha tobacco use in the Middle East: much to learn. Tob Control 25, 236–241, https://doi.org/10.1136/tobaccocontrol-2013-051530 (2016).
Shihadeh, A. et al. Toxicant content, physical properties and biological activity of waterpipe tobacco smoke and its tobacco-free alternatives. Tob Control 24(Suppl 1), i22–i30, https://doi.org/10.1136/tobaccocontrol-2014-051907 (2015).
Abdulle, A. et al. The UAE healthy future study: a pilot for a prospective cohort study of 20,000 United Arab Emirates nationals. BMC Public Health 18, 101, https://doi.org/10.1186/s12889-017-5012-2 (2018).
Maziak, W. et al. The global epidemiology of waterpipe smoking. Tob Control 24(Suppl 1), i3–i12, https://doi.org/10.1136/tobaccocontrol-2014-051903 (2015).
Bik, E. M. et al. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J 4, 962–974, https://doi.org/10.1038/ismej.2010.30 (2010).
Takeshita, T. et al. Bacterial diversity in saliva and oral health-related conditions: the Hisayama Study. Sci Rep 6, 22164, https://doi.org/10.1038/srep22164 (2016).
Takeshita, T. et al. Distinct composition of the oral indigenous microbiota in South Korean and Japanese adults. Sci Rep 4, 6990, https://doi.org/10.1038/srep06990 (2014).
Huang, S. et al. Preliminary characterization of the oral microbiota of Chinese adults with and without gingivitis. BMC Oral Health 11, 33, https://doi.org/10.1186/1472-6831-11-33 (2011).
Contreras, M. et al. The bacterial microbiota in the oral mucosa of rural Amerindians. Microbiology 156, 3282–3287, https://doi.org/10.1099/mic.0.043174-0 (2010).
Clemente, J. C. et al. The microbiome of uncontacted Amerindians. Sci Adv 1, https://doi.org/10.1126/sciadv.1500183 (2015).
Al-Hebshi, N. N., Abdulhaq, A., Albarrag, A., Basode, V. K. & Chen, T. Species-level core oral bacteriome identified by 16S rRNA pyrosequencing in a healthy young Arab population. J Oral Microbiol 8, 31444, https://doi.org/10.3402/jom.v8.31444 (2016).
Joshi, V. et al. Smoking decreases structural and functional resilience in the subgingival ecosystem. J Clin Periodontol 41, 1037–1047, https://doi.org/10.1111/jcpe.12300 (2014).
Kumar, P. S., Matthews, C. R., Joshi, V., de Jager, M. & Aspiras, M. Tobacco smoking affects bacterial acquisition and colonization in oral biofilms. Infect Immun 79, 4730–4738, https://doi.org/10.1128/IAI.05371-11 (2011).
Bostrom, L., Bergstrom, J., Dahlen, G. & Linder, L. E. Smoking and subgingival microflora in periodontal disease. J Clin Periodontol 28, 212–219 (2001).
Gomes, S. C. et al. The effect of a supragingival plaque-control regimen on the subgingival microbiota in smokers and never-smokers: evaluation by real-time polymerase chain reaction. J Periodontol 79, 2297–2304, https://doi.org/10.1902/jop.2008.070558 (2008).
Haffajee, A. D. & Socransky, S. S. Relationship of cigarette smoking to the subgingival microbiota. J Clin Periodontol 28, 377–388 (2001).
Nagler, R. et al. Effect of cigarette smoke on salivary proteins and enzyme activities. Arch Biochem Biophys 379, 229–236, https://doi.org/10.1006/abbi.2000.1877 (2000).
Zuabi, O. et al. The effect of smoking and periodontal treatment on salivary composition in patients with established periodontitis. J Periodontol 70, 1240–1246, https://doi.org/10.1902/jop.1999.70.10.1240 (1999).
Rad, M., Kakoie, S., Niliye Brojeni, F. & Pourdamghan, N. Effect of Long-term Smoking on Whole-mouth Salivary Flow Rate and Oral Health. J Dent Res Dent Clin Dent Prospects 4, 110–114, https://doi.org/10.5681/joddd.2010.028 (2010).
Mason, M. R. et al. The subgingival microbiome of clinically healthy current and never smokers. ISME J 9, 268–272, https://doi.org/10.1038/ismej.2014.114 (2015).
Griffen, A. L. et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J 6, 1176–1185, https://doi.org/10.1038/ismej.2011.191 (2012).
Abusleme, L. et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J 7, 1016–1025, https://doi.org/10.1038/ismej.2012.174 (2013).
Costalonga, M. & Herzberg, M. C. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol Lett 162, 22–38, https://doi.org/10.1016/j.imlet.2014.08.017 (2014).
Paropkari, A. D., Leblebicioglu, B., Christian, L. M. & Kumar, P. S. Smoking, pregnancy and the subgingival microbiome. Sci Rep 6, 30388, https://doi.org/10.1038/srep30388 (2016).
Rinke, C. et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437, https://doi.org/10.1038/nature12352 (2013).
Campbell, J. H. et al. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc Natl Acad Sci USA 110, 5540–5545, https://doi.org/10.1073/pnas.1303090110 (2013).
Camanocha, A. & Dewhirst, F. E. Host-associated bacterial taxa from Chlorobi, Chloroflexi, GN02, Synergistetes, SR1, TM7, and WPS-2 Phyla/candidate divisions. J Oral Microbiol 6, https://doi.org/10.3402/jom.v6.25468 (2014).
Peralbo-Molina, A., Calderon-Santiago, M., Jurado-Gamez, B., Luque de Castro, M. D. & Priego-Capote, F. Exhaled breath condensate to discriminate individuals with different smoking habits by GC-TOF/MS. Sci Rep 7, 1421, https://doi.org/10.1038/s41598-017-01564-z (2017).
Charlson, E. S. et al. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One 5, e15216, https://doi.org/10.1371/journal.pone.0015216 (2010).
Shchipkova, A. Y., Nagaraja, H. N. & Kumar, P. S. Subgingival microbial profiles of smokers with periodontitis. J Dent Res 89, 1247–1253, https://doi.org/10.1177/0022034510377203 (2010).
Shaikh, R. B. et al. Acute effects of dokha smoking on the cardiovascular and respiratory systems among UAE male university students. Asian Pac J Cancer Prev 13, 1819–1822 (2012).
Walters, M. S. et al. Waterpipe smoking induces epigenetic changes in the small airway epithelium. PLoS One 12, e0171112, https://doi.org/10.1371/journal.pone.0171112 (2017).
Jaffri, S. B., Yousuf, A. & Qidwai, W. Water pipe smoking amongst the university and college students of Karachi, Pakistan. Pakistan Journal of Chest Medicine 18, 13–19 (2012).
Zaura, E. et al. Same Exposure but Two Radically Different Responses to Antibiotics: Resilience of the Salivary Microbiome versus Long-Term Microbial Shifts in Feces. MBio 6, e01693–01615, https://doi.org/10.1128/mBio.01693-15 (2015).
Dar, N. A. et al. Hookah smoking, nass chewing, and oesophageal squamous cell carcinoma in Kashmir, India. Br J Cancer 107, 1618–1623, https://doi.org/10.1038/bjc.2012.449 (2012).
Nuwayhid, I. A., Yamout, B., Azar, G. & Kambris, M. A. Narghile (hubble-bubble) smoking, low birth weight, and other pregnancy outcomes. Am J Epidemiol 148, 375–383 (1998).
Cobb, C. O., Sahmarani, K., Eissenberg, T. & Shihadeh, A. Acute toxicant exposure and cardiac autonomic dysfunction from smoking a single narghile waterpipe with tobacco and with a “healthy” tobacco-free alternative. Toxicol Lett 215, 70–75, https://doi.org/10.1016/j.toxlet.2012.09.026 (2012).
Staley, J., Irgens, R. & Brenner, D. Enhydrobacter aerasaccus gen. nov., sp. nov. a Gas-Vacuolated, Facultatively Anaerobic, Heterotrophic Rod. International Journal of Systematic Bacteriology 37, 289–291 (1987).
van Amsterdam, J. et al. Effect of ammonia in cigarette tobacco on nicotine absorption in human smokers. Food Chem Toxicol 49, 3025–3030, https://doi.org/10.1016/j.fct.2011.09.037 (2011).
Bergmann, G. T. et al. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem 43, 1450–1455, https://doi.org/10.1016/j.soilbio.2011.03.012 (2011).
Haas, B. J. et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21, 494–504, https://doi.org/10.1101/gr.112730.110 (2011).
Siqueira, J. F., Jr., Fouad, A. F. & Rocas, I. N. Pyrosequencing as a tool for better understanding of human microbiomes. J Oral Microbiol 4, https://doi.org/10.3402/jom.v4i0.10743 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336, https://doi.org/10.1038/nmeth.f.303 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267, https://doi.org/10.1093/bioinformatics/btp636 (2010).
Oksanen, J. et al. vegan: Community Ecology Package (2017).
Lozupone, C. A., Hamady, M., Kelley, S. T. & Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73, 1576–1585, https://doi.org/10.1128/AEM.01996-06 (2007).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 57, 289–300 (1995).
Team, R. C. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Viena, Austria. (2014).
Acknowledgements
The authors are indebted to the support of the funding organization New York University Abu Dhabi Research Institute. In addition, the authors would like to acknowledge New York University Abu Dhabi Bioinformatics Core (CGSB) and the sequencing core for their support in the development of the pipeline for sequence processing.
Author information
Authors and Affiliations
Contributions
Y.V., C.K.I., R.B.H. and J.A. designed the study, conducted data collection, analyzed the data interpreted the results and drafted the manuscript. B.A.P. analyzed the data and reviewed the manuscript. L.A.W., H.A., F.A.A., A.A.D., D.G., M.H., A.A.H., A.A.H.O., M.A.H., A.A.J., M.K., W.A.M., F.A.M., A.A.L., N.O. and E.A.Z. conducted data collection, and reviewed the manuscript. R.A., A.A., T.K., R.R., A.M.S., M.W. and S.S. designed the study, conducted data collection and reviewed the manuscript. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vallès, Y., Inman, C.K., Peters, B.A. et al. Types of tobacco consumption and the oral microbiome in the United Arab Emirates Healthy Future (UAEHFS) Pilot Study. Sci Rep 8, 11327 (2018). https://doi.org/10.1038/s41598-018-29730-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29730-x
This article is cited by
-
Association of cigarette smoking with oral bacterial microbiota and cardiometabolic health in Chinese adults
BMC Microbiology (2023)
-
A literature review addressing midwakh and e-cigarette use in the Gulf region
Journal of the Egyptian Public Health Association (2023)
-
Smoking and salivary microbiota: a cross-sectional analysis of an Italian alpine population
Scientific Reports (2023)
-
Alteration of oral bacteriome of smokeless tobacco users and their association with oral cancer
Applied Microbiology and Biotechnology (2023)
-
The role of the oral microbiome in smoking-related cardiovascular risk: a review of the literature exploring mechanisms and pathways
Journal of Translational Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
