
Abstract
Molecular techniques are not routinely employed for malaria surveillance, while cross-sectional, community-based parasite surveys require significant resources. Here, we describe a novel use of malaria rapid diagnostic tests (RDTs) collected at a single facility as source material for sequencing to esimtate malaria transmission intensity across a relatively large catchment area. We extracted Plasmodium falciparum DNA from RDTs, then amplified and sequenced a region of the apical membrane antigen 1 (pfama1) using targeted amplicon deep sequencing. We determined the multiplicity of infection (MOI) for each sample and examined associations with demographic, clinical, and spatial factors. We successfully genotyped 223 of 287 (77.7%) of the samples. We demonstrated an inverse relationship between the MOI and elevation with individuals presenting from the highest elevation villages harboring infections approximately half as complex as those from the lowest (MOI 1.85 vs. 3.51, AOR 0.25, 95% CI 0.09–0.65, p = 0.004). This study demonstrates the feasibility and validity of using routinely-collected RDTs for molecular surveillance of malaria and has real-world utility, especially as the cost of high-throughpout sequencing continues to decline.
Similar content being viewed by others
Introduction
To acheieve malaria elimination, there is a need for tools that accurately measure transmission intensity to identify target areas for public health interventions1,2,3. The multiplicity of infection (MOI), defined as the number of concurrent parasite clones per Plasmodium falciparum-infected host, has shown promise as a surrogate measure of malaria transmission intensity4. In the more than twenty years since its first descriptions5,6,7, the MOI has been employed in a variety of roles to include estimating malaria transmission intensity between differing areas8,9, assessing changes in transmission intensity over time10,11, and evaluating the impact of interventions ranging from chemoprophylaxis12,13 to vaccines14,15.
In malaria endemic areas, the presence of polyclonal infections is common, and caused by infection from multiple mosquitoes or infection from a single mosquito harboring multiple parasite clones16. The MOI is generally considered to be positively associated with the intensity of transmission. As shown in Table 1, significant differences in the MOI between sites of varying transmission intensity have been consistently demonstrated across sub-Saharan Africa17,18,19,20,21,22,23,24,25,26,27,28,29,30,31. Host factors such as premunition, however, also impact the MOI5,7,32. Efforts have been made to correlate the MOI with age, parasite density, and disease severity, all of which reflect host immunity, with mixed results (Table 1).
To estimate MOI, investigators have historically collected dried blood spots (DBS), which allow long-term storage of malarial DNA. However, DBS are not collected in routine clinical practice, and require additional supplies for sampling and storage, which generally precludes estimation of MOI outside of research studies. Moroever, the MOI is traditionally determined using nested PCR (nPCR) with gel electrophoresis to detect polymorphisms in the highly variable surface antigens of the merozoite surface proteins (msp-1 & -2) or the glutamate-rich protein (glurp)33. However, these techniques are labor intensive and often fail to detect all sequence polymorphisms and low-abundance variants34,35. Newer methods utilizing targeted amplicon deep sequencing of malaria infections provide a high-throughput, highly sensitive approach for detecting clones in polyclonal infections as well as more accurate quantitative estimates of clonal frequency36.
In this study, we investigated the use of a targeted amplicon deep sequencing approach to describe the clonal diversity of the P. falciparum apical membrane antigen 1 (pfama1) across a geographically diverse area of western Uganda. To achieve this, we extracted DNA from malaria rapid diagnostic tests (RDTs) collected during routine care at a single rural health facility and stored at room temperature under tropical conditions. Our primary objective was to identify spatial differences in the MOI as a surrogate marker of malaria transmission intensity over a highland area using only a facility-based sample of routinely-collected rapid tests. While RDTs have previously been used as a source of malarial DNA for PCR and the sequencing of drug resistance and mitochondrial polymorphisms, they have not been employed to estimate transmission intensity37,38,39,40,41. Our relatively efficient sampling strategy precludes the need for more resource-intensive, population-based approaches to mapping transmission intensity and parasite diversity. Secondary objectives included an investigation of associations between the MOI, age, and the clinical spectrum of malaria, as well as an exploration of the spatial micro-epidemiology of parasites based on the pfama1 haplotypes.
Materials and Methods
Study setting
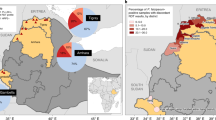
Samples were collected from patients presenting with febrile illness to the Bugoye Level III Health Center in the Kasese District of western Uganda. The geography of the study area is highly varied. The westernmost villages of the sub-county are characterized by deep river valleys and steep hillsides with elevations up to 2,000 meters. In contrast, villages located to the east are defined by low-lying, level terrain (Fig. 1). Three large rivers (the Mubuku to the north, Sebwe bisecting the sub-county, and the Nambiaji to the south) flow down valleys from west to east converging in the low-lying basin area near the health center. Like much of Uganda, the climate in Bugoye permits year-round malaria transmission marked by semi-annual transmission peaks typically following the end of the rainy seasons42. The two most recent malaria indicator surveys undertaken in the region found declining parasitemia prevalence from 48.4% to 17.6% in 2009 and 2014, respectively43,44.

Map of the study area shaded by elevation quartiles. Map created using ArcGIS, Version 10.4.1 (ESRI, Redlands, CA) available at http://desktop.arcgis.com/en/.
Sample Selection
Rapid diagnostic tests were performed as part of the Rapid Diagnostic Tests for Severe Malaria (RDTSM) study, a prospective, observational cohort study of patients with a parasitological diagnosis of malaria conducted from May to November 2015. We have reported full details of the study methodology elsewhere45.
In brief, initial testing for malaria was performed using the Standard Diagnostics 05FK60 Malaria Ag P.f/Pan RDT assay (Standard Diagnostics, Hagal-Dong, Korea). Study RDT were obtained directly from the manufacturer, stored in the original packaging at room temperature, and utilized in accordance with the manufacturer’s instructions prior to the expiry date. Each day, study staff packaged completed RDTs in a polyethylene bag with desiccant. RDTs were stored at room temperature for approximately two years prior to extraction. Study staff with training in laboratory medicine prepared thin and thick blood smears for all patients with a positive RDT result. Hemoglobin levels were measured using the Hemocue Hb 201+ analyzer (Brea, CA), and venous lactate values were obtained using the Abbott iStat analyzer (Princeton, NJ).
We first stratified the RDT-positive results by village. From each village-based strata, we sorted by level of parasitemia as determined by microscopy and systematically sampled starting from the highest density sample to achieve a balance of high and low parasitemias within each village-based strata. A convenience sample of approximately sixteen RDTs from each village was selected. Post hoc, we found that this number was more than sufficient to power the primary analysis at a level of 0.80 (α = 0.05) to detect the observed mean difference in MOI between the highest and lowest quartiles of elevation, even accounting for failed genotyping in nearly a quarter of samples.
Laboratory and Bioinformatic Methods
We removed a 1 cm section of the internal filter paper from the RDTs according to a previously described protocol (Supplementary Fig. S1)40. DNA was extracted using a previously described Chelex extraction method in a final volume of 100 μL46. The concentration of extracted P. falciparum DNA in individual samples was determined using a quantitative real-time PCR (qPCR) for P. falciparum lactate dehydrogenase (pfldh)47. Pfama1 was amplified from individual samples in duplicate in a hemi-nested PCR modified from Miller et al., which was shown to reproducibly and accurately determine the frequency and haplotype of variants in known mixtures in our laboratory using Ion Torrent sequencing48. In this approach, the inner forward primers are labeled with a unique barcode (Supplementary Table S1), referred to as an MID, which allows for multiple PCR products to be pooled prior to sequencing library preparation (Supplemental Fig. S2)48. We employed 22 unique MIDs, allowing 11 samples to be sequenced in duplicate in each Illumina library. Each PCR within a final sequencing library contained a unique MID, thus allowing the PCR replicate for each clinical sample to be de-convoluted bioinformatically as described below.
Modifications to the PCR protocol included using 2.5 units of Roche FastStart high fidelity Taq polymerase, and final concentrations of 1.8 mM for MgCl2, 200 uM for each dNTP (dATP, dCTP, dGTP, dTTP), and 167 nM for forward and reverse primers for each step of the hemi-nested PCR. Thus, all samples were amplified and uniquely barcoded in duplicate. Successful amplification was confirmed by visualizing the qPCR products on a 1% agarose gel and the product was quantified using Quantifluor dsDNA System (Promega, Fitchburg, WI) on a Synergy HT Multi-Mode Microplate Reader (BioTek, Winooski, VT). Barcoded samples were then pooled in equal amounts based on concentration. Pools of MID labeled PCR products were cleaned using KAPA Pure Beads (Illumina, San Diego, CA) and multiple indexed libraries were prepared for sequencing using KAPA Hyper Library Prep kits (Illumina, San Diego, CA) using NEXTflex DNA barcodes (BiooScientific, Austin, TX). The indexed pools were then combined in equimolar concentration into a final pool that was sequenced on a part of a MiSeq (2 × 300 bp chemistry) at the University of North Carolina High Throughput Sequencing Facility. Unlike Ion Torrent, Illumina sequencers have problems handling samples with low heterogeneity. We typically use two approaches to increase thje heterogeneity on the sequencing runs. First, we include 10% PhiX spike in all of our amplicon deep sequencing runs. Second, we often run libraries for other projects on the same run as the amount of sequencing necessary for a small project is not sufficient to warrant a complete run. This library comprised an estimated 80% of the run which also contained amplicons from other studies.
For these experiements, we included a positive control comprised of a known mixture of three malaria strains: 3D7 (MRA-102G, BEI Resources, Manansas, VA), FCR3 (MRA-321G, BEI Resources), and Dd2 (MRA-150G, BEI Resources). Using this control mixture, pfama1 amplicons were generated for 5 controls, representing 10 MID labeled PCR reactions, and sequenced alongside the clinical samples. To ensure that we correctly estimated the frequency of the three strains in the mixture, we amplified the mixture using the same conditions using only the second round reaction for 30 cycles. We cloned the PCR product using a TOPO TA cloning kit (Invitrogen) and performed Sanger sequencing of 22 colonies using the M13F primer at GENWIZ (RTP, NC). Sequences were analyzed using Geneious R10 (Biomatters Inc., Newark, NJ). We estimated allele frequencies and confidence intervals by the bootsrap method with 1,000 replicate bootstraps. This was compared to the allele frequency determined by the four replicate control deep sequencing reactions.
The deep sequencing reads were de-multiplexed and clustered using the software package, SeekDeep (http://baileylab.umassmed.edu/SeekDeep), as previously described49. This approach uses the PCR replicates for each sample to reduce PCR and sequencing errors as shown in the SeekDeep Process Cluster step of Fig. 1 of Hathaway et al., and has been shown to provide accurate frequency and genotype determinations with as few as 200 reads per sample49. As haplotypes in each PCR are called independently and then only haplotypes conserved between replicate PCRs are used, this provides a conservative approach for haplotype detection to determine MOI. Here, samples were included in the final analysis if they had ≥250 total reads combined between the replicates. The low read coverage in some samples did not influence the MOI estimates as seen in Supplemental Fig. S5. Haplotypes were included in the analysis if ≥80% of haplotype reads had a Phred Quality Score of ≥30 and if they occurred in both sequencing replicates and were above an averaged frequency cutoff of 2.5% between the two PCRs, similar to our previous work (i.e. if the within-sample average frequency across duplicate runs was above this cutoff)48. Haplotypes that were marked as likely chimeric were excluded. Haplotypes in each sample were compared to all identified haplotypes in order to provide population-level statistics. All data generated or analyzed during this study are included in this published article and the Sequence Read Archive available at www.ncbi.nlm.nih.gov/sra (SRA Accession Number Pending).
Statistical analysis
We first summarized demographic, clinical, and laboratory characteristics of the cohort using Student’s t-test for continuous variables and Pearson’s Chi squared test for categorical variables. We defined severe malaria in accordance with the WHO guidelines for research and epidemiological studies using a threshold of ≥250,000 parasites/μl to define hyperparasitemia50.
The MOI was calculated as the number of concurrent parasite clones per P. falciparum-positive sample. We performed ordinal logistic regression to explore the demographic, spatial, clinical and laboratory parameters associated with the MOI, the primary outcome measure of interest. Age categories were set to be comparable with prior studies that found significant associations between age and MOI22,30. All variables that were significant in univariate models with a pre-specified P-value of <0.25 were included in the subsequent multivariate analysis51. We compared the results of the regression analysis to the RDT positivity rate, defined as the number of positive malaria tests per 100 suspected cases examined.
To assess the potential relationship between the MOI and disease severity, we selected three outcomes of interest: (a) lactic acid levels, (b) hemoglobin levels, and (c) severe malaria. For the continuous outcomes of lactic acid and hemoglobin levels, we first performed linear regression with MOI serving as the primary explanatory variable. We then utilized negative binomial general linear regression model with a log-link function and robust standard errors to explore associations between the categorical outcomes of (a) lactic acidosis (venous lactate ≥5 mmol/L), (b) anemia (hemoglobin <7 g/dL), and (c) severe malaria. Data were analyzed with Stata 12.1 (College Station, TX).
To estimate associations between geographic factors and the MOI, we completed village-level geographic information system (GIS) mapping of the sub-county and surrounding environs, comprising an area of approximately 55 square kilometers. These data were entered into ArcGIS, Version 10.4.1 (ESRI, Redlands, CA) to create a reference map, from which we calculated the mean elevation and area of each village and subsequently divided the data into quartiles of elevation.
Ethics statement
Ethical approval of the study was provided by the institutional review boards of the University of North Carolina at Chapel Hill, the Mbarara University of Science and Technology, and the Uganda National Council for Science and Technology. Written informed consent was obtained from all adult study participants and the caregivers of participating children. All research was performed in accordance with relevant guidelines and regulations.
Results
Validation of control mixtures
Allele frequency calls for all five control mixtures are shown in Supplemental Fig. S3. Using a previously described measure of concordance for allele frequency calls between PCR replicates (do), the replicate PCRs showed a high level of agreement of allele frequencies in each of the five controls with a mean do of 0.0252. Using data from the cloned Sanger sequences as the reference standard, we evaluated the accuracy of the deep sequencing frequency estimates. We bootstrapped the frequency estimates to generate confidence intervals for the Sanger sequence data and compared it to the variation in replicate deep sequencing reactions (Supplementary Fig. S4). The amplicon deep sequencing method provided accurate estimates of haplotype frequency, suggesting that haplotype frequencies in the clinical samples were representative. No false haplotypes were detected in the control mixtures.
Extraction of genomic DNA from RDTs and amplification of samples
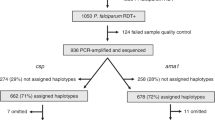
We detected pfldh malarial DNA and were subsequently able to amplify pfama1 in 287 of 299 (96.0%) of RDT positive samples. Of the twelve RDTs from which we were unable to detect pfldh or pfama1, four were negative on microscopy while another four had parasite densities of <250 parasites/μl. We successfully sequenced 223 of the 287 (77.7%) samples with detectable malarial DNA. Demographic and clinical characteristics of the cohort are summarized in Table 2. There were no significant differences in age (p = 0.28), sex (p = 0.85), parasitemia (p = 0.29), or disease severity (p = 0.70) between those samples successfully genotyped and those that were excluded from the analysis.
Multiplicity of Infection
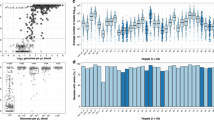
After filtering in SeekDeep, we used 5.56 million of 8.15 million sequencing reads to call AMA haplotypes and determine multiplicity of infection in each sample. The mean number of reads used per sample was 22,763 (range 254–120,213). Read depth for each sample stratified by MOI is shown in Supplementary Fig. S5. The mean and median MOI was 3.09 (95% CI 2.74–3.44) and 2.0 (IQR 1.0–4.0), respectively. The crude monthly mean MOI peaked in May (MOI = 4.68, 95% CI 3.22–6.14), which is the last month of the traditional rainy season, and then declined significantly, reaching a nadir in October, (MOI = 2.06, 95% CI 1.06–3.07, p = 0.003 compared to May), which marks the beginning of the second rainy season.
The MOI varied significantly with geographic factors, including village elevation and river valley of residence. The MOI demonstrated an inverse relationship with elevation (Table 3). On average, individuals presenting from the highest elevation villages harbored infections approximately half as complex as those from the lowest elevation villages (MOI 1.85 vs 3.51, OR 0.24, 95% CI 0.10–0.57, p = 0.001) (Fig. 2). This finding was robust in the final model, which was adjusted for age and disease severity. We also observed a significantly lower MOI among individuals presenting from the Sebwe (MOI 2.28, aOR 0.55, 95% CI 0.31–0.97, p = 0.04) and Nambiaji (MOI 1.75, aOR 0.31, 95% CI 0.11–0.85, p = 0.02) river valleys compared to the basin area (MOI 3.25) where the rivers converge. These results were generally consistent with the RDT positivity rates observed in each valley with both the Sebwe (aOR 0.84, 95% CI 0.72 – 0.0.97, p = 0.02) and Nambiaji (aOR 0.64, 95% CI 0.50–0.81, p < 0.001) river valleys being significantly lower than the basin area.

Multiplicity of infection (MOI) stratified by elevation quartiles showing that mono-infections comprised the smallest proportion of infections in the lowest elevation villages (Quartile 1) and the highest proportion in the highest villages (Quartile 4).
The highest MOI was found in children age 3 to 4 years of age (MOI = 3.47, 95% CI 1.99–4.96) compared to those aged 8–11 years of age, who had the lowest MOI (2.97, 95% CI 2.21–3.74). Differences by age, however, were not significant as shown in Table 3 or when age groups were broadened to include children <5 years of age, 5 to 15 years of age, and ≥15 years of age (Supplementary Table S2). No clinical factors were associated with the degree of infection complexity. While there were trends towards lower MOI with severe disease in the univariate regression analysis, these findings were not significant in adjusted models.
When we defined severe malaria as the outcome measure of interest, we found strong correlations with male sex, age less than five years, and parasite density greater than 100,000 parasites/μl in the 21 cases of severe disease (Table 4). There was, however, no significant difference in the proportion of patients with polyclonal infections among patients with uncomplicated versus severe malaria (66.1 vs. 57.1%, p = 0.41). Increasingly complex infections (≥3 identified haplotypes) demonstrated a trend towards a reduced risk of severe malaria although this finding was not significant in the multivariate model (aIRR 0.65, 95% CI 0.25–1.73, p = 0.39), which was adjusted for age. Of note, when the analysis was stratified by age, we found that risk of severe malaria was non-significantly higher with more complex infections (IRR = 1.67, 95% CI 0.34–8.06, p = 0.53) in children <5 years of age, while the risk of severe malaria trended lower with more complex infections in children and adults ≥5 years of age (IRR 0.24, 95% CI 0.05–1.12, p = 0.07).
Similarly, there was no association between the MOI and disease severity when we defined lactic acidosis (>5 mmol/L) or anemia (Hb < 7 g/dL) as the outcome measure of interest. While lactic acidosis and anemia were relatively rare outcomes in the cohort, we did not observe any trend between lactic acid (β coefficient = 0.02, 95% CI −0.03–0.07, p = 0.40) or hemoglobin levels (β coefficient = 0.07, 95% CI −0.04–0.17 p = 0.20) and the MOI in the linear regression models.
Haplotype Analysis
A total of 39 unique haplotypes were identified [SRA Accession Number Pending]. The most common haplotype (UgandaMOI.00) was found in 122 of 223 (54.7%) of the included samples and accounted for 24.6% of the population fraction. The distribution of haplotypes in the population are shown in Supplementary Fig. S6. Approximately two thirds of included samples (n = 139, 62.3%) demonstrated polyclonal infections with the highest proportion observed in individuals 18 to 30 years of age (n = 28, 68.3%). In contrast, monoclonal infections were most common in children <3 years of age (n = 11, 57.9%).
In the clustering analysis, we did not identify any evidence of population structure based on either haplotype prevalence (the proportion of infections containing the respective haplotype), or haplotype relative abundance (the proportion of the respective haplotype within an individual infection), when plotting by elevation, river valley or the presence/absence of severe malaria. (Fig. 3).

Principle component analyses (PCA) depicting the absence of population structure between individual haplotypes and quartiles of elevation (3.A), river valleys (3.B), or disease severity (3.C).
Discussion
We carried out targeted amplicon deep sequencing from DNA extracted from routinely-collected RDTs among a facility-based cohort of symptomatic individuals and were able to demonstrate a significant, inverse correlation between village elevation and the MOI, consistent with previous observations27. We also found a lower MOI in two of the river valleys, which correlated with the RDT positivity rate, a crude measure of transmission intensity. Our findings are proof-of-concept that it is possible to estimate transmission intensity across a wide catchment area using only a facility-based sample of routinely-collected RDTs, thus negating the need for additional DBS collection or labor-intensive cross-sectional sampling. Such an approach has relatively broad programmatic application and may even be cost-effective when considering the labor costs associated with large cross-sectional surveys.
In contrast, we found no evidence of population structure by elevation or river valley based on the pfama1 haplotypes. This outcome is not entirely unexpected, as it likely reflects the high intensity of transmission and resulting mixing of parasite sub-populations across the study site. It is also supported by our recent work showing no population structure based on this gene between distant geographic sites in Africa48. We had hypothesized that the high ridgelines separating the three major rivers could serve as a physical barrier to vector migration, isolating each valley from the next. Our results, however, could suggest that this barrier is inadequate to maintain population structure, most likely at the lower elevation areas where the rivers merge into the basin area. In addition, human movement into lower elevation areas, where most commercial activity occurs, could also facilitate the mixing of parasite populations between river valleys. Alternatively, the lack of structure could also result from the choice of marker. Antigens like pfama1 may be under convergent evolution and thus similar patterns of diversity emerge in distant populations, as supported by our previous work53,54.
We did not identify any associations between the MOI and age or the MOI and markers of disease severity (i.e. anemia, lactate, severe malaria). However, given the few cases of severe malaria (n = 21), our analysis was only powered to a level of 0.54 to detect a mean difference of one parasite clone between cases of uncomplicated and severe malaria. Interestingly, we did observe a divergent effect of infection complexity on the risk of severe malaria among children <5 years of age, who had a relatively negligible risk of severe disease with a more complex infection (≥3 identified haplotypes), compared to adults and children ≥5 years of age, who had a clear trend (p = 0.07) towards a reduced risk of severe disease with more complex infections. This finding supports the hypothesis of age-dependent immune response mechanisms and is consistent with a report from Tanzania in which children <3 years of age experienced a greater risk of a subsequent malarial episode with increasing infection complexity, whereas in older children, more complex infections were associated with a decreased risk of clinical malaria,55.
Our study has several limitations. First, we did not utilize traditional measures of transmission intensity such as the entomological inoculation rate (EIR) or parasite prevalence against which we could compare our results. However, the relationship between elevation and transmission intensity is well established56 and thus we are confident that our findings in regard to the association between the MOI and elevation are valid. Second, we did not perform genotyping from samples stored on DBS, which would have allowed direct comparison between the two methods of sample collection, although previous studies have reported similar PCR success rates with each approach38,40. Lastly, we were unable to genotype 64 (22.3%) of our samples. This is likely a result of our conservative duplicate reading thresholds. We are reassured that there were no significant differences in demographic, clinical, or laboratory parameters between those samples that were successfully genotyped and those that were not.
Conclusions
Using routinely-collected malaria RDTs from a single health facility as source material, we were able to deep sequence and estimate malaria transmission intensity across a large and geographically diverse catchment area. To our knowledge, this is the first study to demonstrate the feasibility and validity of such an approach, which we believe has practical implications for malaria surveillance programs, especially as the cost of high-throughput sequencing continues to decline. Similar techniques may also be applicable to other disease conditions where lateral flow assays are commonly utilized for point-of-care diagnosis.
References
Hay, S. I., Smith, D. L. & Snow, R. W. Measuring malaria endemicity from intense to interrupted transmission. Lancet Infect Dis 8, 369–378, https://doi.org/10.1016/s1473-3099(08)70069-0 (2008).
Marsh, K. Research priorities for malaria elimination. Lancet 376, 1626–1627, https://doi.org/10.1016/s0140-6736(10)61499-7 (2010).
The malERA Consultative Group on Diagnoses and Diagnostics. A research agenda for malaria eradication: diagnoses and diagnostics. Plos Med 8, e1000396 10.1371/journal.pmed.1000396 (2011).
Tusting, L. S., Bousema, T., Smith, D. L. & Drakeley, C. Measuring changes in Plasmodium falciparum transmission: precision, accuracy and costs of metrics. Adv Parasitol 84, 151–208, https://doi.org/10.1016/b978-0-12-800099-1.00003-x (2014).
Ntoumi, F. et al. Age-dependent carriage of multiple Plasmodium falciparum merozoite surface antigen-2 alleles in asymptomatic malaria infections. Am J Trop Med Hyg 52, 81–88 (1995).
Contamin, H. et al. Different genetic characteristics of Plasmodium falciparum isolates collected during successive clinical malaria episodes in Senegalese children. Am J Trop Med Hyg 54, 632–643 (1996).
Farnert, A., Rooth, I., Svensson, Snounou, G. & Bjorkman, A. Complexity of Plasmodium falciparum infections is consistent over time and protects against clinical disease in Tanzanian children. J Infect Dis 179, 989–995, https://doi.org/10.1086/314652 (1999).
Arnot, D. Unstable malaria in Sudan: the influence of the dry season. Clone multiplicity of Plasmodium falciparum infections in individuals exposed to variable levels of disease transmission. Trans R Soc Trop Med Hyg 92, 580–585 (1998).
Schoepflin, S. et al. Comparison of Plasmodium falciparum allelic frequency distribution in different endemic settings by high-resolution genotyping. Malar J 8, 250, https://doi.org/10.1186/1475-2875-8-250 (2009).
Niang, M. et al. Spatio-temporal analysis of the genetic diversity and complexity of Plasmodium falciparum infections in Kedougou, southeastern Senegal. Parasit Vectors 10 https://doi.org/10.1186/s13071-017-1976-0 (2017).
Daniels, R. F. et al. Modeling malaria genomics reveals transmission decline and rebound in Senegal. Proc Natl Acad Sci USA 112, 7067–7072, https://doi.org/10.1073/pnas.1505691112 (2015).
Beck, H. P. et al. Effect of iron supplementation and malaria prophylaxis in infants on Plasmodium falciparum genotypes and multiplicity of infection. Trans R Soc Trop Med Hyg 93(Suppl 1), 41–45 (1999).
Liljander, A. et al. Influences of intermittent preventive treatment and persistent multiclonal Plasmodium falciparum infections on clinical malaria risk. Plos One 5, e13649, https://doi.org/10.1371/journal.pone.0013649 (2010).
Beck, H. P. et al. Analysis of multiple Plasmodium falciparum infections in Tanzanian children during the phase III trial of the malaria vaccine SPf66. J Infect Dis 175, 921–926 (1997).
Haywood, M. et al. Reduction in the mean number of Plasmodium falciparum genotypes in Gambian children immunized with the malaria vaccine SPf66. Trans R Soc Trop Med Hyg 93(Suppl 1), 65–68 (1999).
Talisuna, A. O., Okello, P. E., Erhart, A., Coosemans, M. & D’Alessandro, U. Intensity of malaria transmission and the spread of Plasmodium falciparum resistant malaria: a review of epidemiologic field evidence. Am J Trop Med Hyg 77, 170–180 (2007).
A-Elbasit, I. et al. Allelic polymorphism of MSP2 gene in severe P. falciparum malaria in an area of low and seasonal transmission. Parasitol Res 102, 29–34, https://doi.org/10.1007/s00436-007-0716-3 (2007).
Amodu, O. K. et al. Complexity of the msp2 locus and the severity of childhood malaria, in south-western Nigeria. Ann Trop Med Parasitol 102, 95–102, https://doi.org/10.1179/136485908x252340 (2008).
Apinjoh, T. O. et al. Plasmodium falciparum merozoite surface protein 1 block 2 gene polymorphism in field isolates along the slope of mount Cameroon: a cross - sectional study. BMC Infect Dis 15, 309, https://doi.org/10.1186/s12879-015-1066-x (2015).
Bendixen, M., Msangeni, H. A., Pedersen, B. V., Shayo, D. & Bodker, R. Diversity of Plasmodium falciparum populations and complexity of infections in relation to transmission intensity and host age: a study from the Usambara Mountains, Tanzania. Trans R Soc Trop Med Hyg 95, 143–148 (2001).
Durand, R. et al. Analysis of circulating populations of Plasmodium falciparum in mild and severe malaria in two different epidemiological patterns in Madagascar. Trop Med Int Health 13, 1392–1399, https://doi.org/10.1111/j.1365-3156.2008.02156.x (2008).
Engelbrecht, F. et al. Analysis of Plasmodium falciparum infections in a village community in Northern Nigeria: determination of msp2 genotypes and parasite-specific IgG responses. Acta Trop 74, 63–71 (2000).
Francis, D. et al. Geographic differences in antimalarial drug efficacy in Uganda are explained by differences in endemicity and not by known molecular markers of drug resistance. J Infect Dis 193, 978–986, https://doi.org/10.1086/500951 (2006).
Kateera, F. et al. Malaria case clinical profiles and Plasmodium falciparum parasite genetic diversity: a cross sectional survey at two sites of different malaria transmission intensities in Rwanda. Malar J 15, 237, https://doi.org/10.1186/s12936-016-1287-5 (2016).
Kiwuwa, M. S. et al. Genetic diversity of Plasmodium falciparum infections in mild and severe malaria of children from Kampala, Uganda. Parasitol Res 112, 1691–1700, https://doi.org/10.1007/s00436-013-3325-3 (2013).
M A Hamid, M. et al. Multiplicity of infection and genetic diversity of Plasmodium falciparum isolates from patients with uncomplicated and severe malaria in Gezira State, Sudan. Parasit Vectors 9, 362, https://doi.org/10.1186/s13071-016-1641-z (2016).
Manjurano, A. et al. Association of sub-microscopic malaria parasite carriage with transmission intensity in north-eastern Tanzania. Malar J 10, 370, https://doi.org/10.1186/1475-2875-10-370 (2011).
Mayor, A. et al. Plasmodium falciparum multiple infections in Mozambique, its relation to other malariological indices and to prospective risk of malaria morbidity. Trop Med Int Health 8, 3–11 (2003).
Mockenhaupt, F. P. et al. Plasmodium falciparum multiplicity correlates with anaemia in symptomatic malaria. Trop Med Int Health 8, 857–859 (2003).
Peyerl-Hoffmann, G. et al. Genetic diversity of Plasmodium falciparum and its relationship to parasite density in an area with different malaria endemicities in West Uganda. Trop Med Int Health 6, 607–613 (2001).
Shigidi, M. M., Hashim, R. A., Idris, M. N., Mukhtar, M. M. & Sokrab, T. E. Parasite diversity in adult patients with cerebral malaria: a hospital-based, case-control study. Am J Trop Med Hyg 71, 754–757 (2004).
Felger, I., Genton, B., Smith, T., Tanner, M. & Beck, H. P. Molecular monitoring in malaria vaccine trials. Trends Parasitol 19, 60–63 (2003).
World Health Organization. Methods and Techniques for Clinical Trials on Antimalarial Drug Efficacy: Genotyping to Identify Parasite Populations. (World Health Organization, Geneva, 2008).
Juliano, J. J. et al. Misclassification of drug failure in Plasmodium falciparum clinical trials in southeast Asia. J Infect Dis 200, 624–628, https://doi.org/10.1086/600892 (2009).
Juliano, J. J., Kwiek, J. J., Cappell, K., Mwapasa, V. & Meshnick, S. R. Minority-variant pfcrt K76T mutations and chloroquine resistance, Malawi. Emerg Infect Dis 13, 872–877, https://doi.org/10.3201/eid1306.061182 (2007).
Juliano, J. J. et al. Exposing malaria in-host diversity and estimating population diversity by capture-recapture using massively parallel pyrosequencing. Proc Natl Acad Sci USA 107, 20138–20143, https://doi.org/10.1073/pnas.1007068107 (2010).
Veron, V. & Carme, B. Recovery and use of Plasmodium DNA from malaria rapid diagnostic tests. Am J Trop Med Hyg 74, 941–943 (2006).
Cnops, L., Boderie, M., Gillet, P., Van Esbroeck, M. & Jacobs, J. Rapid diagnostic tests as a source of DNA for Plasmodium species-specific real-time PCR. Malar J 10, 67, https://doi.org/10.1186/1475-2875-10-67 (2011).
Ishengoma, D. S. et al. Using rapid diagnostic tests as source of malaria parasite DNA for molecular analyses in the era of declining malaria prevalence. Malar J 10, 6, https://doi.org/10.1186/1475-2875-10-6 (2011).
Morris, U. et al. Rapid diagnostic tests for molecular surveillance of Plasmodium falciparum malaria -assessment of DNA extraction methods and field applicability. Malaria journal 12, 106, https://doi.org/10.1186/1475-2875-12-106 (2013).
Nag, S. et al. High throughput resistance profiling of Plasmodium falciparum infections based on custom dual indexing and Illumina next generation sequencing-technology. Sci Rep 7, 2398, https://doi.org/10.1038/s41598-017-02724-x (2017).
Yeka, A. et al. Malaria in Uganda: challenges to control on the long road to elimination: I. Epidemiology and current control efforts. Acta tropica 121, 184–195, https://doi.org/10.1016/j.actatropica.2011.03.004 (2012).
Uganda Bureau of Statistics (UBOS) and ICF International. Uganda Malaria Indicator Survey 2009. (Calverton, Maryland, USA, 2010).
Uganda Bureau of Statistics (UBOS) and ICF International. Uganda Malaria Indicator Survey 2014-15., (Kampala, Uganda, and Rockville, Maryland, USA, 2015).
Boyce, R. et al. Use of a Dual-Antigen Rapid Diagnostic Test to Screen Children for Severe Plasmodium falciparum Malaria in a High-Transmission, Resource-Limited Setting. Clin Infect Dis 65, 1509–1515, https://doi.org/10.1093/cid/cix592 (2017).
Plowe, C. V., Djimde, A., Bouare, M., Doumbo, O. & Wellems, T. E. Pyrimethamine and proguanil resistance-conferring mutations in Plasmodium falciparum dihydrofolate reductase: polymerase chain reaction methods for surveillance in Africa. Am J Trop Med Hyg 52, 565–568 (1995).
Taylor, S. M. et al. High-throughput pooling and real-time PCR-based strategy for malaria detection. J Clin Microbiol 48, 512–519, https://doi.org/10.1128/jcm.01800-09 (2010).
Miller, R. H. et al. A deep sequencing approach to estimate Plasmodium falciparum complexity of infection (COI) and explore apical membrane antigen 1 diversity. Malar J 16, 490, https://doi.org/10.1186/s12936-017-2137-9 (2017).
Hathaway, N. J., Parobek, C. M., Juliano, J. J. & Bailey, J. A. SeekDeep: single-base resolution de novo clustering for amplicon deep sequencing. Nucleic Acids Res 10.1093/nar/gkx1201 (2017).
WHO. Guidelines for the Treatment of Malaria., (World Health Organization, Geneva, 2015).
Hosmer, D., Lemeshow, S. Applied Logistic Regression. (Wiley, 2000).
Mideo, N. et al. Ahead of the curve: next generation estimators of drug resistance in malaria infections. Trends in parasitology 29, 321–328, https://doi.org/10.1016/j.pt.2013.05.004 (2013).
Aragam, N. R. et al. Diversity of T cell epitopes in Plasmodium falciparum circumsporozoite protein likely due to protein-protein interactions. PLoS One 8, e62427, https://doi.org/10.1371/journal.pone.0062427 (2013).
Miller, R. et al. A Deep Sequencing Approach to Estimate Plasmodium falciparum Complexity of Infection (COI) and Explore Apical Membrane Antigen 1 Diversity. In Press. Malar J (2017).
Henning, L. et al. A prospective study of Plasmodium falciparum multiplicity of infection and morbidity in Tanzanian children. Trans R Soc Trop Med Hyg 98, 687–694, https://doi.org/10.1016/j.trstmh.2004.03.010 (2004).
Githeko, A. K. et al. Topography and malaria transmission heterogeneity in western Kenya highlands: prospects for focal vector control. Malar J 5, 107, https://doi.org/10.1186/1475-2875-5-107 (2006).
Acknowledgements
We wish to thank the clinical staff and patients of the Bugoye Health Centre for their continued support. We also appreciate the efforts of Corinna Keeler, who provided cartography assistance. RMB is supported by the National Institutes of Health (T32AI007151). In addition, he received support for this work from the Harvard Global Health Initiative and the Thrasher Research Fund. MJS receives support from the National Institutes of Health (K23MH099916). JJJ received support from National Institute of Health (K24AI134990, R01AI121558, R21AI121465). EM, MN, and MM received grant support for this work from Abbott Point of Care. Standard Diagnostics provided the rapid diagnostic tests for the study at no cost. Abbott Point of Care provided the iStat analyzers and the associated cartridges to the study. Neither Standard Diagnostics nor Abbott Point of Care had any role in the design or conduct of the study or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
R.M.B. and J.J.J. conceived the study. R.M.B., R.R., M.M., M.N. and E.M. led the clinical study and oversaw data collection. T.F., N.H., A.W., J.A.B. and J.J.J. performed the laboratory and bibliometric analysis. R.M.B. and M.J.S. designed and carried out the statistical analysis. All authors contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boyce, R.M., Hathaway, N., Fulton, T. et al. Reuse of malaria rapid diagnostic tests for amplicon deep sequencing to estimate Plasmodium falciparum transmission intensity in western Uganda. Sci Rep 8, 10159 (2018). https://doi.org/10.1038/s41598-018-28534-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-28534-3
This article is cited by
-
Plasmodium falciparum population structure inferred by msp1 amplicon sequencing of parasites collected from febrile patients in Kenya
Malaria Journal (2023)
-
Comparing field-collected versus remotely-sensed variables to model malaria risk in the highlands of western Uganda
Malaria Journal (2023)
-
Comparative analysis of targeted next-generation sequencing for Plasmodium falciparum drug resistance markers
Scientific Reports (2022)
-
Malaria prevalence and long-lasting insecticidal net use in rural western Uganda: results of a cross-sectional survey conducted in an area of highly variable malaria transmission intensity
Malaria Journal (2021)
-
Genotyping cognate Plasmodium falciparum in humans and mosquitoes to estimate onward transmission of asymptomatic infections
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
