
Abstract
We sequenced the genome of Raoultella ornithinolytica strain Marseille-P1025 that caused a rare case of prosthetic joint infection in a 67-year-old immunocompetent male. The 6.7-Mb genome exhibited a genomic island (RoGI) that was unique among R. ornithinolytica strains. RoGI was likely acquired by lateral gene transfer from a member of the Pectobacterium genus and coded for a type IVa secretion system found in other pathogenic bacteria and that may have conferred strain Marseille-P1025 an increased virulence. Strain Marseille-P1025 was also able to infect, multiply within, and kill Acanthamoaeba castellanii amoebae.
Similar content being viewed by others

Introduction
In 2001, the analysis of the 16S rRNA and rpoB gene sequences enabled reclassification of some Klebsiella species within the genus Raoultella1. Formerly known as Klebsiella ornithinolytica, Raoultella ornithinolytica is a Gram-negative, non-motile and encapsulated bacillus1 that inhabits aquatic environments and can also be found in hospital water circuits2. Reports of human R. ornithinolytica infections, initially rare, are increasing and mostly include biliary or urinary tract infections, and bacteremias3,4,5,6,7,8,9. Bone and joint infections caused by R. ornithinolytica are seldom reported10. We recently reported a case of chronic prosthetic joint infection caused by R. ornithinolytica in a 67-year-old immunocompetent male11. In this study, the causative strain, Marseille-P1025, was isolated from the peri-prosthetic pus11.
Herein, in order to determine whether this strain had specific virulence factors, we sequenced its genome and compared it to those of other R. ornithinolytica strains available in public databases.
Results
General genomic features
The draft genome sequence of R. ornithinolytica strain Marseille-P1025 consisted of 38 scaffolds after assembly and finishing. No putative plasmid sequence was detected. The chromosome size, G + C content, and CDS content were 5,644,584 bp, 55.6% and 5,260, respectively. A total of 86 RNA genes were identified, including one complete rRNA operon, a second 23S rRNA, eight other 5S rRNAs and 74 tRNAs. Of the 5,260 predicted CDSs, 4,391 genes were assigned a putative function (83.48%) and 869 (16.52%) were annotated as hypothetical proteins. A total of 4,438 (84.37%) genes were assigned a COG functional category.
Genome comparison
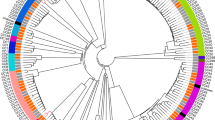
The genomic comparison is summarized in Table 1. Strain Marseille-P1025, with 5,260 CDs, had a smaller genome than those of strains 10–5246, 2–156_04_S1_C1, 2-156-04_S1_C2, TNT, 811_RORN and BAL286 (5,288, 5,281, 5,284, 5,281, 5,314 and 5,646 CDs, respectively) but larger than those of strains NBRC 105727, B6, A14, CMUL058, CB1 and Yangling l2 (5,108, 4,907, 4,933, 5,202, 4,953 and 5,033 CDs, respectively) (Table 1). Strain Marseille-P1025 exhibited 95 specific genes (Fig. 1, Table 2) when compared to all other studied R. ornithinolytica strains. In contrast, 37 genes present in at least 7 strains were absent in strain Marseille-P1025 (Fig. 1, Table S1).

Pan-genome analysis of R. ornithinolytica whole-genome sequences. A maximum likelihood tree was constructed from the accessory genome elements (left). The presence (blue) and absence (white) of accessory genome elements is presented on the right.
The thirteen studied strains exhibited a pangenome and a core genome of 9,815 and 3,822 genes, respectively (Fig. 1). Figure 1 shows the dispersion of the pangenome of R. ornithinolytica. The phylogenetic analysis based on accessory genes clustered strains Marseille-P1025 and NBRC 105727.
Functional annotation
The COG functional classification of the 95 genes specific of strain Marseille-P1025 demonstrated that 23 of the Marseille-P1025-specific genes were grouped in a 11,473-kb genomic island located in scaffold 21 (Fig. 2). This genomic island, which we named RoGI, exhibited a G + C content of 49.5% (vs 55 to 56% for the genomes of R. ornithinolytica strains, Table 1), and was absent from other R. ornithinolytica (Fig. 2). Of these 23 genes, nine coded type IVa secretion system proteins (Table 2), including seven VirB proteins (VirB 4 to 11, Table 2) and two proteins related to bacterial conjugation, including a type IVa secretion system conjugative DNA transfer protein and a conjugal transfer protein (Table 2). Moreover, the RoGI island contained a gene coding a second CP4-57 prophage integrase (intA) (Table 2). The genes coding the VirB1, VirB2, VirB3 and VirD4 proteins were identified at other locations of the genome from strain Marseille-P1025, thus supporting the assumption that this strain had a complete and putatively functional type IVa secretion system.

Comparison of sequences of the scaffold 21 from R. ornithinolytica strain Marseille-P1025 with those of R. ornithinolytica strains NBRC 105727 (A), 2-156-04_S1_C1 (B) and 2-156-04_S1_C2 (C). Figure 2D shows an alignment of all four compared genomes. Common and specific genes are displayed in orange and red, respectively.
In addition, seven (7.4%) proteins were involved in intracellular trafficking and secretion, seven (7.4%) in replication and repair, four (4.2%) in cell wall/membrane/envelop biogenesis and four (4.2%) had a general functional prediction only (Table 2). Finally, three genes coded integrases including a CP4-57 prophage integrase (intA), two genes coded integrating conjugative element proteins, and two genes coded a CP4-57 prophage regulatory protein AlpA and a transposase, respectively (Table 2).
ClonalframeML and Phylogenetic Analysis
To verify whether the RoGI island was acquired by lateral gene transfer, we used a recombination and phylogenetic analysis. Figure 3 shows the recombination events of external origin marked by a dark blue horizontal line. ClonalFrameML identified 170 recombination events on all branches of the clonal genealogy, including 23 recombination events in the genome of strain Marseille-P1025 (Fig. 3). These 23 regions appeared to be possible recombination hotspots (Fig. 3). Three of these recombination hotspots (red circle) were located in scaffold 21 of strain Marseille-P1025 (located from nucleotides 5,425,000 to 5,612,500) (Fig. 3), close to the RoGI island that coded the type IVa secretion system (located from nucleotides 5,504,317 to 5,515,790, Fig. 3).

Analysis of genomic recombinations in the R. ornithinolytica species based on the alignment of 13 genomes including 12 genomes mapped against that of strain Marseille-P1025, using ClonalFrameML. Recombination events are shown by dark blue horizontal bars. For a given branch, light blue sites mean no substitution. Any other color from white to red indicates a substitution. White indicates non-homoplasic substitutions and the increasing level of redness indicates the increasing degree of homoplasy. The arrow shows recombination events in scaffold 21 where the RoGI genomic island is located.
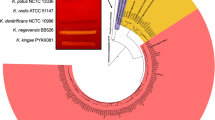
The phylogenetic analysis of nucleotide sequences from the RoGI island supported the assumption that it was acquired by lateral gene transfer by identifying close phylogenetic neighbours in Pectobacterium atrosepticum strain JG10-08, Pectobacterium sp. strain SCC3193, two Pectobacterium wasabiae (strain CFBP-3304 and strain RNS08.42.1 A), Cedecea neteri strain ND14b and Citrobacter amalonaticus strain Y19 (Fig. S1).
Conjugative pilus
It is known that type IVa secretion systems elaborate pili to establish a host contact for substrate secretion or bacterial conjugation12. In order to confirm that strain Marseille-P1025 elaborates a conjugative pilus, electron microscopy was performed on cells after 24 h of incubation. Figure 4 shows that strain Marseille-P1025 possesses a conjugative pilus.

Transmission electron microscopy of R. ornithinolytica strain Marseille-P1025 using a Morgagni 268D transmission electron microscope (Philips) at an operating voltage of 60 kV. The scale bar represents 2 µm.
Interaction of R. ornithinolytica with A. castellanii trophozoites
Acanthamoeba castellanii is a free-living amoeba that has previously been used as a eukaryote model to study the virulence of pathogenic microorganisms, including Acinetobacter baumannii, mycobacteria and streptococci13,14,15,16. To determine whether strain Marseille-P1025 can multiply in eukaryotic cells, triplicate co-culture assays were performed with Acanthamoeba castellanii amoebae. Raoultella ornithinolytica strain P2310, isolated from the feces of a healthy individual, was used as a control for this experiment (Figs 5 and 6). We observed that the numbers of both uninfected and infected A. castellanii trophozoites incubated into PAS at 32 °C decreased over time. However, the mean percentages of remaining live amoebae at day 3 were 45.0+/−1.33%, 19.69+/−1.44% and 27.83+/−4.82% for uninfected amoebae, amoebae infected with strain P2310 and amoebae infected with strain Marseille-P1025, respectively. Therefore, the number of infected amoebae decreased significantly more than those of uninfected amoebae (p < 0.05) in presence of both R. ornithinolytica strains (Figs 5A, S2), and strain Marseille-P1025 caused a higher amoebal mortality than strain P2310, although this difference was not statistically different (p = 0.17).

Co-culture of R. ornithinolytica and A. castellanii amoebae. (A) Rate multiplication of Raoultella ornithinolytica strains P2310 and Marseille-P1025 within A. castellanii in PAS at 32 °C. (B) Percentage of live A. castellanii infected with R. ornithinolytica strains P2310 and Marseille-P1025. Each bar represents the mean of triplicate wells, and the standard errors are represented by error bars. *P < 0.05.

Optical microscopy observation of A. castellanii trophozoites infected with R. ornithinolytica strain Marseille-P1025 and stained with the Gimenez staining. The presence of R. ornithinolytica was monitored for 3 days: (A) day 0, co-culture after 5 hours of incubation; (B) day1, after 24 hours of incubation; (C) day 2, after 48 hours of incubation; (D) day 3, after 72 hours of incubation.
We also evaluated the numbers of CFUs obtained from intra-amoebal bacteria at H0 and H72 of co-culture (Figs 5B, S2). At H0 and H72, a mean 3.57 × 105 CFUs/mL and 9.67 × 107 CFUs/mL, respectively, were cultivated for strain Marseille-P1025 versus 5.73 × 105 CFUs/mL and 9.33 × 107 CFUs/mL, respectively, for strain P2310. The growth rate of both strains (270+/−97.5 and 174+/−29.3, respectively) was significantly higher for strain Marseille-P1025 (p < 0.05). Hence, these experiment demonstrated that strain Marseille-P1025 exhibited a higher pathogenicity for amoebae than the control strain (Fig. 5B). To confirm these observations, we examined bacteria within amoebae by Gimenez staining. Optical microscopy observations were consistent with the CFU evaluations. We observed that after 5 h of co-culture, most A. castellanii cells were infected by R. ornithinolytica strain Marseille-P1025 (Fig. 6A). Not only was R. ornithinolytica strain Marseille-P1025 able to survive within A. castellanii, but it began to multiply after 24 hours of co-culture (Fig. 6B). At day 2 of co-culture, strain Marseille-P1025 continued to multiply within amoebae. Furthermore, at day 3 of co-culture, infected amoebae started to lyse (Fig. 6D) whereas strain Marseille-P1025 kept multiplying. The lysis of A. castellanii amoebae was complete after 5 days of co-culture with bacteria (Fig. S3). We also observed that R. ornithinolytica survived in PAS medium without amoebae but did not multiply from day 0 to day 3.
Discussion
Infections due to R. ornithinolytica are under-reported, possibly because this bacterium is difficult to identify using conventional phenotypic methods17. Raoultella ornithinolytica is currently regarded as an emerging hospital-acquired infection agent, particularly after invasive procedure10. Few pathogenic factors are recognized in R. ornithinolytica compared to other members of the family Enterobacteriaceae10. These include the ability to adhere to human tissues, to form biofilms in urinary catheters and to convert histidine to histamine in scombroid fishes, thus causing redness and flushing of the skin10.
By comparing the genome of strain Marseille-P1025 that had caused a chronic prosthetic joint infection in an immunocompetent patient, to those of other R. ornithinolytica strains, we identified a unique 11-kb genomic island (RoGI) among R. ornithinolytica strains. This genomic island coded nine proteins from the type IVa secretion system (SS) (Fig. 2, Table 2), four proteins related to bacterial conjugation and two integrases. In addition, the genome from strain Marseille-P1025 contained another four components of the type IVa SS, which suggests that this system was complete. The presence of a conjugative pili and the ability of strain Marseille-P1025 to invade, survive and multiply in an amoeba (Acanthamoeba castellanii) confirms the presence of type IVa secretion system.
The type IVa SS is involved in various mechanisms of bacterial pathogenesis such as the transfer of Agrobacterium tumefaciens oncogenic DNA into plants leading to tumours18. The Type IVa SS is also involved in conjugation and thus plays a crucial role in genomic plasticity, notably by enabling the transfer of plasmids carrying antibiotic resistance or virulence genes among pathogenic bacteria19. In addition, conjugation systems may also contribute significantly to the development of infections by promoting surface pili adhesion-mediated attachment, colonization and biofilm formation20. It is also reported that the type IVa secretion system, particularly the virB operon, is essential for survival and intracellular multiplication21. Siddiqui et al. have shown that pathogenic bacteria, but not those who are weakly or not pathogenic, can survive within A. castellanii cysts16. The ability of R. ornithinolytica to grow and cause cytopathic effect in A. castellanii seems to be correlated with its virulence14,15. We demonstrated that strain Marseille-P1025 was not only able to survive within amoebae but could also multiply more efficiently than the control strain and kill amoebae, supporting its virulent behavior.
We also demonstrated that the RoGI genomic island of strain Marseille-P1025 was closely related to sequences from the Pectobacterium atrosepticum strain JG10-08, Pectobacterium sp. strain SCC3193, two Pectobacterium wasabiae (strains CFBP-3304 and RNS08.42.1A), Cedecea neteri strain ND14b and Citrobacter amalonaticus strain Y19 (Fig. S1). As the genomes of P. wasabiae strains CFBP-3304 and RNS08.42.1 A, P. atrosepticum strain JG10-08 and Pectobacterium sp. strain SCC3193 exhibit genomic G + C contents of 50.6%, 50.4%, 51.1% and 50.4%, respectively, which is closer to that of the RoGI island (49.5%) than that of strain Marseille-P1025 (55.6%), we assume that it may have been transferred from a Pectobacterium or a phylogenetically-close species. Pectobacterium species (P. wasabiae and P. atrosepticum) are phytopathogens22,23 that belong to the Enterobacteriaceae family like R. ornithinolytica.
We also detected the presence of three specific genes carried by the CP4-57 prophage, including two integrases (intA) and alpA, a transcriptional regulator of intA24. The IntA integrase has been shown to intervene in biofilm formation. In E. coli, the deletion of the intA gene reduces early biofilm formation24 whereas the increased synthesis of IntA leads to excision of the CP4–57 prophage24 which, in turn, increases biofilm formation24. Therefore, IntA may play a role in biofilm formation in strain Marseille-P1025, thus facilitating its adhesion to foreign material such as the patient’s joint prosthesis.
In conclusion, R. ornithinolytica strain Marseille-P1025, that caused a rare case of chronic prosthetic joint infection in a 67-year-old immunocompetent male, exhibited a complete type IVa secretion system that was unique among R. ornithinolytica strains and was able to infect, multiply within, and kill amoebae. These properties may explain its particular virulence. In addition, this type IVa SS was mostly coded by a genomic island (RoGI) that was probably acquired by lateral gene transfer from Pectobacterium species.
Material and Methods
DNA extraction and Genome sequencing
Strain Marseille-P1025 was cultivated on Columbia agar (bioMérieux, Marcy-l’Etoile, France) at 37 °C in aerobic atmosphere for 24 hours. Then, after a pre-treatment with lysozyme at 37 °C for 2 hours, the DNA was extracted using an EZ1 biorobot and the EZ1 DNA tissue kit (Qiagen, hilden, Germany). The elution volume was 50 µL. Genomic DNA (gDNA) was quantified by a Qubit assay with the high sensitivity kit (Life technologies, Carlsbad, CA, USA) to 8 ng/µl, prior to being sequenced on a MiSeq sequencer (Illumina, San Diego CA, USA) with the Paired-End and barcode strategy in order to be mixed with 20 other projects constructed according to the Nextera XT library kit (Illumina).
One ng of gDNA was used as input and tagmented for the fragmentation step. Then, limited cycle PCR amplification completed the tag adapters and introduced dual-index barcodes. The libraries were then normalized on specific beads according to the Nextera XT protocol (Illumina), pooled into a single library and then loaded onto the reagent cartridge. Automated cluster generation and Paired-End sequencing with dual index reads was performed in a single 39-hour run in a 2 × 251-bp.
Total information of 9.8 Gb was obtained from a 1,165 K/mm 2 cluster density with 88% (18,993,000 clusters) of the clusters passing quality control filters. Within this pooled run, the index representation of R. ornithinolytica strain Marseille-P1025 was determined to be 5.51%. The 1,046,713 Paired-End reads were filtered according to the read qualities.
Genome annotation and comparison
The sequencing reads were assembled using the A5 assembler25. Then, a step of finishing was done using the Mauve software26 and CLC bioserver. After assembly and finishing, the genome size was 5.6 Mb. Open reading frames (ORFs) were predicted using the Prodigal tool (http://prodigal.ornl.gov) with defaults parameters. The prediction of protein function was performed by searching against the GenBank database using BLASTP algorithm27. Functional classification of gene families (COG ID and Letters) was obtained using COGnitor against the COG database28. tRNAs and rRNAs were detected using tRNAscan-SE v.1.2129 and RNAmmer v.1.230, respectively. The presence or absence of plasmids was verified both by searching the gene annotation for any plasmid-related gene and by mapping all contigs against previously published Raoultella sp. plasmid sequences.
We compared the genome sequence of R. ornithinolytica strain Marseille-P1025 to those of other strains of this species found in public databases. As of August 30th, 2016 13 R. ornithinolytica genomes were available in public databases. Of these, we used 12 genomes for comparative analysis and excluded that of strain S12 due to its insufficient quality. The twelve comparator genomes were those from strains 10-5246 (AGDM00000000), NBRC 105727 (BCYR00000000), B6 (CP004142), A14 (CP008886), CMUL058 (CVRH00000000), TNT (JHQH00000000), 2-156-04_S1_C1 (JNPC00000000), 2-156-04_S1_C2 (JNPD00000000), 811_RORN (JURX00000000), BAL286 (JXXF00000000), CB1 (LFBW00000000) and Yangling I2 (CP013338). All genomes were re-annotated using the Prokka software, version 1.1131. Comparisons between all selected genomes were done using Roary, a tool that rapidly builds large-scale pangenomes32, with a blast identity cut-off of 97% for the comparison between R. ornithinolytica species. In addition, Roary identified the specific and missing genes from strain Marseille-P1025. Specific genes were checked by BLASTP and TBLASTN against the other studied genomes. Missing genes were checked by TBLASTN against the genome of strain Marseille-P1025, using a coverage and identity of 60% and 80% as thresholds, respectively, as described by Kuenne et al.33,34. Easyfig.35 was used to visualise the coding regions and colour the specific genes of strain Marseille-P1025.
Recombination and Phylogenetic analysis
The genome of strain Marseille-P1025 was used as a reference for whole-genome alignment36 using Mugsy37. Then, a phylogenetic tree based on whole genome sequence alignment was done using the FastTree software38 and the maximum likelihood method (Fig. 1). ClonalFrameML was used to search recombination hotspots in R. ornithinolytica genomes by analyzing both the whole genome alignment and the phylogenetic tree39.
Unique sequences were detected by a BLASTN search for homologous sequences and multiple sequence alignment using the Mafft software algorithm40. Phylogenetic analysis of these unique sequences was performed using MEGA version 741 and the maximum likelihood (ML) algorithm, with 1,000 bootstrap replicates.
Electron microscopy
Electron microscopy was performed with detection Formvar coated grids. Forty 40 μL of bacterial suspension were deposited on a grid and incubated at 37 °C for 30 min, followed by a 10 sec incubation on ammonium molybdate 1%. Grids were then observed using a Morgagni 268D transmission electron microscope (Philips) at an operating voltage of 60 kV.
Culture of R. ornithinolytica and A. castellanii
Raoultella ornithinolytica strain P2310, isolated from the feces of a healthy individual, was used as a control in co-culture experiments. Raoutella ornithinolytica strains Marseille-P1025 and P2310 were grown on 5% sheep blood-enriched Columbia agar (BioMérieux) at 35 °C for 24 hours in anaerobic atmosphere. Bacteria were then harvested, centrifuged at 4,000 × g during 5 minutes, washed twice and suspended in Page’s modified Neff’s amoeba saline (PAS). The PAS medium was prepared as follows: solution A (for 100 mL of sterile distilled water), 1.2 g NaCl + 0.04 g MgSO4.7H2O + 1.42 g Na2HPO4 + 1.36 g KH2PO4; solution B (for 100 mL of sterile distilled water), 0.04 g CaCl2.2H2O; PAS solution −10 mL of solution A + 10 mL of solution B + 980 mL of sterile distilled water). The inoculum density was determined by the McFarland method.
Acanthamoeba castellanii strain Neff (ATCC 30010) was grown in 175 cm² culture flasks containing 30 mL peptone-yeast extract-glucose (PYG) at 28 °C. When a monolayer was formed, A. castellanii trophozoites were harvested by shaking the flasks and centrifuged at 500 × g for 10 min. The pellet was suspended in 30 mL PAS medium. The quantification of the A. castellanii population was performed using a KOVA® slide cell counting chamber.
Co-culture experiments
The amoebal trophozoite suspension (5 × 105 amoeba/mL) was inoculated in 24-well plates and allowed to adhere for 30 minutes at 32 °C. Then, R. ornithinolytica suspensions were inoculated on amoebae to achieve ratios of infection of 10 R. ornithinolytica cells/amoeba. As controls, A. castellanii and R. ornithinolytica strains were cultivated separately in PAS. After incubation for 2.5 h at 32 °C under a 5% CO2 atmosphere, the co-culture wells were washed three times with PAS to remove any remaining extracellular or adherent bacteria. Incubation at 32 °C under 5% CO2 was then performed for 3 days. The presence of viable Raoultella inside amoebal trophozoites was documented by sub-culturing at 0, 24, 48 and 72 h of incubation. For each time point, the A. castellanii monolayer from a well was lysed by three passages through a 25-gauge needle. Serial dilutions of the lysate were carried out, plated onto COS medium and incubated for 2 days at 32 °C under anaerobic atmosphere to determine the numbers of intracellular R. ornithinolytica colony forming units (CFU). Multiplication rate of the bacterial invasion was calculated as follows: recovered R. ornithinolytica (CFU)/R. ornithinolytica (CFU) at time 0. The A. castellanii population was also monitored during the 3-day experiment: counting and viability check of amoebae, cultivated alone and in co-culture, was performed using KOVA® slides after Trypan Blue 0.4% coloration (Sigma-Aldrich, Taufkirchen, Germany). All experiments were reproduced three times, each time in duplicate. The standard error of the mean (SEM) was used to evaluate the experiment value distribution. To compare the intra-amoebal growth of the two tested bacterial strains, we also estimated the dayly multiplication rate of bacteria.
The presence of R. ornithinolytica within amoebae was also monitored for 5 days by Gimenez staining42. The observation was performed with a LEICA DM 2500 LED microscope.
Statistical analyses
Statistical analyses mentioned in this study were performed using the Student’s t-test and Chi-square test, with a significance level of P inferior or equal to 0.05.
Nucleotide sequence accession numbers
The genome sequence from R. ornithinolytica strain Marseille-P1025 was deposited in GenBank under accession number FTLF01000000.
References
Drancourt, M., Bollet, C., Carta, A. & Rousselier, P. Phylogenetic analyses of Klebsiella species delineate Klebsiella and Raoultella gen. nov., with description of Raoultella ornithinolytica comb. nov., Raoultella terrigena comb. nov. and Raoultella planticola comb. nov. Int. J. Syst. Evol. Microbiol. 51, 925–932 (2001).
Kanki, M., Yoda, T., Tsukamoto, T. & Shibata, T. Klebsiella pneumoniae produces no histamine: Raoultella planticola and Raoultella ornithinolytica strains are histamine producers. Appl. Environ. Microbiol. 68, 3462–3466 (2002).
Hadano, Y. et al. Raoultella ornithinolytica bacteremia in cancer patients: report of three cases. Intern. Med. 51, 3193–3195 (2012).
Haruki, Y. et al. Clinical characteristics of Raoultella ornithinolytica bacteremia: A case series and literature review. J. Infect. Chemother. 20, 589–591 (2014).
Mau, N. & Ross, L. A. Raoultella ornithinolytica bacteremia in an infant with visceral heterotaxy. Pediatr. Infect. Dis. J. 29, 477–478 (2010).
Chun, S., Yun, J. W., Huh, H. J. & Lee, N. Y. Clinical characteristics of Raoultella ornithinolytica bacteremia. Infection 43, 59–64 (2014).
Sibanda, M. Primary peritonitis caused by Raoultella ornithinolytica in a 53‐year‐old man. JMM Case Rep. 1, (2014).
Cleveland, K. O., Mazumder, S. A. & Gelfand, M. S. Association of Raoultella bacteremia with diseases of the biliary tract. Scand. J. Infect. Dis. 46, 541–542 (2014).
Jong, Ede et al. Predominant association of Raoultella bacteremia with diseases of the biliary tract. Scand. J. Infect. Dis. 46, 141–143 (2014).
Seng, P. et al. Emerging role of Raoultella ornithinolytica in human infections: a series of cases and review of the literature. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 45, 65–71 (2016).
Seng, P. et al. Raoultella ornithinolytica: An unusual pathogen for prosthetic joint infection. IDCases 5, 46–48 (2016).
Darbari, V. C. & Waksman, G. Structural biology of bacterial type IV secretion systems. Annu. Rev. Biochem. 84, 603–629 (2015).
Da Silva, J. L., Nguyen, J., Fennelly, K. P., Zelazny, A. M. & Olivier, K. N. Survival of pathogenic Mycobacterium abscessus subsp. massiliensein Acanthamoeba castellanii. Res. Microbiol. 169, 56–60 (2018).
Tamang, M. D., Kim, S., Kim, S.-M., Kong, H.-H. & Kim, J. Interaction of Acinetobacter baumannii 19606 and 1656-2 with Acanthamoeba castellanii. J. Microbiol. 49, 841–846 (2011).
Goy, G. et al. The Neff strain of Acanthamoeba castellanii, a tool for testing the virulence of Mycobacterium kansasii. Res. Microbiol. 158, 393–397 (2007).
Siddiqui, R., Lakhundi, S. & Khan, N. A. Interactions of Pseudomonas aeruginosa and Corynebacterium spp. with non-phagocytic brain microvascular endothelial cells and phagocytic Acanthamoeba castellanii. Parasitol. Res. 114, 2349–2356 (2015).
Park, J. S. et al. Evaluation of three phenotypic identification systems for clinical isolates of Raoultella ornithinolytica. J. Med. Microbiol. 60, 492–499 (2011).
McCullen, C. A. & Binns, A. N. Agrobacterium tumefaciens and plant cell interactions and activities required for interkingdom macromolecular transfer. Annu. Rev. Cell Dev. Biol. 22, 101–127 (2006).
Juhas, M. et al. Novel type IV secretion system involved in propagation of genomic islands. J. Bacteriol. 189, 761–771 (2007).
Gonzalez-Rivera, C., Bhatty, M. & Christie, P. J. Mechanism and function of type IV secretion during infection of the human host. Microbiol. Spectr. 4, (2016).
Sieira, R., Comerci, D. J., Sánchez, D. O. & Ugalde, R. A. A homologue of an operon required for DNA transfer in Agrobacterium is required in Brucella abortus for virulence and intracellular multiplication. J. Bacteriol. 182, 4849–4855 (2000).
Nykyri, J. et al. Revised phylogeny and novel horizontally acquired virulence determinants of the model soft rot phytopathogen Pectobacterium wasabiae SCC3193. PLoS Pathog. 8, (2012).
De Boer, S. H., Li, X. & Ward, L. J. Pectobacterium spp. associated with bacterial stem rot syndrome of potato in Canada. Phytopathology 102, 937–947 (2012).
Wang, X., Kim, Y. & Wood, T. K. Control and benefits of CP4-57 prophage excision in Escherichia coli biofilms. ISME J. 3, 1164–1179 (2009).
Tritt, A., Eisen, J. A., Facciotti, M. T. & Darling, A. E. An Integrated pipeline for de novo assembly of microbial genomes. PLoS ONE 7, e42304 (2012).
Darling, A. C. E., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004).
Altschul, S. F. BLAST Algorithm. in eLS(ed. John Wiley & Sons Ltd). https://doi.org/10.1002/9780470015902.a0005253.pub2 (John Wiley & Sons, Ltd, 2014).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 0955–0964 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinforma. Oxf. Engl. https://doi.org/10.1093/bioinformatics/btv421 (2015).
Rychli, K. et al. Genome sequencing of Listeria monocytogenes ‘Quargel’ listeriosis outbreak strains reveals two different strains with distinct in vitro virulence potential. PLoS ONE 9, (2014).
Kuenne, C. et al. Reassessment of the Listeria monocytogenes pan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Genomics 14, 47 (2013).
Sullivan, M. J., Petty, N. K. & Beatson, S. A. Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010 (2011).
Maiden, M. C. J. et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736 (2013).
Angiuoli, S. V. & Salzberg, S. L. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 27, 334–342 (2011).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009).
Didelot, X. & Wilson, D. J. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11, (2015).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Giménez, D. F. Staining rickettsiae in yolk-sac cultures. Stain Technol. 39, 135–140 (1964).
Acknowledgements
This study was funded by the Mediterranee Infection foundation and the French Agence Nationale de la Recherche under reference Investissements d’Avenir Méditerranée Infection 10-IAHU-03.
Author information
Authors and Affiliations
Contributions
Mamadou Beye wrote the manuscript and performed the phenotypic and genomic analyses; Issam Hasni performed the amoebal infection experiments and imaging analyses and edited the manuscript; Piseth Seng took care of the patient and edited the manuscript; Caroline Michelle performed the genomic sequencing and, assembly and edited the manuscript; Bernard La Scola performed the amoebal infection experiments and edited the manuscript; Didier Raoult designed the study, analysed the data and wrote the manuscript; Pierre-Edouard Fournier designed the study, analysed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beye, M., Hasni, I., Seng, P. et al. Genomic analysis of a Raoultella ornithinolytica strain causing prosthetic joint infection in an immunocompetent patient. Sci Rep 8, 9462 (2018). https://doi.org/10.1038/s41598-018-27833-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27833-z
This article is cited by
-
A case report of community-acquired Raoultella ornithinolytica infection in a healthy, young individual
BMC Infectious Diseases (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
