
Abstract
In the era of precision medicine, cancer therapy can be tailored to an individual patient based on the genomic profile of a tumour. Despite the ever-increasing abundance of cancer genomic data, linking mutation profiles to drug efficacy remains a challenge. Herein, we report Cancer Drug Response profile scan (CDRscan) a novel deep learning model that predicts anticancer drug responsiveness based on a large-scale drug screening assay data encompassing genomic profiles of 787 human cancer cell lines and structural profiles of 244 drugs. CDRscan employs a two-step convolution architecture, where the genomic mutational fingerprints of cell lines and the molecular fingerprints of drugs are processed individually, then merged by ‘virtual docking’, an in silico modelling of drug treatment. Analysis of the goodness-of-fit between observed and predicted drug response revealed a high prediction accuracy of CDRscan (R2 > 0.84; AUROC > 0.98). We applied CDRscan to 1,487 approved drugs and identified 14 oncology and 23 non-oncology drugs having new potential cancer indications. This, to our knowledge, is the first-time application of a deep learning model in predicting the feasibility of drug repurposing. By further clinical validation, CDRscan is expected to allow selection of the most effective anticancer drugs for the genomic profile of the individual patient.
Similar content being viewed by others


Introduction
Over the past two decades, we have witnessed remarkable progress in understanding the complexity of genomic landscape of cancer. Exhaustive catalogues of somatic mutations of various cancer types have been created1,2, and major cancer-causing mutations have been identified3. It is not surprising that the expectation towards tailoring cancer treatments to a particular genomic signature of individual tumours is growing rapidly; yet, the current rate of new cancer agents to be approved and used clinically is deemed unsustainable for all stakeholders of healthcare, including cancer patients and pharmaceutical industry4. An efficient and systematic approach to evaluate the link between genomic information and the response of anticancer agents is very much needed.
Several collaborative efforts have been made to catalogue molecular profiling data of cancer cell lines and drug sensitivity data5,6,7 aimed at identifying genomic biomarkers predictive of anticancer drug response. Genomics in Drug Sensitivity in Cancer6 (GDSC, https://www.cancerrxgene.org) is an example of such publicly available databases providing experimentally measured drug sensitivities of 1,001 human cancer cells against 265 anticancer compounds6. Importantly, the molecular profiles of the entire cancer cell lines used in GDSC6 were extensively characterised as a part of COSMIC cell line project1 (CCLP, https://cancer.sanger.ac.uk/cell_lines), including profiling of somatic genomic alterations. These resources are expected to bring great benefit to the realisation of genomic-driven precision cancer medicine. Despite the potential value of such databases, the high-dimensionality and complexity of the datasets poses problems for integrative analysis. A number of approaches have been previously devised for the systematic identification of molecular markers in anticancer drug sensitivity using various machine learning methods. Examples of the methods include regularised regression, elastic net, random forests, kernel based methods, and/or shallow neural network5,8,9,10,11,12,13,14,15,16. However, these methods have their prediction accuracy at a moderate level at best and are often challenged by large-scale and highly intricate genomic data.
An increasingly applied method to enhance the prediction accuracy and to address the technical challenges is a deep learning model, which has received a great deal of attention with recent advances in information technology17. The deep learning method is a branch of machine learning techniques similar to shallow neural network but with multiple hidden layers and more complex parameters used in training. The method enables high level abstraction from a large volume of heterogeneous and high-dimensional raw datasets18. Until recently, the efficacy of learning was directly limited to the availability of relevant data19. Nevertheless, with a methodological improvement and a powerful machine with parallel computing horsepower, a deep learning model can be trained with multiple hidden layers, containing thousands of hidden units20,21. Since it can operate several types of structural information, such as pharmacological, genomic, and transcriptomic data, it is suitable for predicting drug-target interaction with minimal guidance17.
The pharmaceutical industry has begun showing its vested interest in deep learning to exploit these types of data for drug discovery22. Recently, several promising results have been demonstrated using deep learning in drug development23,24,25,26, drug-target profiling27 and drug repurposing/repositioning (i.e., identification of potential new purposes of approved or investigational drugs) with superior prediction accuracy compared to other conventional machine learning models28,29. Nevertheless, majority of the deep learning-based drug development focuses on the prediction of drug-target interaction, which is based on molecular structures, and few studies take genomics into consideration in developing their deep learning models13.
In this study, we have developed the Cancer Drug Response profile scan (CDRscan), a cancer genomic landscape-guided drug response prediction algorithm. By employing a novel dual convergence architecture deep learning model run on accelerated computing, and incorporating comprehensive drug response assay datasets obtained from CCLP1 and GDSC6 databases, the prediction accuracy of CDRscan was further enhanced over that of the previous computational modelling approaches. This accurate and robust drug response prediction model represents an important milestone for the realisation of precision cancer medicine through its application in the drug development processes such as drug repurposing and screening small chemical libraries for new anticancer drug candidate. In the clinical settings, CDRscan is expected to streamline patient-tailored anticancer drug selection as a clinical decision support system with further clinical validation studies.
Results
Overview of CDRscan and structure of datasets
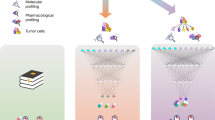
CDRscan is an ensemble of five convolutional neural network (CNN)-based models30 with varied architectures (Table 1, Supplementary Fig. S1, Supplementary Table S1). Each model predicts the half-maximal inhibitory concentration (IC50) values of anticancer compounds from the genomic signature of tumour samples, and the mean is reported as the final prediction value. Input datasets were obtained from two perhaps the most comprehensive public databases related to cancer: CCLP for genomic profiles of human cancer cell lines1; and GDSC for anticancer drug sensitivity assays6 (Fig. 1a). The entire datasets from the databases contained 686,312 mutation positions from 1,001 cell lines and 265 drugs (Fig. 1b), covering 30 cancer types as defined by The Cancer Genome Atlas (TCGA) studies (https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations). We curated a subset of the data to include only gene mutations contained in Cancer Gene Census1,31 (CGC, https://cancer.sanger.ac.uk/census), which is a catalogue of 567 genes strongly associated with cancer pathology (Fig. 1b, Table 2). In addition, the datasets were further screened to exclude the followings: (1) cancer types that were represented by fewer than 10 different cell lines (Table 2, Supplementary Table S2); (2) drugs without PubChem Compound Identifier; (3) drugs with molecular weight greater than 1000 g/mol (Supplementary Table S3, see Methods for more details on exclusion criteria). The final datasets yielded a total of 152,594 instances which contained 787 cell lines across 25 TCGA cancer types, mutation information at 28,328 base positions in 567 genes, and IC50 measurements of cell line-drug treatment in 244 drugs (Table 2, Supplementary Tables S2, S3).

Overview of Cancer Drug Response profile scan (CDRscan). (a) Two main applications of CDRscan and dataset structure. For any given genomic fingerprint (i.e., a list of somatic mutations) of a tumour, CDRscan predicts which of 244 Genomics in Drug Sensitivity in Cancer (GDSC) anticancer drugs would be effective. The input of CDRscan can be molecular information of a particular small molecule for which CDRscan reports the predicted sensitivity of 787 cancer cell lines. The datasets used to train CDRscan were extracted from COSMIC cell line project (CCLP) and GDSC databases which represent 787 cancer cell lines across 25 cancer types defined by TCGA, 28,328 mutation positions in 567 cancer associated genes, and assay results from treatment of 244 anticancer drugs. (b) Data filtering procedure and final datasets. CCLP and GDSC databases contain genomic characterisation of 1,001 cancer cell lines and IC50 values measured from treatment of 1,001 cell lines with 265 anticancer drugs. The datasets were refined to include only the 567 Cosmic Cancer Gene Census genes and the cancer types that have at least 10 cell lines. Drugs without PubChem Compound Identifier or having molecular weight greater than 1000 g/mol were excluded. Totals of 28,328 and 3,072 features were extracted from cell line genomic signatures and drugs, respectively, constituting binary encoding of 31,400 features in total. The graphical image used in Fig. 1a is an original creation by Ye-Bin Jung and is reprinted under a CC BY license with permission from Ye-Bin Jung. All rights reserved.
The input features of the entire instance were represented by 31,400 binary digits. Of these, 28,328 bits represented mutational status of 28,328 genomic positions in each of the 787 cell lines, while 3,072 bits encoded molecular profiles of the individual drugs generated by PaDEL-descriptors32 (Fig. 1b). To effectively process the two distinctive types of inputs, namely the genomic fingerprints of cancer cell lines and the molecular fingerprints of the drugs, a dual convergence architecture was employed in four models (‘master’, ‘fully connected’, ‘shallow’, and ‘tanh’). A series of convolutions was performed independently for each set, thereby generating ‘virtual tumour cells’ and ‘virtual drugs’, respectively. Subsequently, ‘virtual docking’ (i.e., in silico simulation of drug treatment to cells) took place to merge the two separate convoluted features, followed by additional rounds of convolution. In one of the five CDRscan models (‘unified’), a conventional approach was employed, where all 31,400 descriptors in the input layer were convoluted together as one entity. All five models generated predicted IC50 values across the 244 anticancer drugs for each virtual cell line as a final output layer of the models. The average of the five values predicted by each model was then reported as the final outcome of CDRscan.
Training of CDRscan and assessment of prediction accuracy
Of the total of 152,594 instances spanning 25 cancer types, 144,953 instances (i.e., compilation of randomly selected 95% of instances for each cancer type) were selected to train all five models of CDRscan. The remaining 7,641 instances, corresponding to 5% of the total instances, were set aside for evaluation of performance of the models (test set). We then examined the correlation between the observed IC50 values from GDSC6 and the values predicted by CDRscan using the test set. The observed and the predicted IC50 values showed a strong agreement with the mean coefficient of determination (R2) value of 0.843, ranging from 0.838 to 0.853 across five models (Fig. 2a,b, Supplementary Fig. S2). Among the five models, the ‘master’ model exhibited the highest R2 (0.853). The four models that employed the dual convergence architecture had higher R2 values than the ‘unified’ model (Fig. 2b, Supplementary Fig. S2). The averaged root mean squared error (RMSE) value of the five models was 1.069 (s.d. = 0.018, n = 5), confirming that the prediction was accurate in most instances (Fig. 2b, Supplementary Fig. S2). To further confirm the prediction accuracy of CDRscan, we assessed the area under the receiver operating characteristic curve (AUROC) in the test set (n = 7,641). The cell lines were classified as being sensitive against a drug when ln(IC50) <−2 (−2 corresponds to IC50 of approximately 0.135 µM), which was set as a cut-off value for AUROC. The AUROC score of 0.98 was obtained as a result (Supplementary Fig. S3).

Assessment of prediction accuracy of CDRscan. (a) Scatter plots showing correlation between the observed and predicted IC50 values for CDRscan and two other machine learning models to benchmark the prediction accuracy. The test datasets, which correspond to 5% of the total cell line-drug pairs, were used to assess the coefficient of determination (R2). (b) Table summarizing the R2 values and root mean squared errors (RMSE) of CDRscan (mean value of the five models and values for individual models), random forest, and support vector machine.
Across cancer types, a wide variation was demonstrated in the degree of mutation burden (Supplementary Fig. S4), in the total number of associated cell lines of the final datasets, and in the total number of mutation positions (Table 2). Nevertheless, when the performance of CDRscan was assessed against each cancer type, the predicted IC50 values for each of the 25 cancer types strongly agreed with the observed IC50 values across all models (Supplementary Fig. S5), wherein values of R2 ranged from 0.77 (chronic myelogenous leukaemia, LCML, n = 112) to 0.89 (lung squamous cell carcinoma, LUSC, n = 144) (Supplementary Fig. S5). Interestingly, ranks of 25 cancer types based on R2 values were identical across the five models (Supplementary Fig. S5). According to Pearson correlation analysis, we did not find any significant correlation between the prediction accuracy (i.e., R2 values) of individual cancer types and the average mutation burdens or the number of cell lines per cancer type (Supplementary Fig. S4).
Lastly, the R2 values of individual cell lines (785 in total) and drugs (244 in total) were assessed. Two cell lines were excluded in the assessment since their drug treatment data was available for only one or two compounds, the number of which was too small to derive reliable R2 values. Cell line-centric analysis revealed a high mean R2 of five models between the observed and the predicted IC50 values for most cell lines. Amongst all cell lines, BFTC-909 (kidney renal clear cell carcinoma, KIRC) showed the highest R2 value of 0.967 (n = 151), while COR-L32 (small cell lung cancer, SCLC) had the lowest at 0.779 (n = 30) (Fig. 3a, Supplementary Fig. S6). Consistent with the high R2 values, the predicted and observed IC50 values showed strong correlation across all cell lines with p values less than 2.86e−11 (Fig. 3a).

Cell line- and drug-centric correlation analyses. (a) Prediction accuracy assessment for each cell line. Scatter plots show the correlation between observed and CDRscan-predicted ln(IC50) values for the cell lines that showed the strongest (BFTC-909, left) and the lowest agreement (COR-L32, right). The COSMIC IDs of the two cell lines and the corresponding cancer types are indicated above the scatter plots, and the R2 values, Pearson correlation coefficient (r), p values, and the number of instances (n) are shown in the upper left corner of each plot. Histograms on the right show the overall distribution of prediction accuracy assessed for individual cell lines using indicated metrics. (b) Scatter plots showing the strongest and weakest agreement between observed and CDRscan-predicted ln(IC50) in drug-centric correlation analysis. The drug name and its PubCHEM ID are indicated in each plot. The R2 values, Pearson correlation coefficient (r), p values, and the instance counts (n) are also indicated. Histograms on the right show the overall distribution of prediction accuracy (R2) assessed for individual drugs using indicated metrics.
In the drug-centric correlation analysis, dasatinib (tyrosine kinase inhibitor) had the highest mean R2 of 0.902 (n = 288), and bicalutamide (androgen receptor inhibitor) had the lowest at −2.024 (n = 683) (Fig. 3b, Supplementary Fig. S7). Drugs that had negative mean R2 values were characterised by small degrees of variance in the drug activity and in most cases showed little activity [ln(IC50) >−2] across all assayed cell lines. For instance, the observed ln(IC50) values of the bicalutamide-treated cell lines, having the lowest R2, ranged from −0.293 to 4.661 (mean = 2.586, range = 4.954), while those of dasatinib, the drug that was ranked the highest based on R2, ranged from −6.875 to 5.052 (mean = 0.222, range = 11.927) (Fig. 3b, Supplementary Fig. S7). When the Pearson correlation coefficient (r) was computed for all the drugs, even the drugs with low R2 values showed relatively high r values with p values less than 3.86e−37 (Fig. 3b).
One of the previous drug response prediction models has used the datasets obtained from the same databases as in our study (CCLP1 and GDSC6), although there were differences in the total number of datasets and in how the genomic and drug features were expressed13. To address whether and to what degree the application of dual convergence architecture deep learning model can improve the prediction accuracy, we compared the performance of CDRscan and the previously developed prediction model13. For direct comparison, we re-evaluated the prediction accuracy of CDRscan using multi-fold cross validation (five-fold with each fold containing 29,001 instances). The resulting R2 values of the five independent models of CDRscan ranged from 0.843 to 0.852 (Supplementary Table S4, Supplementary Fig. S8), which were comparable to that from the validation using the 5% leave-out test set (R2 = 0.843, Fig. 2a,b, Supplementary Fig. S2). Using the same cross validation method, we confirmed that CDRscan exhibited the performance that is significantly higher than that of the previous model (R2 = 0.72)13. Since the advent of the previous CCLP/GDSC-based machine learning model13, there has been a substantial increase in the amount of GDSC data sets6, which may also have influence the performance of the prediction model in addition to the deep learning architecture. We thus trained two commonly used machine learning models, random forest (RF) and support vector machine (SVM), using the identical instances to train (n = 144,953) and evaluate (n = 7,641) CDRscan, and compared the performances across the three models. The R2 values of RF and SVM models were 0.698 and 0.562, respectively (Fig. 2a,b), showing that their prediction accuracy is significantly compromised compared to CDRscan.
Feasibility of drug repurposing using CDRscan
As a proof of concept in the drug repurposing potential of CDRscan, chemical descriptors of the approved drugs33 (see Methods for more details) were used to predict IC50 values of 787 cell lines. A total of 1,487 compounds were used in the analysis, and 102 of them were classified as oncology drugs according to National Cancer Institute34 (NCI, https://www.cancer.gov/about-cancer/treatment/drugs) (Fig. 4a). As in the AUROC analysis, ln(predicted IC50) < −2.0 was used to define positive anticancer drug response, which is a stringent criteria when compared to other similar studies35. Thirty seven of the 102 approved anticancer drugs had the potential for new caner type indications. In addition, 176 of 1,385 approved non-oncology drugs had the potential anticancer activities in addition to their original drug indications. However, some of the cancer types predicted as a new ‘purpose’ of the approved drugs contained only few drug responder cell lines. When those cancer types with only a few likely sensitive cell lines - less than 10% of the total cell lines showing predicted drug sensitivity - were excluded, the number of approved oncology drugs with repurposing potential was reduced from 37 to 23 (Fig. 4a). Nine of these 23 drugs had CDRscan-predicted anticancer activity against more than 90% (23/25) of the total types (Fig. 4a), suggesting a universal antiproliferative/cytotoxic activity of the compounds. Likewise, 27 of initially selected 176 non-oncology drugs (approximately 2% of 1,385 approved non-oncology drugs) showed strong predicted efficacy for at least one of the 25 cancer types, for which the predicted anticancer response was seen in at least 10% of the cell lines (Fig. 4b). Four of 27 drugs demonstrated strong predicted anticancer activity against more than 90% of all cancer types (Fig. 4b).

Feasibility of drug repurposing using CDRscan (a) Approved anticancer drugs with potential repurposing opportunity. CDRscan predicted that 23 out of 102 approved anticancer drugs have activity against at least one new cancer type in addition to the originally approved indications. Nine of these showed predictive sensitivity of more than 90% cancer types, indicating nonspecific antiproliferative/cytotoxicitc effects. (b) Approved non-oncology drugs with potential repurposing opportunity. Of the 1,385 non-oncology drugs, 27 showed potential anticancer activity. Four of these 27 drugs were predicted to have activity against over 90% of cancer types.
Discussion
Here we describe CDRscan, a dual convergence architecture deep learning model, which predicts somatic mutation profile-based drug responsiveness by linking the tumour genomic fingerprint and its sensitivity to drugs. When the fingerprints of drugs or any small molecules are provided, CDRscan predicts the IC50 values of the CCLP panel of 787 human cancer cell lines (Fig. 1a). From these predictions, the new use of known drugs (i.e., drug repurposing) or novel anticancer agents can be inferred. Two major innovative methods were employed for the design of CDRscan, namely the dual convergence architecture for deep learning and the employment of five independent models. In the former, the cell line genomic fingerprint and the molecular fingerprint of drugs were modelled separately since they contained different types of information, and then the two were merged into one virtual docking model. With the latter, the robustness of the CDRscan was further enhanced by reporting the mean prediction values from multiple models. Of the five individual models of CDRscan, two models, ‘master’ and ‘fully connected’, performed better than the final ensemble model (i.e., mean prediction values). Nevertheless, by reporting the mean value from the ensemble of models, generalisability of CDRscan is ensured by minimizing the artefact that can be derived from a particular model architecture.
It has been shown that the best predictive outcome for estimating drug sensitivity can be obtained using gene expression data36, which has been taken into consideration for most of conventional computational modelling approaches37. However, genomic profile-based drug response prediction algorithms are in greater demand than of the gene expression-based algorithms, since genomic sequencing of a tumour sample is more commonly performed in clinics and for drug development. For instance, genetic testing for the detection of known biomarkers for targeted therapy is a routine clinical practice, and the majority of molecularly targeted cancer drugs are developed against cancer-specific mutant proteins38. The accurate prognosis and prediction of potential relapse by time series genomic analysis of cell free DNA may become a common practice in near future although it is currently costly and technically challenging39,40. We thus reasoned that development of a prediction model utilising genomic profile data including somatic mutations would have a wider array of practical application than a model using only gene expression profiles. Consistently high R2 values across all 25 cancer types in all five models strongly suggest that indeed the somatic mutation signatures have a significant influence on the responsiveness of anticancer drugs. Taken together, our model not only demonstrates high predictive accuracy and robustness, but also supports practicality for future applications.
Identification of new lead molecules for the treatment of diseases, including cancer therapies, is often the result of more than a decade of dedicated efforts from many research groups. Of these painstakingly selected drug candidates, only 5% finally make it to the market41. One solution to this issue is to use already approved drugs for new disease indications. By significantly reducing the time and cost associated with drug development42, drug repurposing benefits both patients and pharmaceutical industry. Despite the promise this method of drug discovery offers, currently there are limited success in repurposing drugs as anticancer agents mainly due to the lack of a systematic approach43. We propose that CDRscan can be an effective tool for this purpose by enabling high throughput in silico screening of drugs against their anticancer activity. When the chemical features of commercially available drugs are provided to CDRscan a subset of the 787 CCLP cell lines with potential sensitivity to the input drugs as well as the corresponding cancer types can be reported (Fig. 1a). The newly predicted cancer types represent a potential new indication for already approved drugs. Notably, CDRscan identified 37 approved drugs (2.5% of total 1,487 approved small molecule drugs in all therapeutic area) with potential new cancer indications. This number may be an underestimation since a stringent set of criteria were applied in our study to operationally define a drug with potential repurposing opportunity: (1) ln(IC50) <−2 for anticancer response; (2) 10% or more cell lines within the cancer type to have the predicted drug response; (3) exclusion of the drugs with pan-cancer type antiproliferative/cytotoxic effect. In vitro and/or clinical evidence supported a subset of the new, CDRscan-predicted indications of multiple oncology drugs (e.g., belinostat44,45,46,47, cabozantinib48, cobimetinib49, etc.) and non-oncology drugs (e.g., pravastatin50,51,52 and ouabain53,54,55). Notably, many of the potentially repurposable drugs with supporting evidence were not in GDSC (i.e., outside the CDRscan training set), strongly arguing for the prediction accuracy as well as the practicality of CDRscan as a systematic in silico screening method for drug repurposing. Although the predicted results require in vitro validation prior to subsequent clinical studies, our results offer a realistic opportunity in drug repurposing. It should also be emphasised that CDRscan can be utilised to screen for new candidates of lead molecules when the chemical features of small compounds from chemical libraries such as the ZINC56,57 are used as an input instead of limiting the input to approved drugs.
It is important to note that CDRscan predicts a drug response at the level of cancer cell lines, not of the cancer types. We considered the cancer types as new indications when they contained the susceptible cell lines with ln(predicted IC50) less than −2. This criteria was chosen for a practical reason since the current guideline for anticancer drug treatment is primarily based on the site of a tumour22. However, one should keep in mind that the genomic fingerprints of the predicted drug responder cell lines may not be truly representative of the cancer type that they belong to. It is important to evaluate how frequently drug response-predictive mutation patterns appear between the genomes of various cancer types. In a hypothetical scenario where a predictive mutational pattern is present in many cancer patients but independent of cancer types, new indication of the drug to be repurposed should be determined by genomic biomarkers, not by cancer types. Ideally, the identification of a minimal number of mutations that appear specifically in sensitive cell lines irrespective of cancer types would represent a good candidate of ‘genomic biomarker’ for indication. In line with this, for the first time, the U.S. Food and Drug Administration has recently approved Keytruda (pembrolizumab) for tumours with a certain genetic biomarker regardless of their origin58. The drug response-associated genomic markers have already been included in drug labels to serve as guidelines for drug prescription, and the genomic marker based approval of anticancer drug is expected to become a more common practice. The rise in the number of companion diagnostics for detecting specific genomic biomarkers is also expected to gain momentum.
Recent analyses have reported that only a limited number of cancer patients would benefit from genetic screening to select targeted drugs using known biomarkers, since those with positive biomarkers are rare59,60. Even among patients with a well-defined biomarker for targeted therapies, the expected clinical outcome is often lost due to prevalent crosstalk between genes. In the clinical settings, CDRscan can be applied to screen for the drugs with the likely effectiveness for individual tumours. As CDRscan predicts drug sensitivity from genomic fingerprints rather than individual mutation, it further broadens the use of genomic screening results in the identification of effective drugs. Eventually, CDRscan would facilitate individually tailored cancer treatment, guiding clinical decision in the selection of anticancer agent(s) that best suits the genomic signature of the individual patients.
Challenges still remain for routine application of CDRscan. Despite its strong predictive accuracy, the model was built on in vitro data. Cancer cell lines of CCLP1 and GDSC6 represent cancer models that are extensively used in the cancer research community33,61,62,63. While those databases archive a large collection of cancer cell lines across a broad spectrum of cancer types, it remains to be determined whether these cell lines truly represent tumours that are growing in vivo41,64. Cancer cell lines are derived from patient tumour samples, but they are homogenous, subject to additional mutational events, and not surrounded by microenvironment. Thus, further in vivo validation of the predictions made by CDRscan is a mandatory next step. Especially, given that CDRscan is developed using selected mutation positions significant for cancer (28,328 positions from 567 genes), mutations occurring outside this range is not used as inputs for genomic fingerprints. Expanding the input positions by coupling it with a solid analysis for clinical validation can be done in the future by using genomic sequence data of cancer patients receiving chemotherapy. In recent years, patient-derived organoids65 and xenograft mice66 have become widely accepted platforms for drug screening in cancer research. These new platforms open up additional avenues for validating the performance of CDRscan.
By incorporating additional data types into computational models, the predictive prowess can be further strengthened15,33. Among several types of molecular profile data, transcriptomes of a large cohort of cancer cell lines in particular have been extensively characterised by a few large-scale projects such as CCLP1 and Cancer Cell Line Encyclopedia1,5. In this regard, there is no doubt that the performance of CDRscan can be further improved by incorporating additional high quality ‘-omics’ data including transcriptome information. Despite the excellent overall performance of CDRscan, prediction accuracy of a few GDSC compounds was compromised with low R2 values due to limited variance of data where the observed IC50 values had a very narrow range (Fig. 3b, Supplementary Fig. S7). Although most of the drug-centric analysis of CDRscan showed strong prediction of trend/order with high r values, incorporating additional assay results whenever available would also enhance the accuracy of prediction.
In this study, we introduced CDRscan, a novel and robust ensemble of deep learning models to predict drug efficacy based on genomic signatures. A web service will soon be available to allow browsing of the datasets used in CDRscan, the predicted and observed IC50 values, and some of the interesting examples of newly predicted indications for approved drugs. With further improvement, we envision that CDRscan will contribute to the rapidly evolving field of oncology by promoting the use of genome data for precision cancer medicine.
Methods
Software and hardware
To design CDRscan, we have implemented CNN using TensorFlow 1.3.0, Keras 2.0.6., and Ubuntu 16.04.3 LTS. Model design, training, and validation were performed on a workstation equipped with NVidia GTX 1080Ti.
Datasets
The genomic and drug sensitivity datasets of a wide array of cancer cell lines were obtained from CCLP1 (version 82, grch37) and GDSC6 (release 6.0). CCLP1 contains various types of molecular profile data, including whole exome sequencing data of 1,001 human cancer cell lines commonly used in cancer research. We selected sequence variation information at 28,328 positions from 567 genes in CGC31 (COSMIC v82, last obtained in Oct 2017). Five types of cancer (Table 2) were excluded since they had a limited number of available cell lines (<10). As a result, the final dataset contained 787 cell lines.
GDSC6 provides IC50 values from drug sensitivity assays for over 200,000 drug-cancer cell line pairs. In GDSC6, the identical set of 1,001 cell lines genomically characterised by CCLP1 was used, and 265 anticancer therapeutics from various sources, ranging from approved to those under investigation, were included in the assays. A line notation of simplified molecular-input line entry system (SMILES)67 was initially used to extract the structural and chemical features of each drug from PubChem68. However among 265 drugs, 18 drugs were not registered in SMILES, and three drugs had a molecular weight exceeding 1,000 g/mol. These 21 drugs were removed from the dataset. We also noticed that in GDSC, some identical chemicals were counted as two discrete entities6. There were 15 such pairs, but since the IC50 values were different across all pairs, we considered the 15 pairs as 30 distinctive drugs. The final dataset had 244 drugs representing 229 individual small chemicals (Fig. 1b, Supplementary Table S3). A total of 152,594 instances were in the final matrix of cell lines and drugs and employed to develop the deep learning models.
Feature extraction
Two different sources of input were used in CDRscan, which were genomic sequence variations of individual cancer cell lines and chemical properties of the anticancer drugs. The genomic fingerprints of cancer cell lines were expressed as a string of 28,328 binary codes, each representing a somatic mutation status. We have used only the variants that passed a series of stringent quality control filters, which were finally accepted in the COSMIC database. The list of somatic mutations were filtered further to include only those in CGC1. The presence of a somatic mutation was encoded as 1 and absence as 0. In a given mutation position, the same position substituted with a different base was considered identical. Insertions and deletions were not distinguished from base substitutions.
For SMILES of each of the 244 GDSC drugs6, a PaDEL-descriptor (v2.2.1)32 was employed to extract descriptors of three classes of fingerprints: (1) fingerprinter, (2) extended fingerprinter, and (3) graph only fingerprinter69,70,71, totalling 3,072 binary descriptors per drug, termed ‘molecular fingerprints’.
Model architecture
Two main principles were applied in designing CDRscan deep learning models. First, we employed novel CNN architectures, which we designated as a dual convergence deep learning architecture, to effectively process two very distinct types of input information. This architecture was applied to four of the CDRscan models. Second, we aimed to design a generalisable prediction model. To this end, we employed an ensemble of five independent models to minimise artifact derived from a particular model architecture. The architecture of each model was designed so that they were sufficiently distinct from one another. The major differences we introduced to the model architectures were as follows: (1) The number of convolution layers, (2) presence of absence of fully connected layer, (3) normalisation, and (4) inclusion of dual convergence architecture (Table 1, Supplementary Fig. S1, Supplementary Table S1).
One of the main hurdles for designing a robust deep learning model is overfitting. We thus employed the following techniques to prevent overfitting (Supplementary Fig. S1, Supplementary Table S1): (1) Three to four dropout layers were applied in all five models. In these layers, a subset of parameters (10–20% of the total parameters) were randomly selected and ignored during training, making it less likely to overfit the training data. (2) Maxpooling layers, which reduce dimensionality of input, were included in all models. When the mean square errors (MSE) of train and test set were plotted against the number of epochs, we did not observe an increase nor fluctuations of the test set error (Supplementary Fig. S2), which are considered as typical signs of overfitting. In addition, (3) the performance score of CDRscan measured by five-fold cross validation was consistent (described in the next section), indicating that the high performance score of CDRscan was not due to overfitting (Supplementary Table S4, Supplementary Fig. S8).
Model training and performance evaluation
We randomly selected 144,953 instances to train the models (95% of the total 152,594 instances). To ensure that all 25 cancer types are represented equally in the training set, we randomly choose 95% of the instances from each cancer type. As a result, 25 subsets were created and subsequently compiled as a single training set. The remaining 5% of the instances of individual cancer types were set aside to be used as test sets (i.e., train-test split method), both as 25 separate lists and as one consolidated list. Next, the performance of CDRscan and its five individual models was evaluated using the test datasets. For the evaluation, the experimentally obtained (observed) IC50 values and their counterparts predicted by CDRscan were plotted on a natural log scale. A coefficient of determination (R2), a widely accepted measure of prediction accuracy in machine learning72, was then computed using the following formula:
where y1, y2, … yn are observed IC50 values of each test set with n instances; f1, f2, …, fn are the predicted IC50 values for the corresponding instances;\(\,\bar{{y}}\) is the mean of the observed values.
Root mean squared error (RMSE) values were also calculated to evaluate the level of accuracy of prediction. Performance evaluation was also performed on the 25 separate test sets for individual cancer types, and for individual cell lines or for individual drugs. In the drug-centric evaluation of CDRscan performance, Pearson correlation coefficients (r) and p values were assessed for selected subsets of instances using SciPy (https://www.scipy.org; version 0.19.1). AUROC was also computed for the compiled training set. A classification criteria was applied to IC50 values using ln(IC50) of −2 as a cut-off (approximately 0.135 µM), where a drug is deemed active at ln(IC50) <−2 and inactive otherwise.
To benchmark the level of prediction accuracy, random forest and support vector machine models were trained with the identical test sets and parameters used in CDRscan. The prediction accuracy of two machine learning models were assessed using the same evaluation metrics.
In five-fold cross validation of CDRscan, the train set (144,953 instances) was partitioned into five equal-sized subsamples (28,991 instances in each subsample). One of the subsamples was held as a test set, and each model was trained with the remaining four subsamples. This process was repeated four more times, each time leaving a different subgroup as a test set.
Demonstration of predicting potential new indication of already approved drugs using CDRscan
The list of drugs approved in U.S., Canada, or E.U. was obtained from DrugBank33 (https://www.drugbank.ca; last accessed on September 20th, 2017). This list contained small molecules as well as complex molecules, such as biologic drugs, which cannot be properly expressed by 3,072 PaDEL descriptors used in CDRscan. Thus, the drugs that are compatible for CDRscan were selected from the initial list of the approved drugs by excluding the compounds according to the following criteria: (1) having no SMILES and/or no molecular weight information; (2) having a molecular weight <200 g/mol or >650 g/mol; (3) containing two or more parts that are not bonded together (including ‘ . ’ in their SMILES); and (4) being inorganic compounds. As a result, the final list of 1,487 approved drugs was generated. The final list included both non-oncology and oncology drugs, in which the oncology drugs were from the list provided in National Cancer Institute (NCI, https://www.cancer.gov/about-cancer/treatment/drugs). The structure information of these drugs was converted into 3,072 descriptors using the aforementioned PaDEL-descriptor (v2.2.1), which were used as input for CDRscan. To define ‘drug-sensitive’ cell lines, ln(predicted IC50) of −2 was used again as a cut-off, and the cancer types were designated as being sensitive to a given drug when more than 10% of the corresponding cell lines had ln(predicted IC50) less than −2. When counting the number of the potentially novel anticancer drugs, all non-oncology drugs with at least one sensitive cancer type were counted. For oncology drugs, only those with CDRscan-predicted cancer type indications in addition to the originally approved ones were considered as the drugs with repurposing potentials.
References
Forbes, S. A. et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, 777–83 (2017).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 505, 495–501 (2014).
Stratton, M. R., Campbell, P. J. & Futreal, P. A. The cancer genome. Nature. 458, 719–724 (2009).
Williams, S. P. & McDermott, U. The pursuit of therapeutic biomarkers with high-throughput cancer cell drug screens. Cell Chem Biol. 24, 1066–1074 (2017).
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483, 603–607 (2012).
Yang, W. et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, 955–961 (2013).
Keenan, A. B. et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 6, 13–24 (2018).
Basu, A. et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 154, 1151–1161 (2013).
Haider, S., Rahman, R., Ghosh, S. & Pal, R. A copula based approach for design of multivariate random forests for drug sensitivity prediction. PloS One. 10, e0144490, https://doi.org/10.1371/journal.pone.0144490 (2015).
Garnett, M. J. et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 483, 570–575 (2012).
Iorio, F. et al. A landscape of pharmacogenomic interactions in cancer. Cell. 166, 740–754 (2016).
Gönen, M. & Margoliny, A. A. Drug susceptibility prediction against a panel of drugs using kernelized Bayesian multitask learning. Bioinformatics. 30, i556–563 (2014).
Menden, M. P. et al. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS ONE. 8, e61318, https://doi.org/10.1371/journal.pone.0061318 (2013).
Rahman, R., Matlock, K., Ghosh, S. & Pal, R. Heterogeneity aware random forest for drug sensitivity prediction. Sci Rep. 7, 11347 (2017).
Rahman, R., Otridge, J. & Pal, R. IntegratedMRF: random forest-based framework for integrating prediction from different data types. Bioinformatics. 33, 1407–1410 (2017).
Riddick, G. et al. Predicting in vitro drug sensitivity using Random Forests. Bioinformatics, 27, 220–224 (2011).
Vanhaelen, Q. et al. Design of efficient computational workflows for in silico drug repurposing. Drug Discov Today. 22, 210–222 (2017).
Mamoshina, P., Vieira, A., Putin, E. & Zhavoronkov, A. Applications of deep learning in biomedicine. Mol Pharm. 13, 1445–1454 (2016).
Ramsundar, B. et al. Massively multitask networks for drug discovery. arXiv: 1502, 02072 (2015).
Dahl, G. E., Jaitly, N. & Salakhutdinov, R. Multi-task neural networks for QSAR predictions. arXiv: 1406.1231 (2014).
Yuan, Y. et al. DeepGene: an advanced cancer type classifier based on deep learning and somatic point mutations. BMC Bioinformatics. 17, 243–256 (2016).
Smalley, E. AI-powered drug discovery captures pharma interest. Nat Biotechnol. 35, 604–605 (2017).
Baskin, I. I., Winkler, D. & Tetko, I. V. A renaissance of neural networks in drug discovery. Expert Opin Drug Discov. 11, 785–795 (2016).
Gonczarek, A. et al. Interaction prediction in structure-based virtual screening using deep learning. Comput Biol Med. https://doi.org/10.1016/j.compbiomed.2017.09.007 (2017).
Pereira, J. C., Caffarena, E. R. & Dos Santos, C. N. Boosting docking-based virtual screening with deep learning. J Chem Inf Model. 56, 2495–2506 (2016).
Unterthiner, T. et al. Deep learning as an opportunity in virtual screening. Poster session presented at: Deep Learning and Representation Learning Workshop, Neural Information Processing Systems; 2014 Dec 12; Montreal, Canada (2014).
Wen, M. et al. Deep-learning-based drug-target interaction prediction. J Proteome Res. 16, 1401–1409 (2017).
Aliper, A. et al. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol Pharm. 13, 2524–2530 (2016).
Ching, T. et al. Opportunities and obstacles for deep learning in biology and medicine. Preprint at https://doi.org/10.1101/142760 (2017).
Lecun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature. 521, 436–444 (2015).
Futreal, P. A. et al. A census of human cancer genes. Nat Rev Cancer. 4, 177–183 (2004).
Yap, C. W. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem. 32, 1466–1474 (2011).
Law, V. et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42, D1091–1097 (2014).
National Cancer Institute. A to Z List of Cancer Drugs. https://www.cancer.gov/about-cancer/treatment/drugs (2017)
Gupta, S. et al. Prioritization of anticancer drugs against a cancer using genomic features of cancer cells: A step towards personalized medicine. Sci Rep. 6, 23857 (2016).
Costello, J. C. et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol. 32, 1202–1212 (2014).
Huang, C., Mezencev, R., McDonald, J. F. & Vannberg, F. Open source machine-learning algorithms for the prediction of optimal cancer drug therapies. PLoS One. 12, e0186906, https://doi.org/10.1371/journal.pone.0186906 (2017).
Sawyers, C. Targeted cancer therapy. Nature. 432, 294–297 (2004).
Abbosh, C. et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 545, 446–451 (2017).
Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 376, 2109–2121 (2017).
Paul, S. M. et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 9, 203–214 (2010).
Ashburn, T. T. & Karl, B. T. Drug repurposing: identifying and developing new uses for. exisiting drugs. Nat Rev Drug Discov. 3, 673–683 (2004).
Bertolini, F., Sukhatme, V. P. & Bouche, G. Drug repurposing in oncology-patient and health systems opportunities. Nat Rev Clin Oncol. 12, 732–742 (2015).
Dai, Y. et al. Bortezomib interacts synergistically with belinostat in human aml and all cells in association with perturbations in NF-κB and BIM. Br J Haematol. 153, 222–235 (2011).
Savickiene, J., Treigyte, G., Valiuliene, G., Stirblyte, I. & Navakauskiene, R. Epigenetic and molecular mechanisms underlying the antileukemic activity of the histone deacetylase inhibitor belinostat in human acute promyelocytic leukemia cells. Anticancer Drugs. 25, 938–949 (2014).
Havas et al. Belinostat and vincristine demonstrate mutually synergistic cytotoxicity associated with mitotic arrest and inhibition of polyploidy in a preclinical model of aggressive diffuse large B cell lymphoma. Cancer Biol Ther. 17, 1240–1252 (2016).
Kirschbaum, M. H. et al. A Phase 2 Study of Belinostat (PXD101) in Patients with Relapsed or Refractory Acute Myloid Leukemia or Patients Over 60 with Newly-Diagnosed Acute Myloid Leukemia: A California Cancer Consortium Study. Leuk Lymphoma. 55, 2301–2304 (2014).
Fathi, A. T. et al. Cabozantinib is well tolerated in acute myeloid leukemia and effectively inhibits the resistance-conferring FLT3/tyrosine kinase domain/F691 mutation. Cancer. 124, 306–314 (2018).
Abdel-Wahab, O. et al. Efficacy of intermittent combined RAF and MEK inhibition in a patient with concurrent BRAF- and NRAS-mutant malignancies. Cancer Discov. 4, 538–545 (2014).
Advani, A. S. et al. SWOG0919: a Phase 2 study of idarubicin and cytarabine in combination with pravastatin for relapsed acute myeloid leukaemia. Br J Haematol. 167, 233–237 (2014).
Shadman, M. et al. Idarubicin, cytarabine, and pravastatin as induction therapy for untreated acute myeloid leukemia and high-risk myelodysplastic syndrome. Am J Hematol. 90, 483–486 (2015).
Konings, I. R. et al. The add ition of pravastatin to chemotherapy in advanced gastric carcinoma: a randomised phase II trial. Eur J Cancer. 46, 3200–3204 (2010).
Chanvorachote, P. & Pongrakhananon, V. Ouabain downregulates Mcl-1 and sensitizes lung cancer cells to TRAIL-induced apoptosis. Am J Physiol. 304, C263 (2013).
Ninsontia, C., Chanvorachote, P., Ninsontia, C. & Chanvorachote, P. Ouabain mediates integrin switch in human lung cancer cells. Anticancer Res. 34, 5495–5502 (2014).
Trenti, A. et al. Cardiac glycoside ouabain induces autophagic cell death in non-small cell lung cancer cells via a JNK-dependent decrease of Bcl-2. Biochem Pharmacol. 89, 197–209 (2014).
Irwin, J. J. & Shoichet, B. K. ZINC-a free database of commercially available compounds for virtual screening. J Chem Inf Model. 45, 177–182 (2005).
Irwin, J. J. et al. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 52, 1757–1768 (2012).
Food and Drug Administration. FDA approves first cancer treatment for any solid tumor with a specific genetic feature. https://www.fda.gov/newsevents/newsroom/pressannouncements/ ucm560167. htm (2017).
Pauli, C. et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 7, 462–477 (2017).
Rubio-Perez, C. et al. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 27, 382–396 (2015).
Belizário, J. E., Sangiuliano, B. A., Perez-Sosa, M., Neyra, J. M. & Moreira, D. F. Using Pharmacogenomic Databases for discovering patient-target genes and small molecule candidates to cancer Therapy. Front Pharmacol. 7, 312 (2016).
Benstead-Hume, G., Wooller, S. K. & Pearl, F. M. G. ‘Big data’ approaches for novel anti-cancer drug discovery. Expert Opin Drug Discov. 12, 599–609 (2017).
Wang, H. et al. Current situation and future usage of anticancer drug databases. Apoptosis. 21, 778–794 (2016).
Li, A. et al. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol Cancer Res. 6, 21–30 (2008).
Weeber, F., Ooft, S. N., Dijkstra, K. K. & Voest, E. E. Tumor organoids as a pre-clinical cancer model for drug discovery. Cell Chem Biol. 24, 1092–1100 (2017).
Aparicio, S., Hidalgo, M. & Kung, A. L. Examining the utility of patient-derived xenograft mouse models. Nat Rev Cancer. 15, 311–316 (2015).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Model. 28, 31–36 (1988).
Kim, S. et al. PubChem Substance and Compound databases. Nucleic Acids Res. 44, D1202–1213 (2016).
Willighagen, E. L. et al. The Chemistry Development Kit (CDK) v2.0: atom typing, depiction, molecular formulas, and substructure searching. J Cheminform. 9, 33 (2017).
May, J. W. & Steinbeck, C. Efficient ring perception for the Chemistry Development Kit. J Cheminform. 6, 3 (2014).
Steinbeck, C. et al. The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. J Chem Inf Comput Sci. 43, 493–500 (2003).
Alexander, D. L. J., Tropsha, A. & Winkler, D. A. Beware of R2: simple, unambiguous assessment of the prediction accuracy of QSAR and QSPR models. J Chem Inf Model. 55, 1316–1322 (2015).
Acknowledgements
We appreciate Ye-Bin Jung’s assistance in creating the artwork in the Fig. 1. We also thank Jon Kim for proofreading of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.C., H.P., S.L. and J.-M.S. conceived the original idea for the deep learning models of CDRscan. Y.C., H.P. and S.L. obtained and processed the data from CCLP and GDSC. Y.C. developed the models with the assistance from H.P. and S.L. and J.-M.S. and J.J. provided intellectual input into the design and validation of the models. H.-J.Y. and K.-Y.L. designed and wrote the manuscript. J.-M.S., J.J. and T.S.K. participated in editing of the manuscript. H.-J.Y. and Y.C. designed the figures.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, Y., Park, H., Yang, HJ. et al. Cancer Drug Response Profile scan (CDRscan): A Deep Learning Model That Predicts Drug Effectiveness from Cancer Genomic Signature. Sci Rep 8, 8857 (2018). https://doi.org/10.1038/s41598-018-27214-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27214-6
This article is cited by
-
Deep learning in cancer genomics and histopathology
Genome Medicine (2024)
-
DeepAEG: a model for predicting cancer drug response based on data enhancement and edge-collaborative update strategies
BMC Bioinformatics (2024)
-
GPDRP: a multimodal framework for drug response prediction with graph transformer
BMC Bioinformatics (2023)
-
A performance evaluation of drug response prediction models for individual drugs
Scientific Reports (2023)
-
Predicting response to enzalutamide and abiraterone in metastatic prostate cancer using whole-omics machine learning
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
