
Abstract
In fish, protein-coding and noncoding genes involved in muscle atrophy are not fully characterized. In this study, we characterized coding and noncoding genes involved in gonadogenesis-associated muscle atrophy, and investigated the potential functional interplay between these genes. Using RNA-Seq, we compared expression pattern of mRNAs, long noncoding RNAs (lncRNAs) and microRNAs of atrophying skeletal muscle from gravid females and control skeletal muscle from age-matched sterile individuals. A total of 852 mRNAs, 1,160 lncRNAs and 28 microRNAs were differentially expressed (DE) between the two groups. Muscle atrophy appears to be mediated by many genes encoding ubiquitin-proteasome system, autophagy related proteases, lysosomal proteases and transcription factors. Transcripts encoding atrogin-1 and mir-29 showed exceptional high expression in atrophying muscle, suggesting an important role in bulk muscle proteolysis. DE genes were co-localized in the genome with strong expression correlation, and they exhibited extensive ‘lncRNA-mRNA’, ‘lncRNA-microRNA’, ‘mRNA-microRNA’ and ‘lncRNA-protein’ physical interactions. DE genes exhibiting potential functional interactions comprised the highly correlated ‘lncRNA-mRNA-microRNA’ gene network described as ‘degradome’. This study pinpoints extensive coding and noncoding RNA interactions during muscle atrophy in fish, and provides valuable resources for future mechanistic studies.
Similar content being viewed by others


Introduction
Sexual maturation, starvation and several pathological conditions negatively affect muscle mass and fillet quality attributes1,2,3. Improving growth performance and fillet quality by reducing protein turnover requires an understanding of muscle proteolysis, in vivo. Previously, several studies have identified protein-coding genes associated with skeletal muscle atrophy in fish1,2,3. A previous microarray study identified about 200 protein-coding genes that were differentially expressed (DE) during sexual maturation and associated with muscle atrophy in trout1. This study also found upregulated expression of catheptic and collagenase proteolytic pathways during muscle atrophy. Additionally, activation of calpains and the 28S proteasome subunit during starvation induced skeletal muscle atrophy previously was observed4. Albeit, some of the previous findings are inconsistent and do not provide a comprehensive set of protein-coding genes associated with muscle atrophy. As an example, some studies have reported downregulation of ubiquitin-proteasome system during atrophy1, while others have reported its upregulation5,6. These studies either investigated a single protein-coding gene5,6 or limited sets of protein-coding genes1 due to lack of a holistic approach. Moreover, none of the previous studies investigated the role of microRNAs and long noncoding RNAs (lncRNAs) in trout muscle atrophy. A more robust approach is needed to discover all potential candidate genes involved in muscle atrophy.
MicroRNAs bind 3′ -UTR of mRNA that leads to downregulation of the gene by various mechanisms such as translation suppression7, target mRNA cleavage8 and deadenylation9. There is evidence that a single microRNA can regulate hundreds of genes; and, at the same time, a single gene can be regulated by hundreds of microRNAs10. MicroRNAs are known to regulate muscle proteolysis and muscle atrophy in different mammalian species11. For example, mir-486 regulates disease-related muscle atrophy in mice by regulating FOXO1 transcription factor12. MicroRNA, mir-182 indirectly regulates the expression of key muscle atrophy genes including atrogin-1, cathepsins and autophagy related genes by targeting transcription factor FOXO313. Muscle specific microRNA, mir-1, regulates dexamethasone mediated muscle atrophy by targeting heat shock protein 70 (HSP70)14. However, microRNAs that regulate muscle atrophy in salmonids have not been previously investigated.
LncRNAs are a recently discovered class of noncoding RNAs with critical gene regulatory roles15. LncRNAs are known to regulate genes by direct interaction with microRNAs, mRNAs and proteins. Several lncRNAs bind microRNAs by sequence complementarity, and this interaction leads to cellular sequestration of microRNA (sponge effect) and lncRNA-mRNA competition for microRNA binding16. For example, lncRNA H19 binds and sponges away let-7 family microRNAs from repressing its protein-coding targets17. Similarly, muscle specific lncRNA, linc-MD1, competes with MAML1 and MEF2C to bind microRNAs mir-135 and mir-133, respectively18. LncRNA, MALAT1, modulates mir-133 mediated downregulation of serum response factor (SRF) by sharing mir-133 binding site19. LncRNAs can directly bind or physically interact with mRNA leading to mRNA decay20 and translation suppression21. Some lncRNAs hybridize with the 3′ UTR of target mRNA and facilitate Staufen-1 mediated mRNA decay20. On the other hand, lincRNA-p21 directly binds JUNB and CTNNB1 mRNAs and suppresses their translation21. LncRNA’s physical interaction with proteins modulates the stability22, cellular availability (sequestration)23, activity24 and cellular localization24 of proteins. For example, lncRNA, UPAT1, binds to UHRF1 protein and interferes with its ubiquitination and subsequent degradation22. LncRNA, MALAT1, binds to SR splicing factors and regulates their phosphorylation and hence cellular localization24. Both ‘lncRNA-microRNA’ and ‘lncRNA-protein-coding genes’ interactions are known to regulate development18, disease25 and cancers26,27; however, their involvement in skeletal muscle atrophy remains unknown.
To identify coding and noncoding genes involved in muscle atrophy associated with sexual maturation, we sequenced mRNAs, lncRNAs, and microRNAs from atrophying skeletal muscle of gravid fish and normal skeletal muscle of sterile fish. We subsequently performed differential gene expression of the two groups. In addition, we investigated functional interactions between DE lncRNAs, microRNAs and protein-coding genes in terms of expression correlation, genome co-localization and physical interaction to investigate gene-regulatory circuits during muscle atrophy. This study provides the first genome-wide lncRNA-mRNA-microRNA interaction network describing fish muscle degradation, and defining these interactions will clarify how energetic demand at sexual maturation triggers skeletal muscle atrophy.
Result and Discussion
Characteristics of atrophying and normal skeletal muscle
Differential expression of coding and noncoding genes was performed in atrophying skeletal muscle from diploid (2N), gravid fish in comparison to non-atrophying muscle of sterile triploid (3N) fish. This same set of skeletal muscle samples was used in our laboratory for several previous studies1,28,29. Compared to sterile fish, fertile females yielded less separable muscle per whole body weight (49.9% ± 6.7% vs 62.6% ± 2.2%, p = 0.01), muscle protein (16.9% ± 0.7% vs ~19.1% ± 0.7%, p = 0.01) and muscle shear force (178 ± 19 gram/gram vs 240 ± 18 gram/gram, p = 0.01). On the other hand, atrophied skeletal muscle from 2N females had a higher moisture content (80.3% ± 0.7% vs 77.2% ± 0.6%) and pH (6.61 ± 0.03 vs 6.41 ± 0.04) (Fig. 1). Atrophied muscle also had numerically lower crude fat content than normal muscle, but the difference was not statistically significant (p = 0.30). These textural and compositional difference between two groups of muscle result from extensive muscle atrophy in gravid fish triggered by the energetic demand of sexual maturation.

Comparison of different muscle phenotypes between atrophying skeletal muscle from gravid diploid (2N) fish and non-atrophying muscle from sterile triploid (3N) fish. Bar graph shows mean muscle yield (% of whole body weight) (a), muscle shear force (gram/gram) (b), muscle protein content (%) (c), muscle crude fat content (%) (d), muscle moisture content (%) (e) and muscle PH (F) of five sterile and five fertile fish at the age of spawning. Error bar represent standard error.
Differential expression of mRNAs, lncRNAs and microRNAs in atrophying muscle
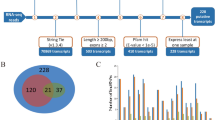
To identify genes likely involved in skeletal muscle atrophy during sexual maturation, we performed deep lncRNA, mRNA and microRNA sequencing, and quantified DE genes between atrophying skeletal muscle of gravid fish and non-atrophying skeletal muscle from sterile fish. A total of 852 mRNAs, 1,160 lncRNAs and 28 microRNAs were DE between these two groups (FDR-p-value < 0.01, fold change: > 3 or <−3) (Fig. 2, Table 1 and Supplementary dataset 1). A total of 1,025 transcripts (352 mRNAs, 661 lncRNAs and 12 microRNAs) were upregulated and 1,015 transcripts (500 mRNAs, 499 lncRNAs and 16 microRNAs) were downregulated in atrophying muscle. Real time PCR validation of 4 transcripts from each DE list of lncRNAs, microRNAs and mRNAs is provided in Supplementary dataset 2. Previously, a microarray based approach performed on the same set of muscle samples identified only 82 upregulated and 120 downregulated protein-coding genes1, suggesting identification, in this study, of a large number of additional candidate genes involved in muscle atrophy. DE protein-coding genes, lncRNAs and microRNAs are described in separate sections hereafter.

Heat map of DE lncRNAs (left) and protein-coding genes (right) between atrophying muscle of gravid fish and non-atrophying muscle of sterile fish. Value of color limit represents normalized expression values (Z scores). Fold change in gene expression was considered significant at: FDR-p-value < 0.01, fold change: >3 or <−3. Darker red and lighter red colors represent higher and lower level of expression, respectively. Transcript annotations are provided in Supplementary dataset 2.
Protein-coding genes
Many genes that promote proteolysis were significantly upregulated in atrophying skeletal muscle. At least 37 genes involved in protein ubiquitination, 22 genes involved in autophagy-related proteolysis, and 15 lysosomal and other proteases (cathepsin D, cathepsin B, cathepsin L and cathepsin Z) showed upregulation in atrophying muscle (Table 2 and Supplementary dataset 1). On the other hand, genes that negatively regulate the ubiquitin-proteasome system (ubiquitin carboxyl-terminal hydrolase 10, ubiquitin-like domain-containing CTD phosphatase 1 and uridine-cytidine kinase 2) and autophagy (CDGSH iron-sulfur domain-containing protein 2) were downregulated. Amino acid and fat biosynthetic genes were downregulated while genes involved in amino acid catabolism and transport were highly upregulated (Supplementary dataset 1). Similarly, genes associated with muscle sarcomere and extracellular matrix were downregulated, consistent with the loss of muscle mass and shear force during atrophy. As an example, 47 collagen-related genes and 24 non-collagen, extracellular matrix protein genes were significantly downregulated. A previous study also showed similar expression pattern of genes involved in protein ubiquitination and associated with the autophagy-lysosome system, extracellular matrix and sarcomere structure during muscle atrophy in mammals30. At least 53 transcription factors (TFs) or transcription regulators were also DE; of these transcription factors or regulators, 28 were upregulated and 25 were downregulated (Supplementary dataset 1). While the proteolytic role for the majority of the TFs was unknown, some transcription regulators, such as zinc finger and BTB domain-containing protein 16 and ddb1- and cul4-associated factor 6, had known function in protein catabolism. On the other hand, development related TFs like myoD were downregulated. These findings suggest that muscle atrophy is triggered by upregulation of proteolytic and catabolic genes with concomitant downregulation of muscle sarcomere, extracellular matrix, muscle development and biosynthetic genes.
The ubiquitin proteasome system appeared to be the major proteolytic system governing muscle atrophy. F-box only protein 32 (FBXO32) (atrogin-1), an E3 ubiquitin ligase, was the most highly upregulated genes in atrophying muscle suggesting that it might be the major player of muscle proteolysis during atrophy. Atrogin-1 genes transcripts, GSONMT00016768001 and GSONMT00031929001, exhibited 378- and 152-fold upregulation, respectively (Table 2). Their expression was validated by real time PCR (Supplementary dataset 2). Overexpression of atrogin-1 during starvation induced skeletal muscle atrophy has been reported previously in rainbow trout5, Atlantic salmon6 and mammals31.
As fish progress from pre-spawning through spawning, severity of skeletal muscle atrophy increases as indicated by loss of muscle mass and muscle protein, and a reduction in muscle shear force,28 as a measure of ultrastructural changes associated with muscle breakdown. To further investigate the potential contribution of DE genes in sexual maturation associated muscle atrophy, we looked at the trends of expression pattern of DE genes over 4 months during pre-spawning (July, November) and spawning (December and January) using RNA sequencing data from our previously described source3. Transcript abundance of ubiquitin-proteasome system genes and autophagy-related proteolytic genes remained constant in July and November, sharply increased in December, and then declined in January (Fig. 3a,b). Expression level of proteases positively correlated with severity of muscle atrophy in the aforementioned timeframes. Late December represents the time of peak sexual maturation associated muscle atrophy. Expression of genes coding for different atrogin-1 isoforms and cathepsin D was highest in December and then declined in January (Fig. 3c,d). Cathepsin D is involved in sexual maturation associated muscle atrophy, and we found an increase in Cathepsin D transcript level. Nonetheless, previously we did not observe a significant change in catalytic activity of cathepsin D during atrophy28. Extracellular matrix protein genes and development related genes showed opposite expression trends (Fig. 3e,f), consistent with the loss of muscle firmness and development during atrophy. These findings suggest that DE genes may serve as reliable candidate(s) critical to sexual maturation associated muscle atrophy in fish. However, the trends of the gene expression showed in Fig. 3 should be taken with caution because a single RNA-Sea library from 10 pooled fish at each time point was used and no replicates were available to run statistical analyses”.

Transcript expression values of different classes of DE genes during pre-spawning and spawning months in atrophying muscle of diploid gravid fish: all ubiquitinating genes combined (a), all autophagy related genes combined (b), atrogin-1 transcripts (c), cathepsin D isoforms (d), all development related genes combined (e), all collagen and extracellular matrix related genes combined (f), all upregulated lncRNAs combined (g) and all downregulated lncRNAs combined (h). Expression level of each gene in gravid fish (2N) was normalized by expression level of respective gene in sterile fish (3N). Gene lists are provided in Supplementary dataset 2.
Long noncoding RNAs (LncRNAs)
For differential lncRNA expression, we initially performed genome-wide discovery of lncRNA transcripts using RNA-Seq reads sequenced from skeletal muscle of gravid and sterile fish. Approximately 15,000 lncRNA transcripts were identified from this assembly and merged with our previously published lncRNA reference32; and both sources were used as a reference for gene expression analysis. A total of 1,160 lncRNAs were DE between atrophying and non-atrophying skeletal muscle. Of 1,160 DE lncRNAs, 225 and 10 lncRNAs had sequence homology with lncRNAs from Atlantic salmon and zebrafish, respectively (sequence identity: >80%, E value: <E-10, query cover: >50 nucleotides) (Supplementary dataset 3), but their functional annotation was not available in any species. Like protein-coding mRNAs, expression level of DE lncRNAs correlated with the severity of muscle atrophy during pre-spawning and spawning. Transcript abundance of upregulated lncRNAs remained constant during pre-spawning, but drastically increased in December and then declined in January (Fig. 3g). On the other hand, transcript abundance of downregulated lncRNAs showed an opposite trend (Fig. 3h). These findings suggest that expression of these DE lncRNAs may be involved in sexual maturation associated muscle atrophy in rainbow trout.
MicroRNAs
A total of 28 microRNAs were DE between skeletal muscle of gravid and sterile fish. Of them, differential expression of mir-1, mir-133, and mir-29 during mammalian muscle atrophy has been previously reported11,33, but the remainder of DE microRNAs was reported for the first time (Table 1). A total of 665 unique mRNA genes were predicted as potential target genes of these 28 DE microRNAs; of these mRNA genes, 17 were also DE. Some of these DE microRNAs and their predicted DE mRNA targets showed reciprocal differential expression (Supplementary dataset 4). As an example, mir-29a predicted target mRNAs encoding collagen alpha, ATP binding cassette subfamily f member 3, alanine tRNA synthase, scavenger receptor class b member 1, and fk506-binding protein 2 were downregulated while mir-29a was upregulated in atrophying muscle. Similarly, mir-125b-1 was downregulated, and its predicted mRNA targets encoding CCAAT enhancer-binding protein delta and pancreatic progenitor cell differentiation and proliferation factor A were upregulated. Out of the DE microRNAs, sixteen were downregulated microRNAs, potentially targeting a total of 206 different protein-coding genes. Twenty-six of the predicted target genes were proteolytic enzymes, including genes involved in ubiquitin-proteasome and autophagy-lysosome mediated proteolysis (Supplementary dataset 4). Twelve upregulated microRNAs were predicted to target 468 different protein-coding genes. Consistent with their upregulated expression during atrophy, 101 predicted target genes were directly involved in extracellular matrix, muscle structure, or development (Supplementary dataset 4). Aforementioned findings suggest that some genes involved in muscle atrophy may not be necessarily regulated at transcription level, and its fate is determined post-transcriptionally by regulated expression of microRNA.
Let-7j was the most highly downregulated microRNA (−1056 × fold) in atrophying muscle (Table 1). It was predicted to target 63 different protein-coding genes that account for a wide range of functions (Supplementary dataset 4). Consistent with its downregulation in atrophying muscle, some of its predicted targets were proteolytic genes such as E3 ubiquitin-protein ligase NRDP1, ubiquitin-conjugating enzyme E2 and protein VPRBP (Supplementary dataset 4). Conversely, six different isoforms of mir-29 were highly upregulated in atrophying muscle (Table 1). Mir-29a, the most highly upregulated microRNA in atrophying muscle (11.7 × fold) was predicted to target 78 genes; the highest number of predicted target genes among DE microRNAs (Supplementary dataset 4). Consistent with its upregulation in atrophying muscle, predicted target genes of mir-29a included genes involved in muscle differentiation (IGF-BP 5), muscle sarcomere (e. g. myosin) structure, extracellular matrix (e. g. collagen), fat biosynthesis (e. g. long-chain-fatty-acid–ligase acsbg2 and acyl-coenzyme a thioesterase 11), protein synthesis (e. g. 60 s ribosomal protein l7) and development (e. g. prospero homeobox protein 1). These findings suggest that DE microRNAs may contribute to muscle atrophy by regulating proteolysis and other genes during muscle atrophy.
Tissue specific and temporal expression of DE lncRNAs and mRNAs
LncRNAs show strict spatial (tissue specific) and temporal (time dependent) expression patterns34. To investigate tissue specificity of DE genes, we studied their expression pattern across 13 vital tissues including red and white muscle (see method section for classification of tissue specific genes). About 40% (462/1,160) of DE lncRNAs and approximately 41% (348/852) of DE mRNAs were ‘specific’ to red or white muscle (Fig. 4). These specificities indicated about 2.5-fold enrichment of muscle specific lncRNAs, and about 5.5-fold enrichment of muscle specific mRNAs in the DE gene list compared to muscle specific expression of approximately 16% (8,460/51,644) of non-DE lncRNAs and approximately 6% (4,583/61,412) of non-DE mRNAs in the trout genome (Supplementary dataset 5). Interestingly, a majority of the most highly upregulated mRNAs were muscle ‘specific’. As an example, 47 out of 61 mRNAs, with fold change greater than 15, had muscle restricted expression patterns that included mRNA encoding atrogin-1. Muscle specific expression of atrogin-1 has also been previously reported in mammals31. In addition, some muscle specific lncRNAs, such as linc-MD1, are known to play an important role in regulation of muscle specific genes18. In addition to tissue specific expression, ~38% (442/1,160) of DE lncRNAs and ~40% (342/852) of DE mRNAs exhibited temporal (time dependent) expression patterns during pre-spawning and spawning (Fig. 4). These findings suggest that a significant proportion of the DE transcriptome in atrophying muscle is comprised of muscle specific gene expression, exhibiting a responsive expression pattern of muscle atrophy during sexual maturation.

Heat map showing tissue specific expression pattern of DE lncRNAs (a), tissue specific expression pattern of DE mRNAs (b) and temporal expression pattern of DE lncRNAs and DE mRNAs during pre-spawning and spawning months (c). Value of color limit represents normalized expression values (Z scores). Darker red and lighter red colors represent higher and lower level of expression, respectively.
Genomic co-localization of DE lncRNA and mRNA genes
Functionally related lncRNAs and mRNAs physically co-localize in the genome35. Out of 1,160 DE lncRNAs, 231 (~20%) unique lncRNAs were either overlapped or were neighbored (<50 kb) by DE mRNA genes. A total of 242 (~28%) unique DE mRNA genes overlapped or neighbored DE lncRNAs (Supplementary dataset 6, tab 1 and 2). These findings suggest that DE lncRNAs and mRNAs tend to co-localize or cluster together in the genome. However, mere physical proximity does not necessarily lead to functional links36,37. To test the functional significance of physical proximity, we computed expression correlation between all neighboring/overlapping lncRNA-mRNA genes. Out of 387 neighboring/overlapping DE lncRNAs-mRNAs pairs, ~36.2% (140) had strongly correlated expression patterns (R > 0.85) compared to ~16.7% (168,207) DE lncRNAs-mRNAs with expression correlation (R > 0.85) regardless of genomic co-localization (Supplementary dataset 6, tab 3). The difference was statistically significant (Chi square p-value < 0.001) suggesting that co-localized lncRNA and protein-coding genes tend to be more correlated in expression than genes with greater separation in the genome. The degree of expression correlation between lncRNAs and protein-coding genes, as a function of physical proximity, was weakly negative (R = −0.35, p value < 0.001) (Supplementary dataset 6, tab 4). Next, we investigated whether ‘strand orientation’ (sense or antisense) was correlated with ‘type of expression correlation’ (negative or positive), and found no significant correlation (Chi square p-value > 0.05) (Supplementary dataset 6, tab 5). These observations suggest that DE lncRNA and mRNA genes tend to co-localize or cluster together in the genome, and often show correlated expression pattern. Further, to investigate the potential mechanistic regulation of all neighboring/overlapping lncRNA-mRNA genes, we scanned promoter sequences of the lncRNA-mRNA for transcription factor (TF) binding cis regulatory motifs. The majority of gene pairs harbored common TF binding motifs in their promoters. Many of the TFs are known to be involved in muscle development (e. g. myoD, myogenin, c-Fos, c-Jun, NF-AT1, Smad3, NF-Y, and NFI/CTF and 24 overlapped/neighboring lnc-mRNA pairs had ER-alpha in their promoters; Supplementary dataset 6, tab 6). Taken together, harboring the same TFs in the promoter regions, may partly, explain the correlated gene expression patterns of the co-localized DE lncRNA-mRNA pairs.
DE lncRNAs acting as microRNA sponges or microRNA precursors
Direct ‘lncRNA-microRNA’ binding has important functional consequences including lncRNA mediated sponging of microRNA and ‘lncRNA-microRNA’ competition for mRNA binding16. To identify DE lncRNAs that potentially interfere with microRNA mediated gene regulation, we searched for high confidence microRNA binding sites in DE lncRNAs and mRNAs. A total of 134 trout microRNAs had binding sites in DE lncRNAs as well as DE mRNAs (Supplementary dataset 7). Some of these microRNAs, such as mir-133, mir-214, and mir-221, are known to regulate muscle atrophy or proteolysis38,39. DE lncRNAs shared microRNA binding sites with mRNAs encoding important proteolytic proteins such as atrogin-1, cathepsins, serine proteases, and several enzymes in the ubiquitin proteasome system (Table 3 and Supplementary dataset 7). For example, atrogin-1 (GSONMT00016768001), the most highly upregulated gene in atrophying muscle, shared mir-22-3p binding sites with Omy200063021 and Omy400145202. Expression patterns of a subset of LncRNAs and mRNAs that shared microRNA binding sites and were strongly correlated is shown in Table 3.
In addition to acting as a microRNA sponge, some lncRNAs serve as precursors of microRNAs and other classes of small noncoding RNA (sRNAs)34,40. DE lncRNA, Omy400148395, harbored mir-27 loci, and Omy200105075 harbored aly-miR-398c-like loci. Expression of mir-27 and aly-miR-398c-like microRNAs was positively correlated with their potential host lncRNAs; correlation R values were 0.60 and 0.81, respectively. This observation suggests that these microRNAs could be generated by post-transcription processing of lncRNA transcripts. Together, these findings suggest that some DE lncRNAs may sequester or generate microRNAs involved in muscle atrophy.
Physical interaction between lncRNAs and protein-coding genes
Direct ‘lncRNA-mRNA’ physical interactions lead to mRNA decay20 and translation suppression21. To investigate potential existence of lncRNA-mRNA physical interactions, we used the IntaRNA tool that considers site accessibility and user defined seed requirement to predict the interaction41. At an interaction energy threshold <−100 Kcal/mole, 1,151 DE lncRNA-DE mRNA pairs exhibited potential physical interactions (Table 4 and Supplementary dataset 8). Interestingly, lncRNA-mRNA pairs, showing evidence of physical interaction, were more strongly correlated in expression (R > 0.85) than lncRNA-mRNA pairs without the potential physical interaction (Table 4 and Supplementary dataset 8).
In addition to interacting with mRNAs, several lncRNAs showed evidence of physical interaction with proteins of DE mRNAs. ‘LncRNA-protein’ physical interaction was computed using CatRapid Omics tool42. A total of 14,602 ‘DE lncRNA- protein’ pairs showed evidence of physical interaction at an interaction strength ≥96% and a discriminative power ≥96% (Table 4 and Supplementary dataset 8). Interestingly, ‘DE lncRNA-DE protein’ pairs showing evidence of direct physical interactions were more strongly correlated in expression (at transcript level) (R > 0.85) than ‘DE lncRNA-DE protein-coding gene’ pairs without the evidence of physical interaction (Chi square p-value < 0.001). Atrogin-1, cathepsin, and several enzymes of the ubiquitin-proteasome system were among the proteins with potential interaction with lncRNAs. ‘LncRNA-protein’ binding regulates protein’s stability22, availability23, activity24 and cellular localization24, suggesting that DE lncRNAs may have an important role in determining fate of DE protein-coding genes during muscle atrophy.
‘LncRNA-mRNA-microRNA’ interactome in atrophying muscle
LncRNAs, microRNAs, and mRNAs comprise interacted gene regulatory networks in the cell43, probably due to mutual regulation between microRNA and lncRNA16. To investigate existence of such gene interaction during muscle atrophy, we computed lncRNA-mRNA-microRNA interaction networks based on their expression pattern across 30 RNA-Seq datasets. At a correlation threshold R > 0.97 or <−0.97, about 50% (1,584) of DE transcripts were components of strongly correlated gene networks (Fig. 5). Interestingly, a majority of the correlated transcripts clustered in one of two major networks; the first comprised of downregulated transcripts and the second comprised of upregulated transcripts. The first network consisted of 430 transcripts (137 lncRNAs, 219 mRNAs and 74 microRNAs). The second network consisted of 960 transcripts (559 lncRNAs, 235 mRNAs and 166 microRNAs). The second network appeared to be an interacted gene “reactome” regulating muscle proteolysis because almost all upregulated proteolytic genes, including enzymes of ubiquitin proteasome system, autophagy related proteolytic genes and other proteases such as cathepsins, were in this network. Similarly, a majority of the upregulated microRNAs including mir-29, let-7, let-7d and mir-132 were in the network. Interestingly, atrogin-1 transcripts (the most highly upregulated transcripts) and mir-29a (the most highly upregulated microRNA) were in the center of this network suggesting a key role in the network. A crucial role of atrogin-1 in muscle atrophy has repeatedly been reported in fish and mammals5,6,31. We identified this network as ‘the rainbow trout muscle degradome’ because it appeared to be a regulatory network of muscle degrading coding and noncoding genes. The network was largely comprised of the genes that showed evidence of physical interaction with each other. A sub-network of the degradome shown in Fig. 5 consist of atrogin-1 transcripts, all DE lncRNAs that bind to atrogin-1 mRNAs, and microRNAs that either bind to lncRNAs and/or atrogin-1 mRNAs. The aforementioned observations suggest that coding and noncoding genes involved in muscle atrophy work in the form of a highly interacted gene network.

Gene expression network of DE lncRNAs (blue node), DE mRNAs (green node) and microRNAs (pink node) (R > 0.97 or <−0.97). Note that most of the DE genes are clustered in one of the two major networks. The larger network (degradome) comprises of upregulated genes and smaller network comprises of downregulated genes. In the network of upregulated transcripts, 4 atrogin-1 transcripts (red nodes) and mir-29 isoforms (black nodes) are in the center of the network. Sub-network drawn from the larger network contains 4 atrogin-1 transcripts, lncRNAs that bind to atrogin-1, and microRNAs that either bind to the lncRNAs and/or atrogin-1. Note: edges that connect nodes (genes) represent correlated expression at R cutoff 0.97 > or <−0.97; the shorter the length, the stronger the expression correlation.
Conclusion
Sexual maturation associated skeletal muscle atrophy serves as an excellent model to study piscine muscle proteolysis1,3,29. Previous efforts to investigate fish muscle proteolysis have provided limited information because these studies relied on individual or a limited set of protein-coding genes1,5,6. In the present study, we used deep lncRNA, mRNA, and microRNA sequencing approaches to investigate genes and gene regulatory networks that regulate muscle proteolysis in fish. Through investigation of the atrophying muscle transcriptome, we elucidated that fish muscle atrophy, like mammalian muscle atrophy, is regulated mainly by the ubiquitin-proteasome system. In addition, many autophagy-lysosomal proteases and transcription factors appeared to participate in muscle proteolysis during atrophication. Atrophying muscle exhibited upregulation of proteolytic genes with concomitant downregulation of genes involved in muscle sarcomere, extracellular matrix, protein and fat biosynthesis, and development. This trend in expression pattern of genes in atrophying muscle correlated well with characterizations of the atrophying muscle phenotype (e. g. muscle mass, protein content and muscle shear force), suggesting essential roles for DE genes in muscle atrophy. The present study identified a large number of new candidate coding and noncoding genes in addition to the genes identified by previous microarray and proteomic approaches1,29 suggesting that the RNA-Seq approach has identified a large number of reliable candidate genes involved in muscle proteolysis/degradation.
In present study, we characterized lncRNAs potentially involved in fish muscle proteolysis, and we investigated lncRNA-mRNA, lncRNA-microRNA and mRNA-microRNA interactions that potentially regulate muscle atrophy. A majority of DE lncRNAs and DE mRNA genes were co-localized in the genome and correlated in expression. DE lncRNAs appeared to physically interact extensively with DE protein-coding genes at transcript and protein levels. Similarly, DE lncRNA also showed potential to bind and sequester cellular microRNAs implicated in muscle proteolysis. LncRNA, mRNA and microRNAs that exhibited the aforementioned interactions are components of a highly correlated gene network in atrophying muscle. This finding indicates that the majority of genes involved in muscle proteolysis are expressed simultaneously by a common gene transcription program. Important to mention, ‘DE lncRNA-DE protein-coding’ gene pairs that either co-localized in the genome or showed evidence of direct physical interaction or competed for a common microRNA binding, were more frequently correlated in expression than random ‘DE lncRNA-DE protein-coding’ gene pairs. Perhaps this work is the first genome wide study that provides links between expression correlation and potential functional interactions between lncRNA and mRNA in fish. The present study has investigated potential coding and noncoding RNA interactions during muscle atrophy and contributes to our understanding of how energetic demand of sexual maturation triggers skeletal muscle atrophy in fish.
Materials and Methods
Ethics statement
Fish muscle tissues were obtained commercially from a private farm; therefore, Institutional Animal Care and Use Committee (IACUC) approval was not required.
Fish population and muscle sampling
We previously described the fish population in another study1. Briefly, mature sterile (3N: triploid) and fertile (2N: diploid) female rainbow trout (about 500 gram) were obtained from Flowing Springs Trout Farm (Delray, WV) during spawning season; these fish were cultured in identical raceways. Water from a common spring was circulated in raceways at temperature 13 ± 3 °C. Both groups of fish were fed ad libitum (Zeiglar Gold; Zeigler Bros., Gardeners, PA) via demand feeder until sampling. At the time of muscle sampling gonado-somatic index (GSI) of fertile fish was 15.8 ± 0.3 (n = 5) compared to 0.3 ± 0.2 (n = 5) in sterile fish confirming the gravid stage of fertile fish. White muscle tissue of 8 fish (4 fertile and 4 sterile) was collected from the dorsal musculature, flash frozen in liquid nitrogen and stored at −80 °C until RNA extraction. Total RNA was extracted from the muscle using TRIzol method (Invitrogen, Carlsbad, CA). For phenotype measurement, boneless and skinless muscle fillet was obtained in a manual filleting procedure. Muscle yield was measured as percentage of whole body weight. A representative portion of the muscle fillet was used for shear force, pH, and proximate analyses (e.g. crude protein content, crude fat content, and moisture content).
Library construction and sequencing
Sequencing libraries were prepared using Illumina TruSeq stranded total RNA with Ribo-Zero gold protocol following the manufacturer’s recommendations (Illumina Inc, CA, USA). One sequencing library was prepared from each fish and was provided a unique barcode. Equal amount of the barcoded libraries from all fish were pooled and sequenced using an Illumina HiSeq. 2000 sequencing platform in a single lane (2 × 100 reads). Similarly, for microRNA sequencing, Illumina’s TruSeq small RNA library preparation kit was used to prepare one barcoded library from each 8 fish and libraries were pooled and sequenced in a single lane of Illumina HiSeq. 2000 sequencing platform. Sequence data are available through the NCBI Sequence Read Archive (SRA) accession: SRP131630.
Discovery of lncRNAs
LncRNAs from sequencing reads were identified by using the pipeline we described previously32. Briefly, reads were mapped to rainbow trout reference genome44 and assembled using TopHat and Cufflinks, respectively. Transcripts shorter than 200 nucleotides were filtered out. Protein-coding transcripts were removed by their sequence homology with NCBI protein entries. In addition, Coding Potential Calculator (CPC)45 tool was used to remove any transcripts with protein-coding potential (index value < −0.5). Other classes of noncoding RNAs were removed based on their sequence homology with noncoding RNA transcripts reported in public noncoding RNA databases including miRbase, genomic tRNA database, SSU (small subunit ribosomal RNA) and LSU (large subunit ribosomal RNA) databases. Putative lncRNA transcripts from the assembly are available at https://www.animalgenome.org/repository/pub/MTSU2017.1228/.
Identification of DE mRNA, lncRNAs and microRNAs
Read mapping and identification of DE genes were performed using CLC genomics workbench. For protein-coding genes, sequencing reads from every fish were mapped to a mRNA reference from rainbow trout genome44 and our transcriptome assembly46. The expression value of each transcript was calculated in terms of TPM (transcript per million), and DE mRNAs between gravid and sterile fish were identified using EDGE test (FDR-P -value < 0.01, fold change: > 3 or <−3). For lncRNAs, previously published rainbow trout lncRNAs32 and additional lncRNAs assembled from this sequencing project were used as a reference. Read mapping and identification of DE lncRNA was done as described for mRNAs. For microRNAs, sequencing adapters were trimmed and reads were mapped to miRBase microRNA reference (release 21) (mismatch ≤ 2, additional/missing upstream/downstream bases ≤ 2). The total read count for each microRNA was calculated, and used to identify DE microRNAs by EDGE test (FDR-p-value < 0.01, fold change: > 3 or <−3).
Real time PCR validation of DE transcripts
Total RNA from the same 8 fish used for sequencing was used to make template cDNA for qPCR analysis. Contaminating DNA in RNA sample was removed by DNAse treatment and cDNA was synthesized using Verso cDNA Synthesis Kit (Thermo Scientific, Hudson, NH). Transcript abundance of mRNA and lncRNA was quantified per manufacturer’s instruction using DyNAmo Flash SYBR Green Master Mix (Thermo Scientific, Hudson, NH) in Bio Rad CFX96™ System (Bio Rad, Hercules, CA). For microRNAs, miScript II RT kit (Qiagen, Valencia, CA, USA) was used to synthesize cDNA, and miScriptR SYBRR green (Qiagen, Valencia, CA, USA)47 was used to quantify microRNA in Bio Rad CFX96™ System. The endogenous controls used for normalization were B-actin for mRNA and lncRNAs, and U6 for microRNA. None of the endogenous control genes was differentially expressed in this study. Fold changes in gene expression was calculated by using ΔΔCt method as described previously35,48. Mann-Whitney U test was used to check if the transcript level between atrophying and control muscle was statistically significant (p < 0.05). All 12 transcripts subjected to qPCR validation had Mann Whitney U test p-value < 0.05.
Identification of tissue specific genes
The expression pattern of DE genes was investigated across 13 vital tissues: red muscle, white muscle, spleen, liver, skin, testis, brain, intestine, stomach, kidney, head kidney, gill and fat44,46,49. To identify tissue specific genes, we used a statistical approach described by50. The normalized expression value (z score) of every gene was calculated in each tissue from TPM counts. Each gene was classified as ‘specific’ to a tissue if z score was greater than 1.5 in that tissue and less than 1.5 in remaining 12 tissues which is similar to a previous study50. We used the same approach to identify genes ‘specific’ to a particular month during pre-spawning and spawning3, as described in the results section.
LncRNA, mRNA and microRNA co-expression
For mRNA-ncRNA correlation analysis, 8 RNA-Seq samples from gravid and sterile fish and 22 RNA-Seq samples sequenced from muscle of selectively bred fish families developed at USDA/NCCCWA were used. Detailed description of these 22 samples, fish population, and sampling procedure has been previously described51. TPM values (for lncRNAs and mRNAs) and the total count (for microRNAs) were calculated in all 30 samples (8 samples from 2 ploidy groups and 22 samples from fish families) by mapping reads to corresponding mRNA, lncRNA, and microRNA references as described above. TPM and total count values were normalized by using a scaling method as previously described52, and normalized values were used to calculate expression correlation coefficients using Pearson correlations. A lncRNA-mRNA-microRNA expression network was constructed using Expression Correlation in cytoscape53.
Identification of cis regulatory promoter motifs
Promoter regions of all neighboring/overlapping lncRNA-mRNA genes were scanned for putative Transcription factor (TF) binding motifs using ALGGEN PROMO TF motif search tool54. Maximum dissimilarity rate was set to 5%, and RE equality/query was set to <0.05.
Identification of microRNA-harboring lncRNAs
The rainbow trout genome reference44 was annotated with rainbow trout lncRNA reference sequences mentioned above. Rainbow trout pre-microRNA sequences (~64 nts long) from a recently published source55 were aligned to the lncRNA-annotated genome assembly. Pre-microRNA sequences that perfectly align (with no mismatch or gap) within annotated lncRNA loci in the genome were reported.
LncRNA and mRNA targets of microRNAs
In the case of mRNAs, microRNA binding sites were searched in 3′ UTR; whereas for lncRNAs, target sites were searched for throughout the entire sequence length. Three target prediction algorithms: miRanda, PITA and TargetSpy were used to find target genes using sRNAtoolbox56. If the same target site is predicted by all 3 tools, it was considered as a potential microRNA target site. For all tools, minimum energy threshold was chosen as −20 Kcal/mole. Threshold scores chosen were 150 for miRanda and 0.99 for TargetSpy.
Prediction of lncRNA-mRNA and lncRNA-protein interaction
LncRNA-mRNA direct physical interaction was predicted using IntaRNA-RNA-RNA interaction tool41. All lncRNA-mRNA interactions were recorded at an interaction energy threshold < −100 Kcal/mole. LncRNA-protein interactions were predicted using CatRapid Omics tool42. Interaction strength and discriminative power (a measure of predictability of interaction) were set at a ≥96% to consider lncRNA-protein interaction as putative interaction.
References
Salem, M., Kenney, P. B., Rexroad, C. E. & Yao, J. Microarray gene expression analysis in atrophying rainbow trout muscle: a unique nonmammalian muscle degradation model. Physiol Genomics 28, 33–45, https://doi.org/10.1152/physiolgenomics.00114.2006 (2006).
Aussanasuwannakul, A. et al. Effect of sexual maturation on thermal stability, viscoelastic properties, and texture of female rainbow trout, Oncorhynchus mykiss, fillets. J Food Sci 77, S77–83, https://doi.org/10.1111/j.1750-3841.2011.02512.x (2012).
Salem, M. et al. Effect of sexual maturation on muscle gene expression of rainbow trout: RNA-Seq approach. Physiol Rep 1, e00120, https://doi.org/10.1002/phy2.120 (2013).
Salem, M., Silverstein, J., Rexroad, C. E. & Yao, J. Effect of starvation on global gene expression and proteolysis in rainbow trout (Oncorhynchus mykiss). BMC Genomics 8, 328, https://doi.org/10.1186/1471-2164-8-328 (2007).
Cleveland, B. M. & Evenhuis, J. P. Molecular characterization of atrogin-1/F-box protein-32 (FBXO32) and F-box protein-25 (FBXO25) in rainbow trout (Oncorhynchus mykiss): Expression across tissues in response to feed deprivation. Comp Biochem Physiol B Biochem Mol Biol 157, 248–257, https://doi.org/10.1016/j.cbpb.2010.06.010 (2010).
Tacchi, L. et al. Ubiquitin E3 ligase atrogin-1 (Fbox-32) in Atlantic salmon (Salmo salar): sequence analysis, genomic structure and modulation of expression. Comp Biochem Physiol B Biochem Mol Biol 157, 364–373, https://doi.org/10.1016/j.cbpb.2010.08.004 (2010).
Olsen, P. H. & Ambros, V. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol 216, 671–680, https://doi.org/10.1006/dbio.1999.9523 (1999).
Bagga, S. et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122, 553–563, https://doi.org/10.1016/j.cell.2005.07.031 (2005).
Wu, L., Fan, J. & Belasco, J. G. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA 103, 4034–4039, https://doi.org/10.1073/pnas.0510928103 (2006).
Krek, A. et al. Combinatorial microRNA target predictions. Nat Genet 37, 495–500, https://doi.org/10.1038/ng1536 (2005).
Wang, X. H. MicroRNA in myogenesis and muscle atrophy. Curr Opin Clin Nutr Metab Care 16, 258–266, https://doi.org/10.1097/MCO.0b013e32835f81b9 (2013).
Xu, J. et al. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int 82, 401–411, https://doi.org/10.1038/ki.2012.84 (2012).
Hudson, M. B. et al. miR-182 attenuates atrophy-related gene expression by targeting FoxO3 in skeletal muscle. Am J Physiol Cell Physiol 307, C314–319, https://doi.org/10.1152/ajpcell.00395.2013 (2014).
Kukreti, H. et al. Muscle-specific microRNA1 (miR1) targets heat shock protein 70 (HSP70) during dexamethasone-mediated atrophy. J Biol Chem 288, 6663–6678, https://doi.org/10.1074/jbc.M112.390369 (2013).
Rinn, J. L. & Chang, H. Y. Genome regulation by long noncoding RNAs. Annu Rev Biochem 81, 145–166, https://doi.org/10.1146/annurev-biochem-051410-092902 (2012).
Yoon, J. H., Abdelmohsen, K. & Gorospe, M. Functional interactions among microRNAs and long noncoding RNAs. Semin Cell Dev Biol 34, 9–14, https://doi.org/10.1016/j.semcdb.2014.05.015 (2014).
Kallen, A. N. et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell 52, 101–112, https://doi.org/10.1016/j.molcel.2013.08.027 (2013).
Cesana, M. et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147, 358–369, https://doi.org/10.1016/j.cell.2011.09.028 (2011).
Han, X., Yang, F., Cao, H. & Liang, Z. Malat1 regulates serum response factor through miR-133 as a competing endogenous RNA in myogenesis. FASEB J 29, 3054–3064, https://doi.org/10.1096/fj.14-259952 (2015).
Gong, C. & Maquat, L. E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature 470, 284–288, https://doi.org/10.1038/nature09701 (2011).
Yoon, J. H. et al. LincRNA-p21 suppresses target mRNA translation. Mol Cell 47, 648–655, https://doi.org/10.1016/j.molcel.2012.06.027 (2012).
Taniue, K. et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc Natl Acad Sci USA 113, 1273–1278, https://doi.org/10.1073/pnas.1500992113 (2016).
Hirose, T. et al. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol Biol Cell 25, 169–183, https://doi.org/10.1091/mbc.E13-09-0558 (2014).
Tripathi, V. et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39, 925–938, https://doi.org/10.1016/j.molcel.2010.08.011 (2010).
Li, N., Ponnusamy, M., Li, M. P., Wang, K. & Li, P. F. The Role of MicroRNA and LncRNA-MicroRNA Interactions in Regulating Ischemic Heart Disease. J Cardiovasc Pharmacol Ther, https://doi.org/10.1177/1074248416667600 (2016).
Wang, J. et al. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res 38, 5366–5383, https://doi.org/10.1093/nar/gkq285 (2010).
Ma, M. Z. et al. Long non-coding RNA CCAT1 promotes gallbladder cancer development via negative modulation of miRNA-218-5p. Cell Death Dis 6, e1583, https://doi.org/10.1038/cddis.2014.541 (2015).
Salem, M., Kenney, P. B., Rexroad, C. E. & Yao, J. Molecular characterization of muscle atrophy and proteolysis associated with spawning in rainbow trout. Comp Biochem Physiol Part D Genomics Proteomics 1, 227–237, https://doi.org/10.1016/j.cbd.2005.12.003 (2006).
Salem, M., Kenney, P. B., Rexroad, C. E. & Yao, J. Proteomic signature of muscle atrophy in rainbow trout. J Proteomics 73, 778–789, https://doi.org/10.1016/j.jprot.2009.10.014 (2010).
Llano-Diez, M., Gustafson, A. M., Olsson, C., Goransson, H. & Larsson, L. Muscle wasting and the temporal gene expression pattern in a novel rat intensive care unit model. BMC Genomics 12, 602, https://doi.org/10.1186/1471-2164-12-602 (2011).
Gomes, M. D., Lecker, S. H., Jagoe, R. T., Navon, A. & Goldberg, A. L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 98, 14440–14445, https://doi.org/10.1073/pnas.251541198 (2001).
Al-Tobasei, R., Paneru, B. & Salem, M. Genome-Wide Discovery of Long Non-Coding RNAs in Rainbow Trout. PLoS One 11, e0148940, https://doi.org/10.1371/journal.pone.0148940 (2016).
Georgantas, R. W. et al. Inhibition of myogenic microRNAs 1, 133, and 206 by inflammatory cytokines links inflammation and muscle degeneration in adult inflammatory myopathies. Arthritis Rheumatol 66, 1022–1033, https://doi.org/10.1002/art.38292 (2014).
Pauli, A. et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res 22, 577–591, https://doi.org/10.1101/gr.133009.111 (2012).
Paneru, B., Al-Tobasei, R., Palti, Y., Wiens, G. D. & Salem, M. Differential expression of long non-coding RNAs in three genetic lines of rainbow trout in response to infection with Flavobacterium psychrophilum. Sci Rep 6, 36032, https://doi.org/10.1038/srep36032 (2016).
Guttman, M. et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300, https://doi.org/10.1038/nature10398 (2011).
Cabili, M. N. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25, 1915–1927, https://doi.org/10.1101/gad.17446611 (2011).
Eisenberg, I. et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci USA 104, 17016–17021, https://doi.org/10.1073/pnas.0708115104 (2007).
Ardite, E. et al. PAI-1-regulated miR-21 defines a novel age-associated fibrogenic pathway in muscular dystrophy. J Cell Biol 196, 163–175, https://doi.org/10.1083/jcb.201105013 (2012).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, https://doi.org/10.1038/nature11247 (2012).
Busch, A., Richter, A. S. & Backofen, R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 24, 2849–2856, https://doi.org/10.1093/bioinformatics/btn544 (2008).
Agostini, F. et al. catRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics 29, 2928–2930, https://doi.org/10.1093/bioinformatics/btt495 (2013).
Jalali, S., Bhartiya, D., Lalwani, M. K., Sivasubbu, S. & Scaria, V. Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PLoS One 8, e53823, https://doi.org/10.1371/journal.pone.0053823 (2013).
Berthelot, C. et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun 5, 3657, https://doi.org/10.1038/ncomms4657 (2014).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35, W345–349, https://doi.org/10.1093/nar/gkm391 (2007).
Salem, M. et al. Transcriptome assembly, gene annotation and tissue gene expression atlas of the rainbow trout. Plos One (2015).
Ramachandra, R. K., Salem, M., Gahr, S., Rexroad, C. E. & Yao, J. Cloning and characterization of microRNAs from rainbow trout (Oncorhynchus mykiss): their expression during early embryonic development. BMC Dev Biol 8, 41, https://doi.org/10.1186/1471-213X-8-41 (2008).
Marancik, D. et al. Whole-body transcriptome of selectively bred, resistant-, control-, and susceptible-line rainbow trout following experimental challenge with Flavobacterium psychrophilum. Front Genet 5, 453, https://doi.org/10.3389/fgene.2014.00453 (2014).
Ali, A., Rexroad, C. E., Thorgaard, G. H., Yao, J. & Salem, M. Characterization of the rainbow trout spleen transcriptome and identification of immune-related genes. Front Genet 5, 348, https://doi.org/10.3389/fgene.2014.00348 (2014).
Li, J. J., Huang, H., Bickel, P. J. & Brenner, S. E. Comparison of D. melanogaster and C. elegans developmental stages, tissues, and cells by modENCODE RNA-seq data. Genome Res 24, 1086–1101, https://doi.org/10.1101/gr.170100.113 (2014).
Al-Tobasei, R. et al. Identification of SNPs associated with muscle yield and quality traits using allelic-imbalance analyses of pooled RNA-Seq samples in rainbow trout. BMC Genomics 18, 582, https://doi.org/10.1186/s12864-017-3992-z (2017).
Bolstad, B. M., Irizarry, R. A., Astrand, M. & Speed, T. P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 (2003).
Lopes, C. T. et al. Cytoscape Web: an interactive web-based network browser. Bioinformatics 26, 2347–2348, https://doi.org/10.1093/bioinformatics/btq430 (2010).
Farre, D. et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res 31, 3651–3653 (2003).
Juanchich, A. et al. Characterization of an extensive rainbow trout miRNA transcriptome by next generation sequencing. BMC Genomics 17, 164, https://doi.org/10.1186/s12864-016-2505-9 (2016).
Rueda, A. et al. sRNAtoolbox: an integrated collection of small RNA research tools. Nucleic Acids Res 43, W467–473, https://doi.org/10.1093/nar/gkv555 (2015).
Acknowledgements
This study was supported by a competitive grant No. 2014-67015-21602 from the United States Department of Agriculture, National Institute of Food and Agriculture (MS). R.A.T trainee’s projects are supported by Grant Number T32HL072757 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
M.S. and B.K. conceived and designed the experiments. B.P., A.A., R.A., M.S. and B.K. performed the experiment. B.P., A.A., R.A., M.S. and B.K. analyzed the data. B.P. and M.S. wrote the paper. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Paneru, B., Ali, A., Al-Tobasei, R. et al. Crosstalk among lncRNAs, microRNAs and mRNAs in the muscle ‘degradome’ of rainbow trout. Sci Rep 8, 8416 (2018). https://doi.org/10.1038/s41598-018-26753-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26753-2
This article is cited by
-
Expression profile of long non-coding RNAs in the intestine of black rockfish Sebastes schlegelii in response to Edwardsiella tarda infection
Journal of Oceanology and Limnology (2023)
-
Gene expression profiling of trout muscle during flesh quality recovery following spawning
BMC Genomics (2022)
-
Long non-coding RNAs in the physiology of aquaculture animals: a perspective update
Reviews in Fish Biology and Fisheries (2022)
-
Genome-wide scan for common variants associated with intramuscular fat and moisture content in rainbow trout
BMC Genomics (2020)
-
Genome-wide identification and comparison of differentially expressed profiles of miRNAs and lncRNAs with associated ceRNA networks in the gonads of Chinese soft-shelled turtle, Pelodiscus sinensis
BMC Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
