
Abstract
Piranhas and pacus (Characiformes: Serrasalmidae) are a charismatic but understudied family of Neotropical fishes. Here, we analyse a DNA barcode dataset comprising 1,122 specimens, 69 species, 16 genera, 208 localities, and 34 major river drainages in order to make an inventory of diversity and to highlight taxa and biogeographic areas worthy of further sampling effort and conservation protection. Using four methods of species discovery—incorporating both tree and distance based techniques—we report between 76 and 99 species-like clusters, i.e. between 20% and 33% of a priori identified taxonomic species were represented by more than one mtDNA lineage. There was a high degree of congruence between clusters, with 60% supported by three or four methods. Pacus of the genus Myloplus exhibited the most intraspecific variation, with six of the 13 species sampled found to have multiple lineages. Conversely, piranhas of the Serrasalmus rhombeus group proved difficult to delimit with these methods due to genetic similarity and polyphyly. Overall, our results recognise substantially underestimated diversity in the serrasalmids, and emphasise the Guiana and Brazilian Shield rivers as biogeographically important areas with multiple cases of across-shield and within-shield diversifications. We additionally highlight the distinctiveness and complex phylogeographic history of rheophilic taxa in particular, and suggest multiple colonisations of these habitats by different serrasalmid lineages.
Similar content being viewed by others


Introduction
Piranhas are one of the iconic animals of the Amazon, and as vividly expressed by Theodore Roosevelt1 they are often demonised in popular culture as voracious man-eaters: “The head with its short muzzle, staring malignant eyes, and gaping, cruelly armed jaws, is the embodiment of evil ferocity”. Yet despite their infamy, piranhas are poorly known in terms of species diversity, and in common with Amazon ichthyofauna in general are insufficiently investigated taxonomically2.
Together with the mostly herbivorous pacus and silver dollars, piranhas comprise the family Serrasalmidae (Ostariophysi: Characiformes), currently with 94 valid species in 16 genera3. Endemic to the Neotropics, extant serrasalmids are native to all major drainages east of the Andes including the Amazon, Orinoco, São Francisco, Essequibo (plus smaller coastal Atlantic rivers), Paraná-Paraguay, and also the Maracaibo basin4. While some species such as the tambaqui (Colossoma macropomum) and the piranhas Serrasalmus rhombeus and Catoprion mento are widely distributed and found in most major river systems4, others have highly restricted distributions. Ossubtus xinguense, for example, is endemic to the rapids of the middle Xingu River5, while Mylesinus paucisquamatus is endemic to the rapids of the Tocantins River4,6,7. Other rheophilic species of the genera Tometes and Utiaritichthys are also endemic to the rapids of other rivers8,9,10. Important commercial fisheries, subsistence fisheries, and aquaculture industries exist for Colossoma macropomum, Piaractus spp., and Mylossoma spp.11,12. Serrasalmids are a morphologically, ecologically, and behaviourally diverse group with a variety of feeding strategies and associated morphological adaptations13,14,15; Colossoma and Piaractus are fruit and seed eaters inhabiting large rivers and are important dispersers of seeds in flooded forests13,16; Tometes, Mylesinus, Utiaritichthys and Ossubtus are rheophiles and specialist consumers of aquatic plants (Podostemaceae) growing in rapids5; while Acnodon normani and Catoprion mento specialise in eating the scales of other fishes17,18.
Morphologically the family is characterised by a deep and compressed body, pre-dorsal spine, and abdominal and pelvic spines forming a ventral keel4,19. While the 38 species of piscivorous true piranhas of the genera Serrasalmus, Pygocentrus, Pygopristis and Pristobrycon can be easily diagnosed by their single row of sharp interlocking teeth, the remaining genera–a non-monophyletic group of 56 species commonly known as “pacus” (e.g. Myleus, Myloplus, Piaractus) or “silver dollars” (Metynnis)–possess two rows of molariform or incisiform premaxillary teeth5,8,19,20,21,22. Phylogenetic hypotheses based either on morphological23,24 or molecular25,26 characters have been published. Yet despite these phylogenetic studies and an investigation into broad biogeographic patterns27, only limited effort has been applied to understanding intraspecific variation, save for a limited number of phylogeographic studies of species of commercially important genera such as Colossoma28, Piaractus29 and Mylossoma30. Broad coverage DNA barcoding studies of ichthyofauna including serrasalmids are also lacking in the Neotropics, with the exception of the Paraná River basin31,32.
Molecular techniques are being increasingly used as a tool for biodiversity inventories33,34, and are particularly useful for quickly characterising hyperdiverse tropical communities and flagging new candidate species31. Although single locus methods such as DNA barcoding provide insufficient evidence for the description of an individual species without supporting data35,36,37,38, when viewed in the broader contexts of either higher taxonomic rank or geographic area, DNA-barcode species estimates have been shown to be congruent with traditional taxonomy39,40,41. Methods generating these species-like clusters of sequences–also known as candidate species, OTUs, MOTUs, or mtDNA lineages–are best described in terms of “species discovery” rather than “species delimitation”, as the latter requires multiple character sets42; thus, we refer to them as single-locus species-discovery (SLSD) methods. Despite the limitations of inferences based on a single locus, conceptual advances incorporating coalescent theory33,43,44,45 have improved the theoretical justification of tree-based genealogical methods over simpler distance methods requiring arbitrary or generalised distance thresholds42. Importantly, by using multiple methods it is possible to counter potential biases and the lack of statistical power associated with any individual method46,47, while the provision of confidence intervals enables researchers to capture the genealogical and phylogenetic uncertainty inherent in species delimitation48,49.
Given the lack of a geographically wide-ranging DNA-based assessment of serrasalmids, we aim to provide the first detailed inventory of this group with the ultimate aim of better guiding conservation priorities and highlighting groups in need of taxonomic revisions. Specifically, we will: (1) generate a DNA barcode dataset for serrasalmids from the Brazilian Amazon, but also include data from the Orinoco, São Francisco and Paraná river basins where available; (2) make an overall inventory of molecular variation using SLSD methods; (3) identify taxa and geographic areas harbouring previously unrecognised lineages (with particular reference to rheophilic groups).
Methods
Sample collection
Muscle and fin-membrane tissue samples were taken in the field from the right-hand pectoral-fin base or from the right side of the flank, and were stored in 95% ethanol and deposited in the tissue collection of the Laboratόrio de Evolução e Genética Animal (LEGAL) at the Universidade Federal do Amazonas (UFAM). Voucher specimens were fixed in 10% formalin and deposited in the fish collection of the Instituto Nacional de Pesquisas da Amazônia (INPA). Vouchers were identified to species by taxonomic specialists using available comparative material, identification keys, original descriptions, and redescriptions of species8,50,51,52,53,54. Individuals that could not be identified to species level were reported as “Genus sp.” (possible new/unidentified species) or as “Genus aff. species” (closely related species, possibly new).
Field collections in Brazil were authorised by IBAMA/MMA 045/2008-2011, IBAMA/SISBIO 11325-1, and access to genetic resources was authorised by permit No. 034/2005/IBAMA. IBAMA field collection permits are conditional that collection of organisms be undertaken in accordance with the ethical recommendations of the Conselho Federal de Biologia (CFBio; Federal Council of Biologists), Resolution 301 (December 8, 2012). Field collections in Colombia were authorised by the “Permiso Marco de Investigaciόn” granted by the Ministerio de Ambiente y Desarrollo Sostenible to the Universidad de los Andes, Bogota, Colombia. Collections in French Guiana were authorised under permit APA-973-7 for collections in the core area of the French Guiana Amazonian Park.
DNA barcode sequence generation
Total DNA was isolated from approximately 50 mg of tissue using standard phenol-chloroform extraction methods55. A fragment of 651/657 bp of mitochondrial cytochrome c oxidase subunit I (COI) was amplified using the M13-tailed primer cocktails FishF2/FishR2 and VF2/VR1d respectively56. The 15 μL PCR mix included 1.2 μL of 10 mM dNTPs (2.5 mM each DNTP), 1.5 μL 10× buffer (75 mM Tris HCL, 50 mM KCL, 20 mM (NH4)2SO4), 1.2 μL 25 mM MgCl2, 1.5 μL of primer cocktails (2 pmol each), 0.5 μL of Taq DNA polymerase, 1 μL of template DNA and 6.6 μL ddH2O. PCR conditions were: 94 °C (30 sec); 35 cycles of 94 °C (30 sec), 50 °C (35 sec), 72 °C (90 sec); followed by 72 °C (5 min). Amplicons obtained were purified and then sequenced bidirectionally on an automatic ABI 3500 sequencer (Applied Biosystems).
The forward and reverse chromatograms were assembled into contigs using Geneious 7.0.657 and edited manually where required. The sequences were then aligned using Mafft v7.30758, and checked manually for insertions, deletions or stop codons using translated amino acids in Geneious. The alignment was trimmed to 621 bp to reduce missing data and erroneous base calls at the ends of the contigs59.
Further sequences from the GenBank database were added to the dataset. Using the rentrez_1.0.4 interface60 we searched GenBank in July 2017 using the terms “Serrasalmidae” and “COI”, “cox1” or “CO1”, requesting only sequences between 450 and 1,000 bp in length. Searches for longer sequences (i.e. mitochondrial genomes) did not reveal any species not already sampled. Any sequences generated from specimens collected outside of South America, or those that clearly appeared to be attributed to incorrect species names were removed.
Single locus discovery of species
We used four single-locus species-discovery (SLSD) methods to partition our dataset into putative species-like clusters: (1) GMYC, the general mixed Yule coalescent model33,43,61; (2) bGMYC, a Bayesian implementation of the GMYC48; (3) local minima (locMin), a distance threshold optimising and clustering approach from the spider_1.3-0 software package62; and (4) mPTP, the poisson tree process method44,45. Unless otherwise stated, analyses were carried out in R 3.4.163. Beast 1.8.464 was used to generate a posterior sample of ultrametric trees for the GMYC analyses. The dataset was first collapsed to unique haplotypes65. The Beast analysis was set up as follows: substitution model TN93 + Γ as selected by jModeltest266; single model partition; strict molecular clock (relaxed clock was tested for a priori); fixed arbitrary substitution rate of 0.01; and coalescent tree prior33,65. Three independent chains were run for 20 million generations from random starting topologies, and were sampled every 18,000 generations, resulting in 3,333 trees (of which 333 were discarded as burn-in). The 3,000 post burn-in trees were combined and then subsampled to 1,000 for all downstream analyses. Tracer67 was used to verify the chains had reached stationarity.
GMYC, bGMYC and mPTP analyses were carried out as: (1) a point estimate based on the maximum clade credibility tree created in TreeAnnotator 1.8.4 (node heights “ca”); and (2) confidence intervals calculated from the posterior sample of 1,000 trees. We used the bGMYC_1.0.248, splits_1.0-1943 and ape_4.168 packages. The bGMYC posterior samples were summarised into putative species with a conservative posterior probability of conspecificity at 0.05. For mPTP (single lambda), the Beast chronograms (ultrametric trees with branch lengths scaled by time) were first transformed into phylograms (branch lengths scaled by substitutions per site) using maximum likelihood optimisation in phangorn_2.2.069 under the same substitution model settings as described above. The locMin analyses were again conducted as a point estimate, and also on a set of 1,000 bootstrapped datasets to generate a confidence interval for this method.
Data availability
New sequence data generated here are available from the GenBank nucleotide archive under the accessions MG751915–MG752866, and at the Barcode of Life BOLD database under the project name “PRNHA”. Metadata for all sequences used in this study are presented in Supplementary Table S1 as a comma delimited flatfile following Darwin Core standard vocabulary (http://rs.tdwg.org/dwc/terms/index.htm). The datasets and scripts used in this study are available from a public GitHub repository hosted at https://github.com/legalLab/publications.
Results
Sampling and data description
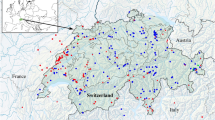
As part of this study a total of 975 serrasalmid individuals were collected from 168 unique localities in 30 major river drainages (Fig. 1). With the addition of data from GenBank this increased to 1,122 specimens from 208 unique localities in 34 major drainages. Upon morphological assessment a total of 68 species-level taxa were identified and we were able to assign taxonomic names to 60 of these (i.e. valid nominal species), with a further eight being identified to genus level only, i.e. putative new species (six pacus, two piranhas). Data for thirteen species were obtained from GenBank, but only one of these (Serrasalmus marginatus) was not already present in our dataset (bringing the total to 69 species). Overall, the sampling covered 61 (19 piranha species and 42 pacu species) of the 94 (65%) valid serrasalmid species representing all 16 genera (100%). Per species, 63 of the total 69 (91%) were represented by more than one individual, with 56 (81%) represented by five or more individuals; median number of individuals per species was ten, mean was 16.3, and maximum was 95 (Serrasalmus rhombeus). Fifty-one species (74%) were collected from more than one locality; 45 (65%) were collected from three or more localities; median number of localities per species was four, mean was 6.2, and maximum was 35 (Serrasalmus rhombeus). Forty species (58%) were collected in more than one drainage; 33 (48%) were collected from three or more drainages; median number of drainages per species was two, mean was 3.6, and maximum was 15 (Serrasalmus rhombeus). The aligned DNA barcode matrix comprised 1,122 taxa by 621 bp. The unaligned sequences varied in length 417–621 bp; 11 (1%) were less than 500 bp, 97% were greater than 530 bp, 37% were the full 621 bp; median sequence length was 609 bp (mean 595 bp). The dataset comprised a total of 444 unique haplotypes. Per species (Table 1), 59 (86%) were represented by more than one haplotype, with 28 (41%) represented by five or more haplotypes; median number of haplotypes per species was four, mean was six, and maximum was 29 (Serrasalmus rhombeus).

Map of unique sampling locations shown by red triangles. Of the total 208 unique sampling locations in the study, 188 with GPS coordinates are shown here. Total includes all data accessed from GenBank in addition to collections made as part of this study. Figure was created in R 3.4.163 from void-filled elevation (30 sec DEM) raster and river network (RIV) shape files obtained from the World Wildlife Fund HydroSHEDS project96, and used under the following license. This product incorporates data from the HydroSHEDS database which is ©World Wildlife Fund, Inc. (2006–2013) and has been used herein under license. WWF has not evaluated the data as altered and incorporated within, and therefore gives no warranty regarding its accuracy, completeness, currency or suitability for any particular purpose. Portions of the HydroSHEDS database incorporate data which are the intellectual property rights of ©USGS (2006–2008), NASA (2000–2005), ESRI (1992–1998), CIAT (2004–2006), UNEP–WCMC (1993), WWF (2004), Commonwealth of Australia (2007), and Her Royal Majesty and the British Crown and are used under license. The HydroSHEDS database and more information are available at http://www.hydrosheds.org.
Single locus discovery of species
Point estimates for the SLSD varied between 76 putative species (locMin) and 99 species (GMYC), with 118 unique molecular delimitations over all methods (Table 2; Fig. 2); confidence intervals (95%) were largest for locMin at 67–140 species and lowest for mPTP at 75–83 species (Table 2). Centers of the confidence interval distributions tended to be lower than the point estimates for the GMYC and bGMYC analyses, and higher for the locMin and mPTP analyses. Of the point estimate delimitations 49% were supported by congruence of all four methods, 60% were supported by three or four methods, and 14% by only one method (Fig. 2).

Maximum clade credibility chronogram from 1,000 posterior trees generated using Beast. Dataset comprised 444 unique haplotypes (from total 1,122) of serrasalmid COI sequences (621 aligned base pairs). Bayesian posterior probabilities above 0.95 are shown as dark nodes. Point estimate species delimitations (total 118 unique delimitations) are shown by method as coloured boxes; due the large number of unique colours, some may appear similar. Rheophilic species are highlighted in bold blue font. Tree was presented using the ggtree_1.6.11 package97.
Among method point estimates, between 14 (20%; mPTP) and 23 (33%; GMYC) of the species were represented by more than one COI lineage, with between nine (13%; locMin, mPTP) and 13 (19%; GMYC) represented by three or more lineages (Table 1). Three or more lineages were reported by all methods for Metynnis luna, Mylesinus paraschomburgkii, Myloplus asterias, Myloplus rhomboidalis, Myloplus schomburgkii, Myloplus aff. rubripinnis, and Mylossoma duriventre. Of the 13 species delimited by all methods as having more than one lineage, six were within the genus Myloplus.
The locMin analysis optimised a divergence threshold of 0.0135 (p-distance) for the dataset. COI lineages delimited by this method varied between 0.023 maximum intraspecific divergence (Myloplus rhomboidalis) and 0.117 (Myloplus schomburgkii), while eight were greater than 0.05 (Table 1). The overall mean maximum intraspecific divergence was 0.0068 with the exclusion of the 15 species showing intraspecific genetic distances above the threshold (Table 1). Of the 69 a priori identified species, 43 (62%) were monophyletic, 20 (29%) were not monophyletic, and six (9%) were singletons (Table 1). Eighteen species (26%) shared haplotypes with another species, and this most commonly occurred in Myloplus (6 spp.) and Serrasalmus (5 spp.). A neighbour-joining tree showing all 1,122 COI sequences coloured by species is presented as Supplementary Fig. S1.
Discussion
In terms of variation in the pacus, of the 13 morphologically identified species of Myloplus, six had multiple lineages with many of these in the Guiana and Brazilian shields; in M. arnoldi, a lineage from the Guiana Shield (Nhamundá River) was identified as divergent from conspecifics in the Brazilian Shield (Araguaia, Tapajόs and Xingu rivers), separated by 0.086 p-distance (Fig. 3; Table 1); in M. rhomboidalis there were two Guiana Shield lineages (Jari and Branco rivers) and one in the Brazilian Shield (Xingu River); in M. schomburgkii (Fig. 3) there was a Guiana Shield lineage (Branco, Negro and Nhamundá rivers) distinct from a Brazilian Shield lineage (Araguaia, Tapajόs and Xingu Rivers), a third lineage found in the Xingu, as well as an intriguing forth lineage from the upper Amazon (Nanay River, Peru) and the Branco River. The species comprising Myloplus asterias/rubripinnis (including M. aff. rubripinnis) was estimated to contain between 11 and 13 lineages, with distinct lineages found in the Araguaia (one), Tocantins (one), Tapajόs (four), Aripuanã (two), Xingu (two), and Jatapu (one) rivers. Examples of within-shield diversification were apparent with two clades of multiple lineages within the Brazilian Shield, and also one lineage showing across-shield conspecificity (Aripuanã, Trombetas, Nhamundá). Extensive non-monophyly of the nominal taxa ascribed to the Myloplus asterias/rubripinnis group indicates problems in current diagnoses of the taxa and/or application of diagnostic characters supporting these taxa, and therefore given this ambiguity and the apparent high levels of within-drainage endemism, a taxonomic revision of this group should be a priority.

Images of freshly caught pacus highlighting groups with significant intraspecific genetic diversity: (a) Myloplus arnoldi, Tapajόs River; (b) Myloplus arnoldi, Nhamundá River; (c) Myloplus asterias, Branco River; (d) Myloplus asterias, Nhamundá River; (e) Myloplus schomburgkii, Nhamundá River; and (f) Myloplus schomburgkii, Tapajόs River. All images were taken by the authors.
Other pacus also displayed large intraspecific divergences. Mylossoma aureum, M. duriventre, and Piaractus brachypomus all revealed lineages in the Orinoco distinct from those of the Amazon basin (see also Escobar et al.29 and Mateussi et al.30). Mylesinus paraschomburgkii showed evidence of distinct lineages in the Guiana Shield rivers (Uatumã, Trombetas and Jari), while a singleton specimen of Mylesinus aff. paraschomburgkii from the Nhamundá River was nested within Myloplus zorroi from the Aripuanã River. The distribution of lineages within Myloplus lobatus and Myleus setiger also indicated a biogeographic link between the Aripuanã and the Nhamundá, Jatapu, Uatumã and Trombetas rivers, reflecting the historical proximity of the mouths of these southerly flowing Guiana Shield rivers before the capture of the north flowing Brazilian Shield Aripuanã River by the Madeira River. Sharing of species and lineages between the Aripuanã and Guiana Shield rivers is not restricted to the serrasalmids, but has also been observed in cichlids of the genus Symphysodon70,71 and loricariid catfishes72. Aside from the three potential lineages of Metynnis luna, there were few new lineages of silver dollars, likely reflecting the recent taxonomic work22 and ongoing studies being carried out on the group (Ota, studies in progress).
Patterns among piranhas were less clear than for the pacus, with greater incongruence among methods. With 16 species considered valid, there were between 12 (locMin) and 22 (GMYC) lineages in Serrasalmus and Pygocentrus. While the distinctiveness of Pygocentrus cariba, P. piraya, Serrasalmus brandtii, S. elongatus, S. gouldingi, S. manueli, S. serrulatus and S. spilopleura were well supported by at least three of the four methods, the Serrasalmus rhombeus group was the primary source of incongruence. Here, the mPTP method recognised only one species in an inclusive clade comprising eight nominal taxa, and the GMYC methods recognised six species. This indicates that this group may be at the limits of resolution for a single mitochondrial locus and the methods employed here. Previous genetic analyses of the group have reported similar observations, with extensive haplotype sharing between species73. Regardless, several of the species within this group were recovered as monophyletic despite the low genetic divergences, including Serrasalmus altispinis, S. compressus, S. hastatus, and S. marginatus. Some individuals of S. gibbus and S. maculatus were nested within S. rhombeus, but these were probably the result of misidentification (Supplementary Fig. S1). Although not supported by all methods, it is possible that given the patterns observed in other taxa, the S. rhombeus individuals from the Xingu River represent a distinct species. Therefore, due to ontogenetic complexities, genetic similarity and the subtle morphological differences among species and lineages of the S. rhombeus clade, we feel it would benefit greatly from a population genomic analyses before any taxonomic treatment of the group is embarked upon. In the genus Pygocentrus, the species P. nattereri was found to comprise up to four lineages. One of these four was represented by GenBank samples nested within Serrasalmus maculatus from the Paraná River, and we believe that these samples were misidentified. Although not supported by all delimitations, the other three lineages of P. nattereri are possibly distinct, with one from the Tocantins/Araguaia/São Bento rivers and another from the Guaporé River, and both distinct from the more widespread Amazonas clade. Serrasalmus maculatus from the upper Paraná River was reported by three of the four methods to form a distinct lineage from S. maculatus of the lower Paraná River. The upper and lower Paraná River were distinct ichthyofaunal provinces separated by the Sete Quedas rapids until the construction of the Itaipú dam, which due to its system of locks, permitted the homogenisation of these faunas74.
Among the 69 a priori identified taxonomic species of serrasalmid analysed herein, up to 23 are represented by more than one COI lineage (Table 1). Despite recent studies and taxonomic revisions describing new species5,21,22, our results show that a number of potential new species may still await a formal morphological diagnosis and description. Many factors contribute to this underestimation of diversity within the family. Only few morphological studies have been published in the last 10 years, reflecting the difficulties of interpreting high levels of ontogenetic variation, allometric growth, sexual dimorphism, and spatial variation in both body shape and colour pattern5,21,25,75,76. The number of possibly unrecognised species observed here in serrasalmids support the conclusions of Reis et al.2, who estimated that 34–42% of Neotropical freshwater fishes remain undescribed, and are mostly concentrated in the Amazon basin. The main explanations for this unrecognised diversity stem largely from (1) historically poorly sampled areas above geological barriers such as rapids; (2) widespread taxa or heterogeneous taxa with insufficient or overwhelming amounts of museum material; or (3) cryptic or pseudocryptic (morphological differences apparent but overlooked) diversity in widespread species. Genetic data are an important instrument in uncovering cases of the latter77.
Of particular importance are the rheophilic taxa inhabiting rapids. Geologically the western Amazon basin is characterised by a sedimentary basin, and in the central and eastern portion by the crystalline Guiana and Brazilian Shields separated by the Amazon River78. As affluents of the Amazon descend the Guiana and Brazilian Shields, they form riffles, rapids and waterfalls inhabited by a distinctive fauna and flora. Aquatic flora of the rapids habitats is characterised by the Podostemaceae79, while perhaps the best known faunal component of these habitats are the loricariid catfishes80. Serrasalmids also are a conspicuous component. Our data indicate that rheophilic pacus classified in the genera Tometes, Ossubtus, Utiaritichthys, Mylesinus and Myloplus represent multiple, apparently evolutionarily independent lineages of rheophilic fishes (Fig. 2). Conversely, we also show lineages and haplotypes to be shared between rheophilic habitats of different rivers–e.g. the Tometes camunani complex–supporting the hypothesis of interconnection of these rheophilic habitats during low-water glacial periods of the Pleistocene81. Serrasalmids present not only a fascinating window into adaptations to extreme environments and the complexity of diversification patterns in this environment82,83,84, but the strictly rheophilic species are also the most threatened of the serrasalmids since hydroelectric projects are developed at sites of rapids and waterfalls, largely destroying these unique habitats and their associated taxa85.
The underestimation of the variation and diversity in the Serrasalmidae–and which can be extrapolated to Amazonian aquatic fauna in general–is directly relevant to the conservation of these groups. Due to high proportions of faunal endemism and increasing anthropogenic threats, the Brazilian Shield rivers and their faunas are of particular conservation concern80,86,87. Reis88 summarised the principal anthropogenic threats of the Amazon River basin as: extensive deforestation of Amazon forest; hydroelectric dam building with the associated transformation of lotic environments into lentic environments resulting in the extirpation or significant reduction of populations of rheophilic species (while concomitantly contributing to the proliferation of lentic-adapted species); alluvial gold mining causing mercury contamination; and overexploitation of most commercial species. Therefore, we reiterate the conclusions of Reis88, who suggested that one of the primary instruments for conservation of Amazon basin fishes is increasing expertise in fish taxonomy and systematics. Additionally, we advocate DNA barcoding and other genetic tools as powerful complementary methods for uncovering fish diversity and highlighting groups in need of taxonomic revisions. Here we demonstrate the utility of DNA barcoding in providing an independent estimate of species alpha diversity, and additionally in providing preliminary data on population subdivision, gene flow, and relative ages of divergences.
However, important caveats need to be considered when interpreting single locus mtDNA data. Our confidence intervals were generally wide, reflecting the influence of phylogenetic uncertainty on our results and its importance in species delimitation as a whole48,49. Furthermore, the failure to congruently discriminate closely related species, such as those in the Serrasalmus rhombeus group, is perhaps a reflection of the limitations of single threshold SLSD methods when faced with situations where species with large effective population sizes have recently diverged in rapid succession (i.e. a young radiation), conditions whereby single locus methods are known to underestimate species diversity43,89,90. While multiple threshold models were developed to accommodate variation in coalescent depths between groups33,45, we were unable to generate realistic results while experimenting with these settings (as evidenced by excessive splitting). This is largely due to multiple threshold models making possibly spurious delimitations by recognising population structuring as speciation events91,92. Therefore, where delimitations are implausible or incongruent we recommend secondary sources of data be (re-)examined. While rates of phenotypic evolution and speciation are correlated over macroevolutionary scale93, there will be situations where local adaptation and fine-scale speciation may change phenotypes at a rate significantly faster than can be identified by neutral loci94, emphasising the need for additional data from morphology, behaviour, distribution, and ecology35,36,37,38 when undertaking systematic revisions.
Geographic scale is another important factor in determining the structure of DNA barcode datasets; Bergsten et al.40 demonstrated the substantial increase in intraspecific diversity and the decrease in interspecific divergence over increasing geographical distances. Fortunately, GMYC methods have been shown to be robust to the presence of singletons and absences of intermediate haplotypes65,89, but where we report putatively new lineages based on low numbers of individuals, effort still needs to be made to source more specimens before more conclusive statements can be made about the distinctiveness of those taxa95. Overall our sampling generated a geographically broad dataset with three quarters of the species having been collected from more than one locality, and over half being collected from more than one major drainage. Despite the positive bias for samples from the eastern and central Brazilian Amazon, and the paucity of samples from the western Amazon, we are confident in having captured a significant proportion of serrasalmid diversity. Inventories from Peru, Colombia and Ecuador would be extremely valuable additions, however.
Our single locus species delimitation results support a notion that piranha and pacu taxonomic diversity is currently underestimated in the Brazilian Amazon. The four methods achieved a high level of congruence (60% of the lineages were supported by three or more methods), indicating they were recognising a common signal of diversification, with great majority of these lineages also supported as allopatric and biogeographically distinct populations. The results particularly highlight: (1) the Guiana and Brazilian Shields as regions of underestimated but high ichthyofaunal endemism and diversity; (2) the existence of both between-shield (e.g. Myloplus schomburgkii, M. arnoldi), and within-shield (Myleus setiger, Mylesinus paraschomburgkii, Myloplus rubripinnis/asterias) diversification patterns in pacus; (3) very recent biogeographic connection between the the Aripuanã (Brazilian Shield) and Guiana Shield rivers; (4) distinct lineages of species shared between the Amazon and Orinoco basins (Mylossoma aureum, M. duriventre, Piaractus brachypomus); (5) the evolutionary uniqueness, distinctness, and apparent independent evolution of rheophilic lineages; and (6) the taxonomic difficulties associated with piranhas. Thus, characterisation of these faunas by traditional taxonomic methods combined with further effort in sequencing more loci is needed to better understand the implications of these results in an explicit and testable biogeographic framework of Neotropical diversification and community assemblage.
References
Roosevelt, T. Through the Brazilian Wilderness (New York, 1914).
Reis, R. E. et al. Fish biodiversity and conservation in South America. Journal of Fish Biology 89, 12–47, https://doi.org/10.1111/jfb.13016 (2016).
Eschmeyer, W. N., Fricke, R. & van der Laan, R. Catalog of Fishes (2017).
Jégu, M. Subfamily Serrasalminae (pacus and piranhas). In Reis, R. E., Kullander, S. O. & Ferraris, C. J. (eds) Check List of the Freshwater Fishes of South and Central America, 182–196 (Edipucrs, Porto Alegre, 2003).
Andrade, M. C., Sousa, L. M., Ota, R. P., Jégu, M. & Giarrizzo, T. Redescription and geographical distribution of the endangered fish Ossubtus xinguense Jégu 1992 (Characiformes, Serrasalmidae) with comments on conservation of the rheophilic fauna of the Xingu River. PLoS ONE 11, e0161398, https://doi.org/10.1371/journal.pone.0161398 (2016).
Jégu, M. & dos Santos, G. M. Une nouvelle espèce du genre Mylesinus (Pisces, Serrasalmidae), M. paucisquamatus, décrite du bassin du Rio Tocantins (Amazonie, Brésil). Cybium 12, 331–341 (1988).
Vitorino Júnior, O. B., Agostinho, C. S. & Pelicice, F. M. Ecology of Mylesinus paucisquamatus Jégu & Santos, 1988, an endangered fish species from the rio Tocantins basin. Neotropical Ichthyology 14, 463–470, https://doi.org/10.1590/1982-0224-20150124 (2016).
Andrade, M. C., Giarrizzo, T. & Jégu, M. Tometes camunani (Characiformes: Serrasalmidae), a new species of phytophagous fish from the Guiana Shield, rio Trombetas basin, Brazil. Neotropical Ichthyology 11, 297–306, https://doi.org/10.1590/S1679-62252013000200008 (2013).
Andrade, M. C., Machado, V. N., Jégu, M., Farias, I. P. & Giarrizzo, T. A new species of Tometes Valenciennes 1850 (Characiformes: Serrasalmidae) from Tocantins-Araguaia River Basin based on integrative analysis of molecular and morphological data. PLoS ONE 1850, e0170053, https://doi.org/10.1371/journal.pone.0170053 (2017).
Pereira, T. N. A. & Castro, R. M. C. A new species of Utiaritichthys Miranda Ribeiro (Characiformes: Serrasalmidae) from the Serra dos Parecis, Tapajós drainage. Neotropical Ichthyology 12, 397–402, https://doi.org/10.1590/1982-0224-20130137 (2014).
Ruffino, M. L. A Pesca e os Recursos Pesqueiros na Amazonia Brasileira (ProVarzea/Ibama, 2004).
Saint-Paul, U. Native fish species boosting Brazilian’s aquaculture development. Acta of Fisheries and Aquatic Resources 5, 1–9, https://doi.org/10.2312/ActaFish.2017.5.1.1-9 (2017).
Goulding, M. The Fishes and the Forest (University of California Press, 1980).
Winemiller, K. O. Ontogenetic diet shifts and resource partitioning among piscivorous fishes in the Venezuelan Llanos. Environmental Biology of Fishes 26, 177–199, https://doi.org/10.1007/BF00004815 (1989).
Sazima, I. & Machado, F. A. Underwater observations of piranhas in western Brazil. Environmental Biology of Fishes 28, 17–31, https://doi.org/10.1007/BF00751026 (1990).
Anderson, J. T., Rojas, S. J. & Flecker, A. S. High-quality seed dispersal by fruit-eating fishes in Amazonian floodplain habitats. Oecologia 161, 279–290, https://doi.org/10.1007/s00442-009-1371-4 (2009).
Sazima, I. Scale eating in characoids and other fishes. Environmental Biology of Fishes 9, 87–101, https://doi.org/10.1007/BF00690855 (1983).
Leite, R. G. & Jégu, M. Régime alimentaire de deux éspecies d’Acnodon (Characiformes, Serrasalmidae) et habitudes lepidophages de A. normani. Cybium 14, 353–359 (1990).
Eigenmann, C. H. The Serrasalminae and Mylinae. Annals of the Carnegie Museum 9, 226–272 (1915).
Machado-Allison, A. Estudios sobre la sistemática de la subfamilia Serrasalminae (Teleostei, Characidae). Parte II. Discusión sobre la condición monofilética de la subfamilia. Acta Biologica Venezolana 11, 145–195 (1983).
Andrade, M. C., Jégu, M. & Giarrizzo, T. A new large species of Myloplus (Characiformes, Serrasalmidae) from the Rio Madeira basin, Brazil. ZooKeys 571, 153–167, https://doi.org/10.3897/zookeys.571.5983 (2016).
Ota, R. P., Rapp Py-Daniel, L. H. & Jégu, M. A new silver dollar species of Metynnis Cope, 1878 (Characiformes: Serrasalmidae) from northwestern Brazil and southern Venezuela. Neotropical Ichthyology 14, 1–12, https://doi.org/10.1590/1982-0224-20160023 (2016).
Machado-Allison, A. Estudios sobre la subfamilia Serrasalminae. Parte III: sobre el estatus genérico y relaciones filogenéticas de los géneros Pygopristis, Pygocentrus, Pristobrycon ySerrasalmus (Teleostei-Characidae-Serrasalminae). Acta Biologica Venezolana 12, 19–42 (1985).
Cione, A. L., Dahdul, W. M., Lundberg, J. G. & Machado-Allison, A. Megapiranha paranensis, a new genus and species of Serrasalmidae (Characiformes, Teleostei) from the upper Miocene of Argentina. Journal of Vertebrate Paleontology 29, 350–358, https://doi.org/10.1671/039.029.0221 (2009).
Freeman, B., Nico, L. G., Osentoski, M., Jelks, H. L. &Collins, T. M. Molecular systematics of Serrasalmidae: deciphering the identities of piranha species and unraveling their evolutionary histories. Zootaxa 1–38 (2007).
Thompson, A. W., Betancur-R., R., López-Fernández, H. & Ortí, G. A time-calibrated, multi-locus phylogeny of piranhas and pacus (Characiformes: Serrasalmidae) and a comparison of species tree methods. Molecular Phylogenetics and Evolution 81, 242–257, https://doi.org/10.1016/j.ympev.2014.06.018 (2014).
Hubert, N. et al. Phylogeography of the piranha generaSerrasalmus and Pygocentrus: implications for the diversification of the Neotropical ichthyofauna. Molecular Ecology 16, 2115–2136, https://doi.org/10.1111/j.1365-294X.2007.03267.x (2007).
Santos, M. C. F., Ruffino, M. L. & Farias, I. P. High levels of genetic variability and panmixia of the tambaqui Colossoma macropomum (Cuvier, 1816) in the main channel of the Amazon River. Journal of Fish Biology 71, 33–44, https://doi.org/10.1111/j.1095-8649.2007.01514.x (2007).
Escobar L., M. D., Andrade-López, J., Farias, I. P. & Hrbek, T. Delimiting evolutionarily significant units of the fish, Piaractus brachypomus (Characiformes: Serrasalmidae), from the Orinoco and Amazon river basins with insight on routes of historical connectivity. Journal of Heredity 106, 428–438, https://doi.org/10.1093/jhered/esv047 (2015).
Mateussi, N. T. B., Pavanelli, C. S. & Oliveira, C. Molecular identification of cryptic diversity in species of cis-Andean Mylossoma (Characiformes: Serrasalmidae). Mitochondrial DNA Part A 1394, 1–3, https://doi.org/10.1080/24701394.2016.1180515 (2016).
Pereira, L. H. G., Hanner, R., Foresti, F. & Oliveira, C. Can DNA barcoding accurately discriminate megadiverse Neotropical freshwater fish fauna? BMC Genetics 14, 20, https://doi.org/10.1186/1471-2156-14-20 (2013).
Díaz, J. et al. First DNA barcode reference library for the identification of south American freshwater fish from the lower Paraná river. PLoS ONE 11, e0157419, https://doi.org/10.1371/journal.pone.0157419 (2016).
Monaghan, M. T. et al. Accelerated species inventory on Madagascar using coalescent-based models of species delineation. Systematic Biology 58, 298–311, https://doi.org/10.1093/sysbio/syp027 (2009).
Miller, S. E., Hausmann, A., Hallwachs, W. & Janzen, D. H. Advancing taxonomy and bioinventories with DNA barcodes. Philosophical Transactions of the Royal Society B: Biological Sciences 371, 20150339, https://doi.org/10.1098/rstb.2015.0339 (2016).
Padial, J. M., Miralles, A., De la Riva, I. & Vences, M. The integrative future of taxonomy. Frontiers in Zoology 7, 16, https://doi.org/10.1186/1742-9994-7-16 (2010).
Muñoz, A. G., Baxter, S. W., Linares, M. & Jiggins, C. D. Deep mitochondrial divergence within aHeliconius butterfly species is not explained by cryptic speciation or endosymbiotic bacteria. BMC Evolutionary Biology 11, 358, https://doi.org/10.1186/1471-2148-11-358 (2011).
Riedel, A., Sagata, K., Suhardjono, Y. R., Tänzler, R. & Balke, M. Integrative taxonomy on the fast track - towards more sustainability in biodiversity research. Frontiers in Zoology 10, 15, https://doi.org/10.1186/1742-9994-10-15 (2013).
Pante, E., Schoelinck, C. & Puillandre, N. From integrative taxonomy to species description: one step beyond. Systematic Biology 64, 152–160, https://doi.org/10.1093/sysbio/syu083 (2015).
Ward, R. D. DNA barcode divergence among species and genera of birds and fishes. Molecular Ecology Resources 9, 1077–1085, https://doi.org/10.1111/j.1755-0998.2009.02541.x (2009).
Bergsten, J. et al. The effect of geographical scale of sampling on DNA barcoding. Systematic Biology 61, 851–869, https://doi.org/10.1093/sysbio/sys037 (2012).
Mutanen, M. et al. Species-level para- and polyphyly in DNA barcode gene trees: strong operational bias in European Lepidoptera. Systematic Biology 65, 1024–1040, https://doi.org/10.1093/sysbio/syw044 (2016).
Collins, R. A. & Cruickshank, R. H. The seven deadly sins of DNA barcoding. Molecular Ecology Resources 13, 969–975, https://doi.org/10.1111/1755-0998.12046 (2013).
Fujisawa, T. & Barraclough, T. Delimiting species using single-locus data and the generalized mixed Yule coalescent approach: A revised method and evaluation on simulated data sets. Systematic Biology 62, 707–724, https://doi.org/10.1093/sys-bio/syt033 (2013).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29, 2869–2876, https://doi.org/10.1093/bioinformatics/btt499 (2013).
Kapli, P. et al. Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 33, 1630–1638, https://doi.org/10.1093/bioinformatics/btx025 (2017).
Carstens, B. C., Pelletier, T. A., Reid, N. M. & Satler, J. D. How to fail at species delimitation. Molecular Ecology 22, 4369–4383, https://doi.org/10.1111/mec.12413 (2013).
Pentinsaari, M., Vos, R. & Mutanen, M. Algorithmic single-locus species delimitation: effects of sampling effort, variation and nonmonophyly in four methods and 1870 species of beetles. Molecular Ecology Resources 17, 393–404, https://doi.org/10.1111/1755-0998.12557 (2017).
Reid, N. M. & Carstens, B. C. Phylogenetic estimation error can decrease the accuracy of species delimitation: a Bayesian implementation of the general mixed Yule-coalescent model. BMC Evolutionary Biology 12, 196, https://doi.org/10.1186/1471-2148-12-196 (2012).
Tang, C. Q., Humphreys, A. M., Fontaneto, D. & Barraclough, T. G. Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods in Ecology and Evolution 5, 1086–1094, https://doi.org/10.1111/2041-210X.12246 (2014).
Zarske, A. & Géry, J. Revision der neotropischen gattung Metynnis Cope, 1878. 1. Evalution der typusexemplare der nominellen arten (Teleostei: Characiformes: Serrasalmidae). Zoologische Abhandlungen Staatliches Museum für Tierkunde Dresden 50, 169–216 (1999).
Zarske, A. & Géry, J. Revision der neotypischen Gattung Metynnis Cope, 1878. II. Beschreibung zweier neuer arten und zum status von Metynnis goeldii Eigenmann, 1903 (Teleostei: Characiformes: Serrasalmidae). Vertebrate Zoology 58, 173–196 (2008).
Jégu, M. & Santos, G. M. Mise au point à propos de Serrasalmus spilopleura Kner, 1858 et réhabilitation de S. maculatus Kner, 1858 (Characidae: Serrasalminae). Cybium 1858, 119–143 (2001).
Jégu, M., Hubert, N. & Belmont-Jégu, E. Réhabilitation de Myloplus asterias (Müller & Troschel, 1844), espèce-type de Myloplus Gill, 1896 et validation du genre Myloplus Gill (Characidae: Serrasalminae). Cybium 28, 119–157 (2004).
Ota, R. P., Röpke, C. P., Zuanon, J. & Jégu, M. Serrasalmidae. In Queiroz, L. J. et al. (eds) Peixes do rio Madeira Vol. II, 12–47 (Santo Antonio Energia, São Paulo, 2013).
Sambrook, J., et al. Molecular Cloning: a Laboratory Manual. Ed. 2 (Cold Spring HarborLaboratory Press, 1989).
Ivanova, N. V., Zemlak, T. S., Hanner, R. H. & Hebert, P. D. N. Universal primer cocktails for fish DNA barcoding. Molecular Ecology Notes 7, 544–548, https://doi.org/10.1111/j.1471-8286.2007.01748.x (2007).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649, https://doi.org/10.1093/bioinformatics/bts199 (2012).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30, 772–780, https://doi.org/10.1093/molbev/mst010 (2013).
Stoeckle, M. Y. & Kerr, K. C. R. Frequency matrix approach demonstrates high sequence quality in avian barcodes and highlights cryptic pseudogenes. PLoS ONE 7, e43992, https://doi.org/10.1371/journal.pone.0043992 (2012).
Winter, D. J. Rentrez: an R package for the NCBI eUtils API. PeerJ Preprints (2017).
Pons, J. et al. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Systematic Biology 55, 595–609, https://doi.org/10.1080/10635150600852011 (2006).
Brown, S. D. J. et al. Spider: an R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Molecular Ecology Resources 12, 562–565, https://doi.org/10.1111/j.1755-0998.2011.03108.x (2012).
R Core Team, R Foundation for Statistical Computing. R: A language and environment for statistical computing https://www.R-project.org/ (2017).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the Beast 1.7. Molecular Biology and Evolution 29, 1969–1973, https://doi.org/10.1093/molbev/mss075 (2012).
Talavera, G., Dincà, V. & Vila, R. Factors affecting species delimitations with the GMYC model: Insights from a butterfly survey. Methods in Ecology and Evolution 4, 1101–1110, https://doi.org/10.1111/2041-210X.12107 (2013).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9, 772–772, https://doi.org/10.1038/nmeth.2109 (2012).
Rambaut, A., Drummond, A. J. &Suchard, M. Tracer v1.6 http://tree.bio.ed.ac.uk/software/tracer/ (2014).
Paradis, E., Claude, J. & &Strimmer, K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290, https://doi.org/10.1093/bioinformatics/btg412 (2004).
Schliep, K. P. phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593, https://doi.org/10.1093/bioinformatics/btq706 (2011).
Farias, I. P. & Hrbek, T. Patterns of diversification in the discus fishes (Symphysodon spp. Cichlidae) of the Amazon basin. Molecular Phylogenetics and Evolution 49, 32–43, https://doi.org/10.1016/j.ympev.2008.05.033 (2008).
Amado, M. V., Farias, I. P. & Hrbek, T. A molecular perspective on systematics, taxonomy and classification Amazonian discus fishes of the genus Symphysodon. International Journal of Evolutionary Biology 2011, 1–16, https://doi.org/10.4061/2011/360654 (2011).
Collins, R. A. et al. Biogeography and species delimitation of the rheophilic suckermouth-catfish genus Pseudolithoxus (Siluriformes: Loricariidae), with the description of a new species from the Brazilian Amazon. Systematics and Biodiversity https://doi.org/10.1080/14772000.2018.1468362 (2018).
Hubert, N., Torrico, J. P., Bonhomme, F. & Renno, J.-F. Species polyphyly and mtDNA introgression among three Serrasalmus sister-species. Molecular Phylogenetics and Evolution 46, 375–381, https://doi.org/10.1016/j.ympev.2007.04.002 (2008).
Júlio Júnior, H. F., Tós, C. D., Agostinho, Â. A. & Pavanelli, C. S. A massive invasion of fish species after eliminating a natural barrier in the upper rio Paraná basin. Neotropical Ichthyology 7, 709–718, https://doi.org/10.1590/S1679-62252009000400021 (2009).
Fink, W. L. Revision of the piranha genus Pygocentrus (Teleostei, Characiformes). Copeia 1993, 665–687 (1993).
Fink, W. L. & Zelditch, M. L. Phylogenetic analysis of ontogenetic shape transformations: A reassessment of the piranha genus Pygocentrus (Teleostei). Systematic Biology 44, 343–360, https://doi.org/10.1093/sysbio/44.3.343 (1995).
Pfenninger, M. & Schwenk, K. Cryptic animal species are homogeneously distributed among taxa and biogeographical regions. BMC Evolutionary Biology 6, 121, https://doi.org/10.1186/1471-2148-7-121 (2007).
Hoorn, C. et al. Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science 330, 927–931, https://doi.org/10.1126/science.1194585 (2010).
Philbrick, C. T. & Novelo, A. R. New World Podostemaceae: ecological and evolutionary enigmas. Brittonia 47, 210–222 (1995).
Sabaj Pérez, M. H. Where the Xingu bends and will soon break. American Scientist November, 395–403, https://doi.org/10.1511/2015.117.395 (2015).
Jégu, M. Taxinomie des Serrasalminae phytophages et phylogenie des Serrasalminae (Teleostei: Characiformes: Characidae). Phd thesis, Muséum National D’Histoire Naturelle, Paris, France (2004).
Farias, I. P. et al. Are rapids a barrier for floodplain fishes of the Amazon basin? A demographic study of the keystone floodplain species Colossoma macropomum (Teleostei: Characiformes). Molecular Phylogenetics and Evolution 56, 1129–1135, https://doi.org/10.1016/j.ympev.2010.03.028 (2010).
Markert, J. A., Schelly, R. C. & Stiassny, M. L. J. Genetic isolation and morphological divergence mediated by high-energy rapids in two cichlid genera from the lower Congo rapids. BMC Evolutionary Biology 10, 149, https://doi.org/10.1186/1471-2148-10-149 (2010).
Roxo, F. et al. Shift from slow- to fast-water habitats accelerates lineage and phenotype evolution in a clade of Neotropical suckermouth catfishes (Loricariidae: Hypoptopomatinae). PLoS ONE 12, e0178240, https://doi.org/10.1371/jour-nal.pone.0178240 (2017).
Winemiller, K. O. et al. Balancing hydropower and biodiversity in the Amazon, Congo, and Mekong. Science 351, 128–129, https://doi.org/10.1126/science.aac7082 (2016).
Hubert, N. & Renno, J.-F. Historical biogeography of South American freshwater fishes. Journal of Biogeography 33, 1414–1436, https://doi.org/10.1111/j.1365-2699.2006.01518.x (2006).
Hrbek, T. et al. A new species of river dolphin from Brazil or: how little do we know our biodiversity. PLoS ONE 9, e0083623, https://doi.org/10.1371/journal.pone.0083623 (2014).
Reis, R. E. Conserving the freshwater fishes of South America. International Zoo Yearbook 47, 65–70, https://doi.org/10.1111/izy.12000 (2013).
Ahrens, D. et al. Rarity and incomplete sampling in DNA-based species delimitation. Systematic Biology 65, 478–494, https://doi.org/10.1093/sysbio/syw002 (2016).
Hickerson, M. J., Meyer, C. P. & Moritz, C. DNA barcoding will often fail to discover new animal species over broad parameter space. Systematic Biology 55, 729–739, https://doi.org/10.1080/10635150600969898 (2006).
Lohse, K. Can mtDNA barcodes be used to delimit species? A response to Pons et al. (2006). Systematic Biology 58, 439–442, https://doi.org/10.1093/sysbio/syp039 (2009).
Sukumaran, J. & Knowles, L. L. Multispecies coalescent delimits structure, not species. Proceedings of the National Academy of Sciences of the United States of America 114, 1607–1612, https://doi.org/10.1073/pnas.1607921114 (2017).
Rabosky, D. L. et al. Rates of speciation and morphological evolution are correlated across the largest vertebrate radiation. Nature Communications 4, 1–8, https://doi.org/10.1038/ncomms2958 (2013).
Malinsky, M. et al. Genomic islands of speciation separate cichlid ecomorphs in an East African crater lake. Science 350, 1493–1498, https://doi.org/10.1126/science.aac9927 (2015).
Lim, G. S., Balke, M. & Meier, R. Determining species boundaries in a world full of rarity: singletons, species delimitation methods. Systematic Biology 61, 165–169, https://doi.org/10.1093/sysbio/syr030 (2012).
Lehner, B., Verdin, K. & Jarvis, A. New global hydrography derived from spaceborne elevation data. Eos, Transactions American Geophysical Union 89, 93–94, https://doi.org/10.1029/2008EO100001 (2008).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T. Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution 8, 28–36, https://doi.org/10.1111/2041-210X.12628 (2016).
Acknowledgements
We thank Pierre-Yves Le Bail for providing samples from French Guiana, Susana Caballero for assisting with sampling in Colombia, and also the many colleagues, fishers, local people and members of indigenous communities for helping us obtain specimens for this study. Collecting on the Uatumã and Xingu rivers was greatly assisted by Lúcia Py-Daniel, José Birindelli, Fernando Jerep and Mark Sabaj-Pérez. Jansen Zuanon and Tiago Pires helped with specimen identification, and José Gregorio Martínez and Maria Dόris Escobar provided COI sequences of Colossoma macropomum and Piaractus brachypomus. This research was supported by the MCT/CNPq/PPG7 557090/2005-9, CNPq/CT-Amazonia 554057/2006-9, CNPq/CT-Amazonia 575603/2008-9 and CNPq/FAPEAM/SISBIOTA (Rede BioPHAM) 563348/2010 to I.P.F. and CNPq 483155/2010-1, CNPq 490682/2010 and CNPq 400813/2012-2 to T.H.
Author information
Authors and Affiliations
Contributions
I.P.F. and T.H. conceived the experiment and obtained funding; V.N.M. conducted the experiment; R.A.C. analysed the results; V.N.M., R.A.C. and T.H. wrote the manuscript; M.A. and R.P.O. morphotyped and revised specimens, all authors conducted fieldwork and collected specimens; all authors contributed to and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Machado, V.N., Collins, R.A., Ota, R.P. et al. One thousand DNA barcodes of piranhas and pacus reveal geographic structure and unrecognised diversity in the Amazon. Sci Rep 8, 8387 (2018). https://doi.org/10.1038/s41598-018-26550-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26550-x
This article is cited by
-
DNA barcoding and phylogeography of the Hoplias malabaricus species complex
Scientific Reports (2022)
-
Kudoa rousseauxii n. sp. (Cnidaria: Multivalvulida) Infects the Skeletal Muscles of the Freshwater Fish Brachyplatystoma rousseauxii in the Amazon River
Acta Parasitologica (2022)
-
Hyperspectral data as a biodiversity screening tool can differentiate among diverse Neotropical fishes
Scientific Reports (2021)
-
From shallow to deep divergences: mixed messages from Amazon Basin cichlids
Hydrobiologia (2019)
-
Revisiting species boundaries and distribution ranges of Nemacheilus spp. (Cypriniformes: Nemacheilidae) and Rasbora spp. (Cypriniformes: Cyprinidae) in Java, Bali and Lombok through DNA barcodes: implications for conservation in a biodiversity hotspot
Conservation Genetics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
