
Abstract
Bacterial induced inflammatory responses cause pain through direct activation of nociceptive neurons, and the ablation of these neurons leads to increased immune infiltration. In this study, we investigated nociceptive-immune interactions in Drosophila and the role these interactions play during pathogenic bacterial infection. After bacterial infection, we found robust upregulation of ligand-gated ion channels and allatostatin receptors involved in nociception, which potentially leads to hyperalgesia. We further found that Allatostatin-C Receptor 2 (AstC-R2) plays a crucial role in host survival during infection with the pathogenic bacterium Photorhabdus luminescens. Upon examination of immune signaling in AstC-R2 deficient mutants, we demonstrated that Allatostatin-C Receptor 2 specifically inhibits the Immune deficiency pathway, and knockdown of AstC-R2 leads to overproduction of antimicrobial peptides related to this pathway and decreased host survival. This study provides mechanistic insights into the importance of microbe-nociceptor interactions during bacterial challenge. We posit that Allatostatin C is an immunosuppressive substance released by nociceptors or Drosophila hemocytes that dampens IMD signaling in order to either prevent immunopathology or to reduce unnecessary metabolic cost after microbial stimulation. AstC-R2 also acts to dampen thermal nociception in the absence of infection, suggesting an intrinsic neuronal role in mediating these processes during homeostatic conditions. Further examination into the signaling mechanisms by which Allatostatin-C alters immunity and nociception in Drosophila may reveal conserved pathways which can be utilized towards therapeutically targeting inflammatory pain and chronic inflammation.
Similar content being viewed by others


Introduction
In recent years there has been a growing body of research investigating the role of the inflammatory response in causing pain during bacterial infections including the discovery of interactions between bacteria, pain-sensing neurons called nociceptors, primary sensory afferents and the innate immune system1. Upon activation by proinflammatory cytokines, bacterial lipopolysaccharides, flagella or a-hemolysin, specific ligand-gated ion channels (TRPA1, FRPR1, ADAM10) open, resulting in an action potential propagating throughout these neurons2. Once the synapse is reached, these nociceptors release various immunomodulatory neuropeptides into the proximal vicinity including somatostatin, substance-P, CGRP and VIP3,4. These neuropeptides have been shown to have a bimodal effect by altering further nociception, as well as having varied effects on inflammation. In fact, ablation of subcutaneous nociceptors has been shown to increase immune infiltration in mice during Staphylococcus aureus infection whereas ablation near respiratory airways has been shown to reduce inflammation in a murine asthma model1,5. Due to bacteria being able to directly activate these nociceptive neurons and many products of these neurons altering systemic immunity, the question then arises as to whether the ability of bacteria to activate nociceptive neurons is beneficial or detrimental to the host6.
The common fruit fly, Drosophila melanogaster, provides an excellent opportunity to investigate these interactions for numerous reasons. Drosophila is a well-established model for probing questions relating to the innate immune response during microbial infection7,8,9,10,11,12,13,14. Moreover, Drosophila possesses primitive nociceptive neurons that are able to respond to noxious temperatures, mechanical stimuli, as well as harmful chemicals via sensory-gated ion channels15,16,17. Interestingly, these neurons can also be activated by the proinflammatory cytokine, Eiger, and bacterially derived LPS, suggesting a greater degree of functional homology to mammalian systems than previously realized18,19.
Activation of nociceptive neurons in Drosophila leads to an aversion to these noxious stimuli primarily through avoidance behaviors20,21,22,23,24. Beyond this behavioral output, nociceptive neurons in Drosophila are also linked with immune cell differentiation. RNAi knockdown of two genes crucial for nociceptor formation, painless and piezo, has been shown to alter lamellocyte differentiation during parasitoid wasp infection, demonstrating that nociceptor activation and cell-based immunity are linked in this invertebrate organism25.
The aim of the current study was to characterize a panel of known nociceptive (TRPA1, ppk, AstA-R1, AstC-R1, AstC-R2) genes in Drosophila (Table 1), and to determine if any of these genes impacted survivorship or immune function during bacterial infection. For this study, loss-of-function fly mutants for each gene were generated and injected with either a non-pathogenic strain of E. coli or the insect-pathogenic bacterium P. luminecens. Following infection, noxious heat threshold, survival, immune gene expression, and bacterial load in each mutant were analyzed. Results showed that genes coding for TRPA1, ppk, AstA-R1, AstC-R1, and AstC-R2 are upregulated during bacterial infection and this upregulation may lead to hyperalgesia. Interestingly, RNAi knockdown of AstC-R2, a receptor for a neuropeptide hormone released from nociceptors that is homologous to mammalian somatostatin (Supplementary Fig. 1), led to a significant decrease in fly survival during P. luminecens infection. Further characterization of this gene’s role during infection with the pathogen suggests an immune deficiency (IMD)-specific suppressive mechanism of action that, when removed, leads to an over-exuberant inflammatory response and subsequently premature death. Our findings indicate that nociceptive-related genes are upregulated during infection of Drosophila with insect pathogenic bacteria and that neuropeptides released from nociceptive neurons play a significant role in the regulation of the host antibacterial immune response.
Results
Nociceptive-related genes are differentially upregulated in response to E. coli or P. luminescens
Due to prior studies showing allodynia following UV radiation in Drosophila, we sought to determine whether bacterial infection could also alter sensitivity to painful stimuli18. Hyperalgesia is the result of increased transcription and subsequent translation of ligand gated ion channels in nociceptive neurons26. Therefore, we determined whether nociceptive-related genes, including TRPA1, ppk, AstA-R1, AstC-R1, and AstC-R2, were upregulated during bacterial infection and whether the expression of these genes differed upon infection. We injected 7–10 day old wild-type flies with E. coli, P. luminescens, or PBS as a septic injury negative control, and monitored their transcript level activity over the course of the infection.
Our results demonstrate that all five nociceptive genes were upregulated during infection with E. coli or P. luminescens as compared to PBS. We found that nociceptive gene upregulation temporally differed between infections (Fig. 1). AstA-R1 expression was significantly upregulated (p = 0.0064) between 0 and 3 hours post infection with E. coli, before decreasing between 3 and 12 hours post infection (Fig. 1a). For P. luminescens, AstA-R1 expression peaked later during the infection, upregulated between 3 and 12 hours and 3 and 18 hours post infection (p = 0.0335 and p = 0.0063, respectively) (Fig. 1a). Transcript levels were higher in the E. coli infected flies at 3 hours, in contrast to P. luminescens infected flies, which peaked at 18 hours. A similar pattern was seen for transcript levels of AstC-R1, AstC-R2, TRPA1, and ppk (Fig. 1b–h).

Nociceptive gene expression in Drosophila differs temporally during bacterial infection. Expression of (a) AstA-R1 (b) AstC-R1 (c) AstC-R2 (d) ppk (e) TRPA1 and (f) Cecropin A1 (CecA1) in Oregon flies responding to non-pathogenic E. coli or pathogenic P. luminescens bacteria at 0, 3, 12, and 18 hours post infection. (g)Upon infection with E. coli, all pain-related genes are upregulated at 3 hours and their mRNA levels decrease to basal levels by 12 hours. (h) Upon P. luminescens infection nociceptive gene transcript levels increase to a peak at either at 12 or 18 hours post infection. Differences in gene expression profiles were analyzed for statistical significance using a student’s t-test (n = 3–4 groups of 10 flies per time point, *p < 0.05, **p < 0.01, ***p < 0.0001).
Expression of nociception-related genes better correlates with bacterial load than with immune induction
Upon attempting to correlate nociceptive gene transcript levels with bacterial load or immune activation as measured by the induction of an antimicrobial peptide gene readout of the IMD pathway, Cecropin A1, it was surprising to find that TRPA1, AstC-R1, and AstC-R2 expression significantly correlated with bacterial load (p = 0.011, p = 0.047, p = 0.0246 respectively) (Fig. 2a) but not Cecropin A1 transcript levels (p = 0.358, p = 0.329, p = 0.457 respectively) (Fig. 2b) during P. luminescens infection, suggesting bacteria may play an active role in their induction. The same analysis with E. coli demonstrated that nociceptive gene transcript levels better correlated with bacterial load (Supplementary Fig. 2a) than immune activation (Supplementary Fig. 2b), yet neither of these correlations were statistically significant (p > 0.05).

Allatostatin-C receptors and TRPA1 expression in Drosophila significantly correlates with bacterial load during P. luminescens infection. Correlation and linear regression lines for nociceptive gene expression in w1118 flies over time plotted against (a) bacterial load and (b) Cecropin A1 expression following P. luminescens infection. Bacterial load significantly correlates with TRPA1, AstC-R1, and AstC-R2 expression upon infection with P. luminescens via two-tailed linear regression analysis (n = 3–4 groups of 10 flies per time point, *p < 0.05).
Drosophila IMD and TRPA1 RNAi mutants display hypoalgesia whereas AstC-R1 and AstC-R2 mutants display hyperalgesia
To better understand as to whether pain sensitization was linked to the immune response or bacterial injection, we tested various immune and nociceptor Drosophila mutants using a noxious heat escape assay to measure hypoalgesia and a withdrawal latency assay to measure hyperalgesia. The noxious heat threshold of w1118 flies was a mean of 94% for the heat escape assay and these flies had a mean withdrawal latency of 7.7 seconds (Fig. 3a,b).

Nociceptive and immune Drosophila mutants display alterations to pain sensing which can be manipulated via bacterial challenge. (a) Noxious heat threshold of Drosophila RNAi mutants for nociceptive and immune related genes. IMD RNAi mutants as well as TRPA1 mutants display a hypoalgesia whereas the Toll 10b and AstC-R1 and AstC-R2 RNAi mutants do not display a reduced noxious heat sensing capacity. (b) AstC-R2 and AstC-R1 RNAi mutants display reduced withdrawal latency. (c) IMD RNAi mutants display hyperalgesia upon infection with E. coli but not upon PBS injection or in uninjected controls. RNAi mutants were generated by crossing UAS-RNAi lines with an Actin5c Gal4 driver in order to knock the gene of interest down ubiquitously. (−) Indicates the use of an RNAi line whereas (+) indicates a line that constitutively expresses the gene of interest. Differences in noxious-related behaviors between Drosophila RNAi mutants were analyzed for statistical significance using a student’s t-test (n = 3–9 groups of 20 female flies, *p < 0.05, **p < 0.01, ***p < 0.0001).
We determined that IMD knockdown mutants had a significantly increased pain threshold compared to wild-type flies (mean = 83% vs 95%, p = 0.0003) (Fig. 3a). We found no significant changes in the pain threshold of Toll 10b flies, a strain that constitutively expresses the Toll pathway (p = 0.99). Further, we found that the pain threshold of IMD knockdown mutants was significantly decreased by injection of E. coli (83% vs 95%, p = 0.003) (Fig. 3c), suggesting that IMD knockdown is not sufficient to abolish inflammatory pain in Drosophila.
TRPA1 knockdown mutants displayed a significantly increased pain threshold as compared to wild-type flies (mean = 54% vs 94%, p < 0.0001) (Fig. 3a). This result is consistent with previous studies demonstrating the importance of TRPA1 in sensing noxious temperatures26. We found a trend towards a decrease in the pain threshold of AstC-R1 knockdown mutants using the heat escape assay (mean = 98% vs 95%, p = 0.219) and a significant decrease in withdrawal latency (mean = 8.4s vs 3.7s, p = 0.006). Similarly, we saw a trend in the pain threshold of AstC-R2 knockdown mutants and wild-type flies (mean = 96% vs 95%, p = 0.86), while there was a significant decrease in their withdrawal latency (mean = 8.4s vs 5.6s, p = 0.009) (Fig. 3a,b).
RNAi knockdown of AstC-R2 increases susceptibility to P. luminescens infection
Bacteria can directly activate nociceptive neurons in Drosophila and the ablation of these neurons in mice alters immune infiltration1,18. We thus determined whether nociception-related genes are beneficial or detrimental to the host upon bacterial infection. To test this, we generated RNAi knockdown mutants for each nociception-related gene and measured the survival of flies over the course of infection with P. luminescens. Our results show that knockdown of nociception-related genes had varying effects on the survival of the flies during bacterial infection (Fig. 4).

Allatostatin-C Receptor Drosophila mutants show increased susceptibility to P. luminescens infection. Survival curves of RNAi mutant flies for (a) AstA-R1 (b) AstC-R1 and AstC-R2 and (c) TRPA1 post injection with PBS, P. luminescens, and E. coli. Although neither AstA-R1 and TRPA1 RNAi mutants displayed a reduced survival during P. luminescens infection, AstC-R1 and AstC-R2 RNAi flies succumbed faster than their controls with AstC-R2 RNAi individuals showing significantly increased sensitivity to the pathogen (p < 0.01). RNAi mutants were generated by crossing UAS-RNAi lines with an Actin5c Gal4 driver in order to knock the gene of interest down ubiquitously. Survival curves were analyzed using survival curve analysis in GraphPad Prism software (n = 3 groups of 20 flies, **p < 0.01).
There was no significant change in survival of AstA-R1 knockdown mutants as compared to wild-type flies during P. luminescens infection (p = 0.543) (Fig. 4a). In contrast, knockdown of AstC-R1 trended towards a decrease in host survival whereas knockdown of AstC-R2 significantly reduced host survival (p = 0.0056) (Fig. 4b). Although knockdown of AstC-R2 reduced survival during P. luminescens infection, it was not sufficient to change susceptibility to infection with a non-pathogenic strain of E. coli (p = 0.15) (Supplementary Fig. 3a). Finally, knockdown of TRPA1, a known point of interaction between bacteria and host-nociceptor in Drosophila, trended towards an increase in survival as compared to wild-type flies during P. luminescens infection (p = 0.1872) (Fig. 4c).
RNAi knockdown of AstC-R2 hyperactivates the IMD pathway without reducing bacterial load
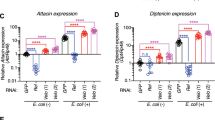
Due to significant decrease in survival of AstC-R2 knockdown flies upon infection with P. luminescens, we sought to determine whether alterations in NF-kB immune pathway activation and bacterial load in these flies could explain this effect. We found a statistically significant hyperactivation of Cecropin A1 and Attacin A, two antimicrobial peptide readouts of the IMD pathway as compared to wild-type flies (Fig. 5a) at 12 and 18 hours post infection with P. luminescens (CecA1: p = 0.0029, p = 0.0040, AttA: p = 0.048, p = 0.014 respectively). We saw a similar hyperactivation in IMD signaling after infection by the non-pathogen, E. coli as well (Supplementary Fig. 3b). However, transcript levels of Cecropin A1 did not differ between AstC-R2 RNAi knockdown flies and wild-type flies in the absence of bacterial injection (Supplementary Fig. 4). Additionally, we observed a modest, yet non-significant increased activation of the Toll pathway as measured by expression of Drosomycin at 18 hours post infection (p = 0.15, Fig. 5b) and a slight decrease in Jak-Stat activation as measured by transcriptional expression of Eiger at 18 hours post infection (p = 0.09, Fig. 5c). Surprisingly, despite this robust increase in IMD signaling, we observed no statistically significant differences in the bacterial load in the AstC-R2 knockdown flies upon P. luminescens infection at any timepoint (p > 0.05, Fig. 5d).

AstC-R2 RNAi Drosophila mutants display hyperactive IMD signaling without altered bacterial load. Immune gene expression of AstC-R2 RNAi mutant and background control flies following infection with P. luminescens bacteria. AstC-R2 RNAi mutant flies display (a) upregulation of the antimicrobial peptide-encoding genes Cecropin A1 (CecA1) and Attacin A (AttaA) which are controlled by the IMD pathway with (b) a modest increase in expression of the antimicrobial peptide-encoding gene Drosomycin (Drs) which is regulated by Toll signaling, and (c) decrease in Eiger (egr) expression. (d) AstC-R2 flies demonstrate no significant decrease in bacterial load over the course of P. luminescens infection. RNAi mutants were generated by crossing UAS-RNAi lines with an Actin5c Gal4 driver in order to knock the gene of interest down ubiquitously. Differences in gene expression profiles were analyzed for statistical significance using a student’s t-test (n = 2–5 groups of 10 flies per time point per genotype, *p < 0.05, **p < 0.01).
Discussion
During bacterial challenge, the host immune response must be mounted in a tightly regulated and quantitatively precise manner. Overproduction of immune effectors results in immune-related pathophysiology, tissue damage, and metabolic cost whereas under-production of these effectors may permit bacterial expansion and subsequently bacterially derived damage27,28,29,30. Recent studies have shown that bacteria can directly interact with nociceptive neurons, and that ablation of these neurons leads to increased lymph drainage during S. aureus infection most likely by suppressing immunomodulatory neuropeptide release. Thus, bacterial activation of nociceptive neurons may be a novel mechanism of immune control. This study represents the first attempt to characterize bacterially induced hyperalgesia and the effects of genes related to this process on host immunity in Drosophila melanogaster. Our study provides support for a newly emerging idea that nociceptive neurons may be crucial to mounting an appropriate immune response during these infections1,2,22.
We investigated the gene kinetics, effect on noxious behavior, and immune consequences of nociceptive gene activation during microbial challenge. We found a robust upregulation of ligand gated ion channels (TRPA1 and ppk) and Allatostatin receptors (AstC-R1, AstC-R2, AstA-R1) upon microbial challenge, the homologs of both of which have been associated with hyperalgesia in mammalian systems31,32,33,34,35,36,37. We found that nociceptive gene activation differed temporally upon infection with E. coli as compared to pathogenic P. luminescens, and that bacterial load better correlated with nociceptive gene activation than immune activation (as measured by the IMD antimicrobial peptide encoding gene, Cecropin A1). Importantly, this correlation supports a recent paper demonstrating that S. aureus bacterial load better correlates with hyperalgesia than paw swelling (immune infiltration) in mice1.
To determine whether the upregulation of these nociception-related genes contributed to hyperalgesia, we generated immune and nociceptive knockdown fly mutants for the genes upregulated, and measured changes to noxious heat sensitization. Upon examining alterations to this behavior, we found that AstC-R1 and AstC-R2 RNAi mutants displayed hyperalgesia whereas IMD and TRPA1 knockdown mutants showed robust hypoalgesia. These results are in agreement with previous studies demonstrating the importance of TRPA1 in noxious heat sensation38. To determine whether we could raise the noxious heat sensitivity of IMD mutants back to wild-type levels by infection with a bacterium, we infected IMD knockdown flies with a non-pathogenic strain of E. coli and found that these mutants displayed hyperalgesia, suggesting IMD activation contributes to, but is not necessary for hyperalgesia during bacterial infections. These results implicate NF-kB activation as a conserved mechanism of hyperalgesia in arthropod and mammalian lineages with the additional hyperalgesia seen upon infection of IMD knockdown mutants being attributed to Toll signaling or direct bacterial activation39,40,41. Indeed, previous studies have found that a transcription factor downstream of IMD activation, Relish, alters thermal nociception as well17,22.
Due to bacteria being able to potentially manipulate the expression of nociceptive genes in their favor, we were curious as to whether any of the nociception-related genes tested played a beneficial or detrimental role to the host during microbial challenge. To test this, we silenced each nociception-related gene ubiquitously in flies and measured their survival upon injection with the insect pathogen P. luminescens. We found a trend towards decreased survival of AstC-R1 knockdown flies and a significant decrease in survival upon knockdown of AstC-R2, suggesting a potential role for Allatostatin-C in modulating host immune processes during bacterial infection. However, when infecting AstC-R2 knockdown flies with the non-pathogen E. coli, we observed no decreased survival over 48 hours hours as compared to wild-type flies suggesting that this effect alone is not sufficient to cause death.
The mammalian homolog of Allatostatin is Somatostatin42,43,44, which has documented effects in reducing systemic inflammation in mammalian systems, and thus we examined whether knockdown of AstC-R2 leads to alterations in immune signaling that could contribute to the decreased survival4,45,46. We observed a robust over induction of IMD signaling with a modest, but non-significant increase in Toll and decrease in Eiger as compared to wild-type flies, suggesting that AstC-R2 reduced IMD signaling independently of the Toll or Jak-Stat pathways respectively. Despite the robust upregulation of the IMD pathway, we observed no changes in bacterial load during P. luminescens infection of AstC-R2 knockdown flies as compared to wild-type controls. These results suggest that antimicrobial peptides related to this pathway are ineffective at controlling this pathogen. Indeed, recent reports have shown that an antimicrobial peptide-resistant sub-population of P. luminescens is responsible for the majority of the virulence during insect infection, and that P. luminescens is able to employ proteases that specifically degrade antimicrobial peptides, rendering them post-translationally ineffective47,48.
By knocking down a receptor for Allatostatin C, which has dual role in inhibiting heat-driven nociception as well as inhibiting the IMD pathway during bacterial challenge, we observed hyperactivation of this immune pathway, hyperalgesia, and reduced survival upon challenge with P. luminescens. The hyperalgesia seen in AstC-R1 and AstC-R2 RNAi knockdown flies in the absence of bacterial challenge most likely is not due to dysregulation of the IMD pathway because we observed similar basal transcript levels of Cecropin A1 in AstC-R2 knockdown mutants as compared to wild-type flies (Supplementary Fig. 3). Indeed, AstC-R1 and AstC-R2 also share structural homology with mammalian opioid receptors22. However, the reduced survival in AstC-R2 knockdown flies may be explained either directly or indirectly by over activated IMD signaling and AstC-R2-IMD double knockdown mutants will be needed in order to confirm this hypothesis. Remarkably, our results recapitulate many of the findings found in a seminal study investigating the importance of somatostatin receptor 4 in the modulation of hyperalgesia and inflammation49. Therefore, Drosophila AstC-R2 may be more functionally similar to mammalian SSTR4 than previously perceived.
Due to the transcriptional upregulation of AstC-R1 and AstC-R2 during infection, it is likely that this upregulation reflects one mechanism of the host fine-tuning the immune response to prevent immune related damage from occurring as well as mediating avoidance behaviors while in a compromised state. Somatostatin regulatory circuits have been documented at sites of chronic inflammation where they have important roles in inhibiting pro-inflammatory cytokine production by macrophages and T-cells yet found processes have not been previously described in Drosophila50,51,52,53. Interestingly, another neuropeptide that acts as a crucial component of this circuit by inhibiting somatostatin release is substance P, an additional molecule released from nociceptive neurons54,55. Thus, immune manipulation during microbial challenge by nociceptive neurons is likely to be a well-orchestrated process that amplifies or suppresses pro-inflammatory cytokine production in a way to best ensure host survival.
Our results imply that nociceptor-immune interactions during microbial infection in Drosophila may be more similar to mammalian systems than previously conceived (Fig. 6). This idea is supported by recent findings demonstrating that nociceptive neurons in flies are sensitive the proinflammatory cytokine Eiger, as well as bacterially derived lipopolysaccharides18,19. Drosophila also possesses homologous genes for other immuno-modulatory substances released from nociceptors including substance P, CGRP and VIP (DTK, DH31, and Pdf respectively), yet their roles in pain sensation and immunity have not been characterized56,57,58. Due to the wealth of transgenic lines available, quick developmental cycle and cheap cost of maintenance, Drosophila could prove to be a valuable tool in deciphering nociceptor-innate immune interactions in the future. Further studies into the interface of pain, immunity, and microbial challenge hold large promise for innovative treatments for inflammatory pain, auto-immune conditions, as well as potential explanations for host-tolerance of the gut microbiota.

Potential role for AstC-R2 in nociceptor-bacterial-immune interactions in Drosophila. Upon bacterial infection by a Gram-negative pathogen, the IMD pathway is activated by DAP-type peptidoglycan, NF- κB is activated and translocates to the nucleus, and transcription of effector antimicrobial peptide-encoding genes related to this pathway (CecA1) occurs. Simultaneously, TRPA1 channels open by direct interaction with bacterial LPS or N-formyl peptides leading to nociceptive neuron firing and the subsequent release of Allatostatin C. In turn, Allatostatin C inhibits the IMD pathway as well as heat driven nociception through binding to AstC-R2 on the fat-body cells and nociceptors respectively, thus completing a negative regulatory circuit controlling IMD activation. Figure was modified from images from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com/.
Materials and Methods
Bacterial preparation
Bacteria were stored as 20% glycerol stocks at −80 °C before use. Bacteria were thawed and then grown in 10 mL of Luria-Bertani broth. Escherichia coli and Photorhabdus luminescens were grown at 30 °C for 18 hours or 22 hours, respectively. After incubation, the bacterial solutions were pelleted by centrifugation for 5 minutes at 4 °C at 3,000 rpm. The pellets were washed twice before resuspension in PBS. Concentrations were adjusted using a Nanodrop 2000 spectrophotometer with an absorbance at 600 nm denoting the respective concentrations. E. coli was used at an optical density (OD) of 0.015, while P. luminescens was used at an OD of 0.10. This OD corresponds to between 300–1000 colony forming units (cfu) of each bacterium.
Drosophila mutants and crosses
The following Drosophila strains were obtained from the Vienna Drosophila Resource Center (Vienna, Austria) or Exelixis at Harvard Medical School (Cambridge and Massachusetts); Oregon and w1118, AstA-R1: v3400 and v3399, AstC-R1: v13560 and v110739, AstC-R2: v50000 and v106146, TRPA1: v37249 and v37250, ppk: v108683, Toll 10b and IMD (−), Actin5c-Gal4. UAS-RNAi Drosophila lines were crossed with the Actin5C Gal4 driver in order to ubiquitously silence the gene of interest in the resulting progeny. Reduced transcriptional activity of each gene silenced via-RNAi was confirmed via quantitative RT-PCR (qRT-PCR) (Supplementary Fig. 5).
Fly injection
Drosophila melanogaster flies aged from 7–10 days were anesthetized using carbon dioxide. 10–12 flies were then injected with 18.4 nl of the standardized bacterial solution using a Nanoject microinjector fitted with capillary needles. PBS was used as a septic injury negative control for all experiments. Flies were collected after injection by freezing at −80 °C.
Gene expression and bacterial load determination
RNA extractions were carried out using PrepEase RNA Spin Kits (USB) or Trizol Reagent (Thermofisher) and eluted using molecular grade H2O. cDNA syntheses were carried out using 300 ng of RNA with High Capacity Reverse Transcription cDNA Synthesis Kit (Applied Biosystems). The cDNA was then diluted 1/10 times before proceeding to qRT-PCR analysis. All qRT-PCR assays were carried out using CFX96 Real-Time System (Bio-Rad). 1 µl of cDNA was used per reaction using gene-specific primers (Table 2) (Eurofins MWG Operon) and SYBR Green Supermix (Bio-Rad). Ct values were analyzed using the Delta Ct method using RpL32 as the control gene, and PBS as the control treatment. Bacterial load was calculated using this method in conjunction with measuring the expression of 16S rRNA.
Noxious escape assays
Twenty female flies between 7–10 days old were collected and placed in single 35 mm petri dish. These flies were left for 30 minutes to acclimate to their new environment. In order to determine the noxious heat threshold, these flies were floated on a heat bath set at 42 °C for 55 seconds, and the noxious heat threshold was determined by the percentage of flies that climbed to the top of the petri dish during this period of time22. Each data point shown on Fig. 3a constitutes the mean of three technical replicates of one group of twenty female flies.
Withdrawal latency assays
Twenty female flies between 7–10 days old were collected and placed in single 35 mm petri dish. These flies were left for 30 minutes to acclimate to their new environment. In order to determine the withdrawal latency, these flies were floated on a heat bath set at 42 °C and the time it took for 75% of flies to reach the top of the petri dish was measured22. Each data point shown on Fig. 3b constitutes the mean of three technical replicates of one group of twenty female flies.
Statistical analyses
All statistical analyses, including student’s t-tests, two-tailed Pearson’s correlations, and Log-Rank (Mantel-Cox) test survival curve analyses were performed using GraphPad Prism 6 software.
Statement of data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Chiu, I. M. et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501, 52–57 (2013).
Yang, N. J. & Chiu, I. M. Bacterial Signaling to the Nervous System through Toxins and Metabolites. Journal of Molecular Biology 429, 587–605 (2017).
Pinho-Ribeiro, F. A., Verri, W. A. & Chiu, I. M. Nociceptor Sensory Neuron–Immune Interactions in Pain and Inflammation. Trends in Immunology 38, 5–19 (2017).
Helyes, Z. et al. Antiinflammatory and Analgesic Effects of Somatostatin Released from Capsaicin-Sensitive Sensory Nerve Terminals in a Freund’s Adjuvant-Induced Chronic Arthritis Model in the Rat. Arthritis and Rheumatism 50, 1677–1685 (2004).
Talbot, S. et al. Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron 87, 341–355 (2015).
Baral, P., Mills, K., Pinho-Ribeiro, F. A. & Chiu, I. M. Pain and Itch: Beneficial or Harmful to Antimicrobial Defense? Cell Host & Microbe 19, 755–759 (2016).
Igboin, C. O., Griffen, A. L. & Leys, E. J. The Drosophila melanogaster host model. Journal of Oral Microbiology, 4, https://doi.org/10.3402/jom.v4i0.10368 (2012).
Gold, K. S. & Brockner, K. Macrophages and cellular immunity in Drosophila melanogaster. Seminars in Immunology 27, 357–368 (2015).
Wang, L., Kounatidis, I. & Ligoxygakis, P. Drosophila as a model to study the role of blood cells in inflammation, innate immunity and cancer. Frontiers in Cellular and Infection. Microbiology 3, 113 (2013).
Ferrandon, D., Imler, J.-L., Hetru, C. & Hoffmann, J. A. The Drosophila systemic immune response: sensing and signaling during bacterial and fungal infections. Nature Reviews Immunology 7, 862–74 (2007).
Hoffmann, J. A. The immune response of Drosophila. Nature 426, 33–38 (2003).
Govind, S. Innate immunity in Drosophila: Pathogens and pathways. Insect Science 15, 29–43 (2008).
Tzou, P., De Gregorio, E. & Lemaitre, B. How Drosophila combats microbial infection: a model to study innate immunity and host-pathogen interactions. Current Opinion in Microbiology 5, 102–110 (2002).
Hoff, J. Drosophila immunity. Trends in Cell Biology 7, 309–316 (1997).
Milinkeviciute, G., Gentile, C. & Neely, G. G. Drosophila as a tool for studying the conserved genetics of pain. Clinical Genetics 82, 359–366 (2012).
Xu, S. Y. et al. Thermal nociception in adult Drosophila: Behavioral characterization and the role of the painless gene. Genes, Brain and Behavior 5, 602–613 (2006).
Manev, H. & Dimitrijevic, N. Drosophila model for in vivo pharmacological analgesia research. European Journal of Pharmacology 491, 207–208 (2004).
Babcock, D. T., Landry, C. & Galko, M. J. Cytokine Signaling Mediates UV-Induced Nociceptive Sensitization in Drosophila Larvae. Current Biology 19, 799–806 (2009).
Soldano, A. et al. Gustatory-mediated avoidance of bacterial lipopolysaccharides via TRPA1 activation in Drosophila. eLife 5, pii: e13133 (2016).
Hwang, R. Y. et al. Nociceptive Neurons Protect Drosophila Larvae from Parasitoid Wasps. Current Biology 17, 2105–2116 (2007).
Robertson, J. L., Tsubouchi, A. & Tracey, W. D. Larval Defense against Attack from Parasitoid Wasps Requires Nociceptive Neurons. PLoS One 8, e78704 (2013).
Neely, G. G. et al. A Genome-wide Drosophila screen for heat nociception identifies α2δ3 as an evolutionarily conserved pain gene. Cell 143, 628–638 (2010).
Tracey, W. D., Wilson, R. I., Laurent, G. & Benzer, S. painless, a Drosophila gene essential for nociception. Cell 113, 261–273 (2003).
Kang, K. et al. Analysis of Drosophila TRPA1 reveals an ancient origin for human chemical nociception. Nature 464, 597–600 (2010).
Tokusumi, Y., Tokusumi, T. & Schulz, R. A. The nociception genes painless and Piezo are required for the cellular immune response of Drosophila larvae to wasp parasitization. Biochemical and Biophysical Research Communications 486, 893–897 (2017).
Treede, R. D., Meyer, R. A., Raja, S. N. & Campbell, J. N. Peripheral and central mechanisms of cutaneous hyperalgesia. Progress in Neurobiology 38, 397–421 (1992).
DiAngelo, J. R., Bland, M. L., Bambina, S., Cherry, S. & Birnbaum, M. J. The immune response attenuates growth and nutrient storage in Drosophila by reducing insulin signaling. Proceedings of the National Academy of Sciences 106, 20853–20858 (2009).
Schneider, D. S. How and Why Does a Fly Turn Its Immune System Off? PLoS Biology 5, e247 (2007).
Medzhitov, R. et al. Disease tolerance as a defense strategy. Science 335, 936–941 (2012).
Soares, M. P., Gozzelino, R. & Weis, S. Tissue damage control in disease tolerance. Trends in Immunology 35, 483–494 (2014).
Pavlov, V. A. & Tracey, K. J. Neural regulation of immunity: Molecular mechanisms and clinical translation. Nature Neuroscience 20, 156–166 (2017).
Bonet, I. J. M., Fischer, L., Parada, C. A. & Tambeli, C. H. The role of transient receptor potential A 1 (TRPA1) in the development and maintenance of carrageenan-induced hyperalgesia. Neuropharmacology 65, 206–212 (2013).
Honda, K. et al. TRPA1 contributes to capsaicin-induced facial cold hyperalgesia in rats. European Journal of Oral Sciences 122, 391–396 (2014).
da Costa, D. S. M. et al. The involvement of the transient receptor potential A1 (TRPA1) in the maintenance of mechanical and cold hyperalgesia in persistent inflammation. Pain 148, 431–437 (2010).
Obata, K. et al. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. Journal of Clinical Investigation 115, 2393–2401 (2005).
Schwartz, E. S. et al. TRPV1 and TRPA1 Antagonists Prevent the Transition of Acute to Chronic Inflammation and Pain in Chronic Pancreatitis. Journal of Neuroscience 33, 5603–5611 (2013).
Helyes, Z. et al. Anti-nociceptive effect induced by somatostatin released from sensory nerve terminals and by synthetic somatostatin analogues in the rat. Neuroscience Letters 278, 185–188 (2000).
Neely, G. G. et al. TrpA1 regulates thermal nociception in Drosophila. PLoS One 6, e24343 (2011).
Niederberger, E. & Geisslinger, G. The IKK-NF-kappaB pathway: a source for novel molecular drug targets in pain therapy? FASEB Journal 22, 3432–3442 (2008).
Nicotra, L., Loram, L. C., Watkins, L. R. & Hutchinson, M. R. Toll-like receptors in chronic pain. Experimental Neurology 234, 316–329 (2012).
Hua, S. & Cabot, P. J. Mechanisms of peripheral immune-cell-mediated analgesia in inflammation: Clinical and therapeutic implications. Trends in Pharmacological Sciences 31, 427–433 (2010).
Birgül, N., Weise, C., Kreienkamp, H. J. & Richter, D. Reverse physiology in Drosophila: Identification of a novel allatostatin-like neuropeptide and its cognate receptor structurally related to the mammalian somatostatin/galanin/opioid receptor family. EMBO Journal 18, 5892–5900 (1999).
Veenstra, J. A. Allatostatin C and its paralog allatostatin double C: The arthropod somatostatins. Insect Biochemistry and Molecular Biology 39, 161–170 (2009).
Bendena, W. G., Donly, B. C. & Tobe, S. S. Allatostatins: a growing family of neuropeptides with structural and functional diversity. Annals of the New York Academy of Sciences 897, 311–329 (1999).
Pintér, E., Helyes, Z. & Szolcsányi, J. Inhibitory effect of somatostatin on inflammation and nociception. Pharmacology and Therapeutics 112, 440–456 (2006).
Elekes, K. et al. Inhibitory effects of synthetic somatostatin receptor subtype 4 agonists on acute and chronic airway inflammation and hyperreactivity in the mouse. European Journal of Pharmacology 578, 313–322 (2008).
Mouammine, A. et al. An antimicrobial peptide-resistant minor subpopulation of Photorhabdus luminescens is responsible for virulence. Scientific Reports 7, 43670 (2013).
Caldas, C., Cherqui, A., Pereira, A. & Simões, N. Purification and characterization of an extracellular protease from Xenorhabdus nematophila involved in insect immunosuppression. Applied and Environmental Microbiology 68, 1297–1304 (2001).
Helyes, Z. et al. Impaired defense mechanism against inflammation, hyperalgesia, and airway hyperreactivity in somatostatin 4 receptor gene-deleted mice. Proceedings of the National Academy of Sciences 106, 13088–13093 (2008).
Ryu, S. Y. et al. Somatostatin and substance P induced in vivo by lipopolysaccharide and in peritoneal macrophages stimulated with lipopolysaccharide or interferon-gamma have differential effects on murine cytokine production. Neuroimmunomodulation 8, 25–30 (2000).
Armani, C., Catalani, E., Balbarini, A., Bagnoli, P. & Cervia, D. Expression, pharmacology, and functional role of somatostatin receptor subtypes 1 and 2 in human macrophages. Journal of Leukocyte Biology 81, 845–855 (2007).
Perez, J. et al. Somatostatin binds to murine macrophages through two distinct subsets of receptors. Journal of Neuroimmunology 138, 38–44 (2003).
Weinstock, J. V. & Elliott, D. The somatostatin immunoregulatory circuit present at sites of chronic inflammation. European Journal of Endocrinology/European Federation of Endocrine Societies 143(Suppl), S15–S19 (2000).
Liu, H., Mantyh, P. W. & Basbaum, A. I. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature 386, 721–724 (1997).
Blum, A. M. et al. Substance P regulates somatostatin expression in inflammation. Journal of Immunology 161, 6316–6322 (1998).
Asahina, K. et al. Tachykinin-expressing neurons control male-specific aggressive arousal in drosophila. Cell 156, 221–235 (2014).
Kunst, M. et al. Calcitonin gene-related peptide neurons mediate sleep-specific circadian output in Drosophila. Current Biology 24, 2652–2664 (2014).
Vosko, A. M., Schroeder, A., Loh, D. H. & Colwell, C. S. Vasoactive intestinal peptide and the mammalian circadian system. General and Comparative Endocrinology 152, 165–75 (2007).
Acknowledgements
We thank members of the Department of Biological Sciences at GWU for critical reading of the manuscript. This research was supported by a start-up fund from the Columbian College of Arts and Sciences at GWU to I.E. and by a start-up fund from the Department of Microbiology, Immunology, and Tropical Medicine to D.F.N., the Luther Rice Undergraduate Research Fellowship from the Center for Undergraduate Fellowships and Research at GWU to N.D.B. and G.A.H.
Author information
Authors and Affiliations
Contributions
N.D.B. designed and executed experiments in coordination with D.F.N. and I.E. G.A.H. performed gene-expression profiling experiments under AstC-R2 knockdown. N.D.B., D.F.N., and I.E. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bachtel, N.D., Hovsepian, G.A., Nixon, D.F. et al. Allatostatin C modulates nociception and immunity in Drosophila. Sci Rep 8, 7501 (2018). https://doi.org/10.1038/s41598-018-25855-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-25855-1
This article is cited by
-
The insect somatostatin pathway gates vitellogenesis progression during reproductive maturation and the post-mating response
Nature Communications (2022)
-
The effect of B-type allatostatin neuropeptides on crosstalk between the insect immune response and cold tolerance
Scientific Reports (2022)
-
The gut hormone Allatostatin C/Somatostatin regulates food intake and metabolic homeostasis under nutrient stress
Nature Communications (2022)
-
Transcriptome analysis reveals the activation of neuroendocrine-immune system in shrimp hemocytes at the early stage of WSSV infection
BMC Genomics (2019)
-
Characterization of G-protein coupled receptors from the blackback land crab Gecarcinus lateralis Y organ transcriptome over the molt cycle
BMC Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
