
Abstract
Cell division during the reproductive phase of the Streptomyces life-cycle requires tight coordination between synchronous formation of multiple septa and DNA segregation. One remarkable difference with most other bacterial systems is that cell division in Streptomyces is positively controlled by the recruitment of FtsZ by SsgB. Here we show that deletion of ylmD (SCO2081) or ylmE (SCO2080), which lie in operon with ftsZ in the dcw cluster of actinomycetes, has major consequences for sporulation-specific cell division in Streptomyces coelicolor. Electron and fluorescence microscopy demonstrated that ylmE mutants have a highly aberrant phenotype with defective septum synthesis, and produce very few spores with low viability and high heat sensitivity. FtsZ-ring formation was also highly disturbed in ylmE mutants. Deletion of ylmD had a far less severe effect on sporulation. Interestingly, the additional deletion of ylmD restored sporulation to the ylmE null mutant. YlmD and YlmE are not part of the divisome, but instead localize diffusely in aerial hyphae, with differential intensity throughout the sporogenic part of the hyphae. Taken together, our work reveals a function for YlmD and YlmE in the control of sporulation-specific cell division in S. coelicolor, whereby the presence of YlmD alone results in major developmental defects.
Similar content being viewed by others

Introduction
In unicellular bacteria, cell division divides a mother cell in two identical daughter cells, each containing a single copy of the chromosome. The control of cell division thereby revolves around finding the mid-cell position, and chromosome segregation and septum synthesis are closely coordinated in time and space to avoid DNA damage by the nascent septum. The cell division scaffold is formed by FtsZ, which is a homologue of tubulin1 and forms a contractile ring (or Z-ring) that mediates the recruitment of the cell division machinery to the division site (reviewed in2,3). Septum-site selection and Z-ring stabilization are mediated by proteins like FtsA and ZipA4,5,6, ZapA7 and SepF8,9. Z-ring (dis-)assembly is thereby actively controlled (reviewed in10).
Streptomycetes are filamentous Gram-positive bacteria that belong to the phylum of Actinobacteria. These bacteria produce over 60% of all known antibiotics and many other bioactive natural products11,12. Exponential growth of the vegetative hyphae is achieved by apical growth and branching. At this stage of the life cycle, cell division does not affect physical separation of the cells, but instead long syncytial cells are formed that are separated by cross-walls13. Hence, streptomycetes are model organisms for the study of multicellularity and bacterial morphogenesis14,15.
Most divisome components except FtsZ are not required for vegetative division, presumably reflecting the lack of constriction16. Spacing between the cross-walls is highly variable, and little is known of the way septum-site selection is controlled. Recently, using cryo-electron tomography, our lab and others showed that intracellular membrane assemblies or cross-membranes are involved in DNA protection during septum synthesis in young vegetative hyphae, suggesting a novel way of cell-division control17,18. In addition, these multicellular bacteria have a complex cytoskeleton, which among others plays a role in the organization of the tip growth machinery19,20.
Canonical division resulting in cell fission occurs during sporulation-specific cell division, which requires all components of the divisome16,21. At this stage of the life cycle up to a hundred septa are formed in a short time span, following a highly complex process of coordinated cell division and DNA segregation, and visualized as long ladders of Z-rings22. Eventually, chains of unigenomic spores are formed, which have a thick protective spore wall facilitating long-term survival in the environment. Though ftsZ null mutants are viable, they fail to produce septa and hence do not sporulate and cell division is not essential for growth of Streptomyces, which provides a unique system for the study of this process23,24.
Sporulation-specific cell division is controlled by the SsgA-like proteins (SALPs), which are exclusively found in morphologically complex actinobacteria25,26. The canonical view is that cell division is negatively controlled by the action of the Min system that inhibits division away from midcell, and by nucleoid occlusion (Noc) to avoid septum synthesis near the chromosome to avoid DNA damage, as seen in B. subtilis and E. coli. In contrast, cell division in streptomycetes is positively controlled by the recruitment of FtsZ to future septum sites by SsgB, in an SsgA-dependent manner27. As a consequence, both SsgA and SsgB are required for sporulation28,29. We recently showed that SepG (formerly called YlmG) assists in docking of SsgB to the membrane, and also plays a major role in maintaining the nucleoid shape in the spores30.
Many of the genes for the components of the divisome and the cell-wall biosynthetic machinery are located in the so-called dcw cluster (division and cell-wall synthesis31). Most of these genes have been studied extensively and their functions have been well characterized. However, little is known of the genes ylmD and ylmE that lie immediately downstream of, and likely form an operon with, ftsZ on the genome of streptomycetes and many other bacteria, including firmicutes. Earlier work on a mutant of S. venezuelae lacking both ylmD and ylmE showed that the double mutant had no obvious phenotypic defects32.
In this study, we show that deletion of ylmE alone results in severe cell division defects, while deletion of ylmD only slightly affects sporulation. Interestingly, the cell division defects of ylmE mutants were rescued by the additional deletion of ylmD, and ylmDE mutants had no obvious sporulation defects. These data strongly suggest that expression of YlmD alone is detrimental for sporulation-specific cell division in streptomycetes, which is counteracted by YlmE. This is consistent with the phylogenetic evidence that some bacteria only harbor an ortholog of ylmE, whereas ylmD never occurs without ylmE.
Results and Discussion
Phylogenetic analysis of YlmE (SCO2080) and YlmD (SCO2081)
Many of the genes in the dcw gene cluster have been extensively studied and their functions are well established. However, this is not the case for ylmD and ylmE, which lie immediately downstream of ftsZ in many bacteria. In all Streptomyces genomes analyzed, ftsZ (SCO2082), ylmD (SCO2081) and ylmE (SCO2080) form an operon, an observation that is supported by high-resolution transcript mapping33; moreover there is apparent translational fusion between ftsZ and ylmD (overlapping start and stop codons) and only 6 nt spacing between ylmD and ylmE. This transcriptional coupling to ftsZ suggests that these genes may play a prominent role in the cell division process. Transcript levels of ftsZ are similar to those of ylmD and ylmE during vegetative growth on MM agar plates; however, transcription of ftsZ is enhanced during sporulation, while that of ylmD and ylmE is not significantly altered34. YlmD and YlmE are wide-spread in Gram-positive bacteria, and particularly in actinobacteria. SCO2081 and SCO2080 share 32% aa identity with YlmD and YlmE of B. subtilis, respectively. Orthologues of these proteins are also found in several genera of Gram-negative bacteria. Phylogenetic analysis of the YlmD and YlmE proteins in actinobacteria shows that while YlmE is widespread, YlmD is often absent in actinobacteria, such as Stackebrandtia, Catenulispora, Salinispora, Micromonospora, Amycolatopsis and Mycobacterium, several of which are spore-forming actinobacteria (Fig. 1). Remarkably, there are no examples of YlmD being present in the absence of YlmE and thus it would appear that loss of YlmD (SCO2081) has occurred on multiple occasions, given that there is wide but patchy distribution of this gene across distinct actinobacteria lineages and the sequences clade tightly within the accepted actinomycete phylogenies.

Phylogenetic analysis of YlmE in actinobacteria and its genetic linkage to YlmD. A phylogenetic tree is shown of YlmE in actinobacteria (left) and genetic linkage of ylmE (grey) to ftsZ (black) and ylmD (white) (right).
Analysis of ylmD using the EMBL String engine35 shows functional linkage in two groups to cell division associated genes (sepF (SCO2079), SCO2085 and ftsZ) along with a group of mainly hypothetical proteins (STRING Data link: http://bit.ly/2gc5kCB). YlmD has a domain that has homology to the multiple-copper polyphenol oxidoreductase laccases, which are oxidoreductases that are widely distributed in both prokaryotes and eukaryotes36. A similar analysis of YlmE also indicates a functional linkage to cell-wall biosynthesis and cell division (STRING Data link: http://bit.ly/2gbPyHK). YlmE is a member of the family of YBL036c-like proteins, which generally contain pyridoxal 5-phosphate dependent enzymes. The structure of YBL036c from Saccharomyces cerevisiae was resolved at 2.0 Å resolution (PDB 1CT537). The protein has homology to the N-terminal domains of alanine racemases but lacks of the β-sandwich domain which would likely limit the activity of YBL036c as alanine or non-specific amino acid racemase38. To test the hypothesis that YlmE may have alanine racemase activity and thus may play a role in determining the amino acid composition of the peptidoglycan, we tested purified YlmE for alanine racemase assays as described previously39, using alanine racemase Alr as positive control. Whereas purified Alr successfully catalyzed the conversion of L-alanine to D-alanine, YlmE could not perform this reaction under the same conditions, and over-expression of YlmE failed to restore a D-Ala prototrophy to alr null mutants (data not shown). These observations coupled with protein structure homology data make it highly unlikely that YlmE functions as an alanine racemase in vivo.
ylmD and ylmE are required for proper sporulation
To analyze the role of ylmD and ylmE, deletion mutants were created in S. coelicolor M145 as detailed in the Materials and Methods section. The +25 to +696 region of ylmE (SCO2080) or the +25 to +705 region of ylmD (SCO2081) were replaced by the apramycin resistance cassette, followed by deletion of the cassette using the Cre recombinase so as to avoid polar effects. For each mutant, four independent mutants were selected and all had the same phenotype. Therefore, one was selected for more detailed analysis, designated GAL47 (S. coelicolor M145 ΔylmD) and GAL48 (S. coelicolor M145 ΔylmE). The M145 ylmDE mutant (GAL130) was created by replacing ylmD in ylmE null mutant GAL48 by the apramycin resistance cassette.
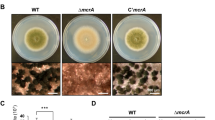
The ylmD null mutant GAL47 had a wild-type-like appearance, while ylmE null mutant GAL48 hardly produced any grey pigment after 5 days incubation on SFM agar plates, indicative of a failure to complete full development. Surprisingly, ylmDE mutant GAL130 had a grey appearance similar to that of the parental strain M145 (Fig. 2A). Phase-contrast light microscopy of impression prints of the top of the colonies demonstrated that while the parent produced typical long spore chains, the ylmD null mutant produced abundant but aberrantly sized spores, the ylmE null mutant produced fewer spores and those produced were unusually large. The ylmDE double mutant produced abundant spores, though some were irregularly sized (Figs 2B and S1). The same defect in sporulation was observed on R5 and MM mannitol agar plates. Antibiotic production was not affected, and the mutants produced normal levels of the pigmented antibiotics actinorhodin and undecylprodigiosin.

Phenotype of the S. coelicolor ylmD and ylmE mutants. (A) sporulation of S. coelicolor M145 and its ylmD, ylmE and ylmDE mutants on SFM agar after 5 days incubation. Note the lack of grey pigmentation of the ylmE mutant, indicative of a sporulation defect. (B) phase-contrast micrographs of impression prints of the strains shown in (A) as well as the complemented mutants. Arrows point at irregularly shaped spores in the ylmD mutant. The sporulation defect of the ylmD mutant could be complemented by introduction of wild-type ylmD and ylmD-eGFP, while complementation of the ylmE mutant by wild-type ylmE or ylmE-egfp restored sporulation, although irregularly sized spores were often produced. Full complementation of ylmE mutants was obtained by introduction of pKR8, which contains part of the S. coelicolor dcw cluster. Bar, 5 μm. (C) Size distributions of wild-type spores and those of ylmD, ylmE and ylmDE mutants presented as boxplots. Data are presented as median and interquartile range in boxplots, with whiskers spread to the maximal and minimal values. The numbers (n) of spores measured for each strain are indicated. Assessment of normality of spore length data was performed by a Kolgomorov-Smirnov test, the results showing that spore length data for all mutants followed non-normal distribution (Dmax > Dcrit). The shown two-tailed P values between each mutant and the parental strain were calculated using a Mann-Whitney U test. The two-tailed P values were far below 0.05, which shows that the sizes of the mutant spores deviated significantly from those of wild-type spores.
To further ascertain that the sporulation defects were indeed solely due to the deletion of the respective genes, we introduced plasmids expressing ylmD or ylmD-egfp in the in the ylmD null mutant and ylmE or ylmE-egfp in the ylmE mutant, in all cases with transcription directed from the native ftsZ promoter region. While the majority of the spores of the complemented ylmD mutant had a regular appearance, those of the complemented ylmE mutants still showed variable lengths (Fig. 2B). This partial complementation suggests that the deletion of ylmE may have polar effect on the expression of its downstream genes. This is supported by the fact that introduction of a plasmid harboring the entire region from murD to divIVA fully restored sporulation to ylmE mutants (Figs 2B and S2). Promoter probing using the redD reporter system40 showed that besides the intergenic promoter between ylmE and sepF, an additional promoter is located within ylmE, suggesting that deletion of the entire ylmE gene negatively affects the transcription of sepF (data not shown). This may explain why ylmE alone failed to fully complement the ylmE null mutant.
To quantify the spore lengths and their size distribution (i.e. variability), approximately 300 spores from the parental strain S. coelicolor M145, its ylmD and ylmE mutants and the genetically complemented strains were measured from light images from phase-contrast microscopy (100x magnification). Spore sizes were compared using a boxplot analysis (Fig. 2C). Wild-type spores typically had lengths between 0.8–1.2 µm, with an average length of 1.01 ± 0.15 µm, while ylmD mutant spores showed a slightly broader distribution (Fig. 2C), with an average length of 1.12 ± 0.27 µm. The average length was reduced to wild-type levels by complementation, namely to 1.03 µm and 1.05 µm by introduction of ylmD and ylmD-egfp, respectively. Spores of the ylmE null mutant were much larger and with a significantly wider variation, showing an average length of 2.23 ± 0.86 µm (Fig. 2C). Genetic complementation of the ylmE mutant restored spore length variation of ylmE mutant to wild-type levels, with an average size of 1.03 ± 0.19 µm. Partial complementation of the ylmE null mutant was seen when constructs expressing ylmE or ylmE-egfp were introduced into the mutant, with spore lengths of 1.62 ± 0.50 µm and 1.85 ± 0.59 µm, respectively. The average spore length of the combined ylmDE mutant was much closer to that of the parental strain, namely 1.23 ± 0.34 µm, thereby showing significantly smaller size variation than ylmE null mutants (Fig. 2C). The statistical validity of the variations in spore sizes between mutants and the parental strain was validated by a Kolgomorov-Smirnov test and a Mann-Whitney U test, which confirmed that the sizes of mutant spores deviated significantly from those of wild-type spores.
To study the spores of ylmD and ylmE null mutants at high resolution, cryo-scanning electron microscopy (SEM) was performed, which again demonstrated the sporulation defects. The parental strain S. coelicolor M145 produced abundant spore chains, with nearly all hyphae fully developed into mature spore chains (Fig. 3A,B). The ylmD null mutant GAL47 frequently produced aberrantly sized spores (Fig. 3C,D), while in ylmE null mutant GAL48, precious few and often aberrant spores were identified (Fig. 3E,F). The same approach was used to create ylmD and ylmE mutants in Streptomyces lividans 66, and these again showed the same morphology, strongly supporting the notion that the observed phenotypes were solely due to the mutations in ylmD or ylmE (data not shown). The highly similar phenotypes of the S. coelicolor and S. lividans mutants support the notion that the genes play a key role in sporulation-specific cell division and are consistent with the conserved function of the genes in sporulation-specific cell division in Streptomyces.

Cryo-scanning electron micrographs of aerial hyphae of S. coelicolor M145 and its ylmD and ylmE mutants. The parental strain produced wild-type spores, the ylmD mutant produced abundant but often irregular spores, while the ylmE null mutant produced occasional spores with highly irregular sizes. Cultures were grown on SFM agar plates for 5 days at 30 °C. Bars: top row, 1 μm; bottom row, 5 μm.
Deletion of ylmD and especially ylmE results in reduced robustness of spores
Viability of the spores was tested by plating around 1000 spores - counted in a hemocytometer - of S. coelicolor M145 and its mutants GAL47 (ΔylmD) and GAL48 (ΔylmE) onto SFM agar plates. While the wild-type strain had close to 100% viability, spores of the ylmD and ylmE mutants had reduced viability, with 50% and 60% viable spores, respectively. Spores of in particular the ylmE mutant were heat sensitive, with only 11 ± 1% viability after 15 min incubation at 58 °C, as compared to 47 ± 6% survival for ylmD null mutant spores and 55 ± 5% for the parental strain. Genetic complementation of the ylmE null mutant restored wild-type survival to heat shock (58 ± 3%). The same was true when ylmD was deleted in the ylmE null mutant (56 ± 4% survival).
To analyze the possible cell-wall defects in more detail, all strains were grown on SFM agar plates for 5 days and analyzed by Transmission Electron Microscopy (TEM) (Fig. 4). The parent produced typical spore chains and thick-walled spores. Conversely, ylmD and ylmE mutant spores were deformed and highly irregular. In particular the spores of the ylmE mutant had a thin wall similar to that of the hyphae, suggesting that spore-wall synthesis was compromised.

Transmission electron micrographs of spores of S. coelicolor M145 and its ylmD and ylmE mutants. Wild-type spores (M145) show regular sizes and appearance (A). In contrast, spores of the ylmD (B) and ylmE (C) null mutants have an irregular appearance. Note the lighter appearance of the spore wall in the ylmD null mutant and the lack of the typical thick spore wall in ylmE mutants. Cultures were grown on SFM agar plates for 5 days at 30 °C. Bar, 500 nm.
YlmD and YlmE are required for peptidoglycan synthesis at the septum
To analyze cell wall and membrane distribution in ylmD and ylmE mutants, fluorescence microscopy was performed on five-days old SFM surface-grown cultures of mutants GAL47 (M145 ΔylmD) and GAL48 (M145 ΔylmE) and compared to the parental strain M145. Peptidoglycan precursors were stained with FITC-WGA or Oregon-WGA and membranes visualized by staining with FM5-95. In wild-type pre-division aerial hyphae, long symmetrical septal ladders were observed when stained for cell wall or membranes (Fig. 5A). In contrast, aerial hyphae of the mutants showed highly disturbed cell wall and membrane distribution, which was more pronounced in ylmE than in ylmD mutants (Fig. 5A). Septation of ylmD mutants was complemented via re-introduction of ylmD; similarly, septation was restored to the ylmE mutant by re-introduction of ylmE, although peptidoglycan synthesis and septum spacing was still irregular (Fig. 5B). These data again show that the consequences of deleting ylmE are much more severe for sporulation-specific cell division then when ylmD is deleted.

Fluorescence microscopy of cell wall, DNA and membranes. (A) Fluorescent micrographs of hyphae stained for cell-wall synthesis (FITC-WGA or Oregon-WGA) or membranes (FM5-95). An overlay of these images is presented in the third column, and the corresponding light image in the last column. Bar, 5 µm. (B) Fluorescence micrographs showing DNA and cell-wall distribution in the complemented ylmD and ylmE mutants. While ladders of septa were formed in both strains, indicative that sporulation was restored to the mutants, in particular the complemented ylmE mutant formed imperfect septa. Cultures were grown on SFM agar plates for 5 days at 30 °C. Bar, 10 µm.
Cellular localization of YlmD and YlmE
Given the impact of ylmDE on sporulation, we analyzed how YlmD and YlmE were localized in the hyphae of S. coelicolor. To this end, constructs based on the low-copy number vector pHJL401 were prepared to allow the expression of YlmD-eGFP and YlmE-eGFP fusion proteins, which were expressed from the natural ftsZ promoter region (see Materials and Methods section for details). These constructs were then introduced into S. coelicolor M145 and (as a control for functionality) in the mutants. The constructs partially restored development to the respective mutants, with significant restoration of the sporulation defects in ylmD mutants, and partial restoration of sporulation to ylmE mutants (Fig. 2B).
In vegetative hyphae, only very weak signals were obtained for YlmD-eGFP and YlmE-eGFP, indicative of low protein expression (Fig. 6A). In early and late aerial hyphae, both YlmD-eGFP and YlmE-eGFP became more visible and showed diffuse localization along the wall of the aerial hyphae (Fig. 6A). During early sporulation, YlmD-eGFP and YlmE-eGFP showed an irregular pattern of varying intensity, with some spores showing bright fluorescence indicative of high concentrations of YlmD or YlmE, while others hardly showed any fluorescence (Fig. 6A). This suggests that YlmD and YlmE are differentially expressed throughout the nascent spore chains, and do not exclusively co-localize with FtsZ and the divisome. To rule out possible proteolysis of YlmD- or YlmE-eGFP fusion proteins, Western analysis was performed on extracts of surface-grown S. coelicolor mycelia, using antibodies against GFP. S. coelicolor M145 with empty vector and S coelicolor M145 expressing freely mobile eGFP41 were included as controls. Only a single band was identified in strains GAL50 and GAL49, corresponding to the expected lengths for YlmD-eGFP and YlmE-eGFP, respectively (Fig. S3). In both cases, the predicted mass of the fusion protein is around 52 kDa, which conforms well to the apparent mobility seen by Western analysis. The strain expressing only eGFP showed the expected band of around 27 kDa for the non-fused protein. Therefore, we conclude that the observed localizations can indeed be ascribed to intact YlmD- or YlmE-eGFP.

Localization of YlmD-GFP and YlmE-GFP. (A) Sporogenic aerial hyphae of S. coelicolor M145 at different stages of development were imaged by fluorescence microscopy visualizing YlmD-eGFP and YlmE-eGFP. Stages were: vegetative growth, early aerial development, late aerial development, early sporulation and mature spores. During spore maturation, YlmD and YlmE had a ‘patchy’ localization, suggesting that at this stage YlmD and YlmE may co-localize with the cell-wall synthetic machinery. Bar, 5 µm. (B) Relative fluorescence intensity of YlmD-eGFP and YlmE-eGFP during Streptomyces development. VEG, vegetative growth; EA, early aerial growth; LA, late aerial growth; ES, early sporulation; MS, mature spores.
The fluorescence intensity of YlmD-eGFP and YlmE-eGFP was measured on images representing different developmental stages (Fig. 6B). As development progressed, the fluorescence intensities increased for both YlmD-eGFP and YlmE-eGFP and reached peak levels during the phase corresponding to late aerial growth and early sporulation. When spores matured, YlmD-eGFP and YlmE-eGFP signals decreased to the level as in vegetative growth. These results suggest YlmD and YlmE proteins are active primarily during the phase of sporulation-specific cell division.
Localization of FtsZ is disturbed in ylmD and ylmE null mutants
To investigate the localization of FtsZ in the mutants, integrative plasmid pKF41 that expresses FtsZ-eGFP from the native ftsZ promoter region42 was introduced into the ylmD and ylmE mutants, to generate strain GAL52 and GAL53, respectively. Prior to sporulation, typical ladders of Z-rings were observed in the parental strain S. coelicolor M145, while ylmD and ylmE null mutants showed abnormal Z-ladders (Fig. 7). In the absence of YlmD, ladders were still observed, but the intensity varied and spacing between the individual rings was less regular, with many neighboring Z-rings either close together or widely spaced. Consistent with the sporulation defect, ylmE null mutants produced very few Z-rings, and the few Z-ladders that where formed were highly irregular or unfinished, and significantly shorter than in the parental strain. Thus, FtsZ localization is irregular in ylmD mutants and highly compromised in ylmE mutants.

Localization of FtsZ-eGFP in S. coelicolor M145 and its ylmD and ylmE mutants. FtsZ-eGFP formed typical ladders in wild-type cells (M145). In contrast, YlmE is required for the formation of ladders of FtsZ, while the absence of YlmD caused irregular spacing between the septa. Cultures were grown on SFM agar plates for 5 days at 30 °C. Bar, 5 μm.
A model for sporulation control by YlmDE in streptomycetes
Our data show that mutants of S. coelicolor lacking ylmE have severe developmental defects, with highly compromised cell-wall synthesis and aberrant cell division. Mutants which lack ylmD sporulated well, though many spores exhibited aberrant shapes and variable lengths. Surprisingly, the additional deletion of ylmD greatly rescued the sporulation-deficient phenotype of ylmE mutants, with the ylmDE null mutant producing abundant and mostly regularly sized spores.
We propose that YlmD and YlmE form a two-part system, reminiscent of toxin-antitoxin systems43. Similarly dysfunction of one protein in the presence of another has also been reported for the sporulation control protein WhiJ, following the observation that media-dependent sporulation defects caused by a point mutation in whiJ (SCO4543) could be rescued by the full deletion of whiJ44. The authors proposed a model wherein repression of developmental genes by WhiJ could be released when WhiJ interacts with a WhiJ-associated protein encoded by the adjacent gene SCO454244. It was suggested that the aberrant protein WhiJ* still binds to operator sequences of developmental genes, leading to their transcriptional repression, but fails to associate with SCO4542, with a permanent block of transcription of developmental genes as a result44. Similarly, the expression of YlmD alone is detrimental for sporulation, suggesting that YlmD acts in a deleterious manner towards sporulation, yet this effect can be relieved by the presence of YlmE. Supporting evidence for this is that deletion of both genes allows Streptomyces to develop normally, and by the observation that ylmD is never found alone in bacterial genomes, while the occurrence of an orphan ylmE is seen frequently. Further support comes from the report that ylmDE double mutants of S. venezuelae also sporulate relatively normally32. This also indicates that this two-part system functions in a similar manner in a range of Streptomyces species. This apparently rules out major divergence between the two morphologically distinct Streptomyces clades, namely those that sporulate in submerged cultures and those that do not26,45. However, it will be very interesting to see if submerged sporulation of S. venezuelae is prevented by the deletion of just ylmE. Currently, we are performing detailed structural and functional analysis of YlmD and YlmE, to elucidate their biochemical function and their precise role in bacterial cell division.
Methods
Phylogenetic analysis of ylmD and ylmE
The amino acid sequence of YlmD and YlmE were extracted from StrepDB (http://strepdb.streptomyces.org.uk) and used to search the NCBI database (www.ncbi.nlm.nih.gov) using BLASTP against the non-redundant protein sequence database. Alignment of YlmD and YlmE was generated using ClustalW46 followed by manual editing in MEGA v. 4.0. The neighbour-joining trees47 were generated with default parameters settings as implemented in MEGA v. 4.048. The maximum-likelihood trees were made using the best fit models predicted by MEGA. Tree reliability was estimated by bootstrapping with 1000 replicates. Trees were drawn with either N-J or ML algorithms gave trees of similar topologies indicating that the phylogenies are likely to reliable.
Bacterial strains and media
All bacterial strains used in this study are listed in Table S1. E. coli JM109 was used for routine cloning and ET1256749 to prepare nonmethylated DNA to bypass the methyl-specific restriction system of S. coelicolor. E. coli strains were propagated in Luria broth, where appropriate supplemented with antibiotics for selection, namely ampicillin (100 µg/ml end concentration), apramycin (50 µg/ml) and/or chloramphenicol (25 µg/ml). S. coelicolor A3(2) M14550 and S. lividans 6651 were obtained from the John Innes Centre strain collection. S. coelicolor strains were grown on soya flour medium (SFM) or minimal media mannitol (MM) agar plates for phenotypic characterization and on R5 agar plates for regeneration of protoplasts52. Antibiotics used for screening Streptomyces were apramycin (20 µg/ml end concentration) and thiostrepton (10 µg/ml).
Plasmids and constructs
All plasmids and constructs described in this study are summarized in Table S2. The oligonucleotides used for PCR are listed in Table S3. PCR reactions were performed using Pfu DNA polymerase as described53. All inserts of the constructs were verified by DNA sequencing, which was performed at BaseClear (Leiden, The Netherlands).
Constructs for the deletion of ylmD and ylmE
The strategy for creating knock-out mutants is based on the unstable multi-copy vector pWHM354 as described previously55. For each knock-out construct roughly 1.5 kb of upstream and downstream region of the respective genes were amplified by PCR from cosmid St4A10 that contains the dcw cluster of S. coelicolor. The upstream region was thereby cloned as an EcoRI-XbaI fragment, and the downstream part as an XbaI-BamHI fragment, and these were ligated into EcoRI-BamHI-digested pWHM3 (for the precise location of the oligonucleotides see Table S3). In this way, an XbaI site was engineered in-between the flanking regions of the gene of interest. This was then used to insert the apramycin resistance cassette aac(3)IV flanked by loxP sites, using engineered XbaI sites. The presence of the loxP recognition sites allows the efficient removal of the apramycin resistance cassette following the introduction of a plasmid pUWL-Cre expressing the Cre recombinase56,57. Knock-out plasmids pGWS728 and pGWS729 were created for the deletion of nucleotide positions +25/+ 696 of ylmE (SCO2080) and +25/+ 705 of ylmD (SCO2081), whereby + 1 refers to the translational start site of the respective genes. This allowed first the replacement by the apramycin resistance cassette. Subsequently the apramycin resistance cassette was removed using expression of pUWLCre. To create a ylmDE double deletion mutant, construct pGWS1044 was created which contains the upstream region of ylmD and downstream region of ylmE flanked by loxP sites, and with the apramycin resistance cassette aac(3)IV inserted in between.
For complementation of the ylmE and ylmD null mutants, the entire coding regions of SCO2080 and SCO2081 (with stop codons) were amplified from the S. coelicolor M145 chromosome using primer pairs ylmE_F + 1 and ylmE_R + 723 and ylmD_F + 1 and ylmD_R + 732, respectively. The PCR products were digested with StuI/XbaI, and inserted downstream of the native ftsZ promoter region in pHJL401, respectively. Thus constructs pGWS1042 and pGWS1043 were generated that express ylmE and ylmD, respectively, under control of the S. coelicolor ftsZ promoter region. Alternatively, construct pKR8 was used for complementation; pKR8 is based on integrative vector pIJ860058 and contains the 2227742-2241015 region of the S. coelicolor genome, encompassing the end of murX (SCO2087), the entire coding sequences of murD, ftsW, murG, ftsQ, ftsZ, ylmD, ylmE, sepF, sepG, divIVA and a large part of SCO2076 (encoding Ile-tRNA synthetase).
Constructs for the localization of YlmD and YlmE
The entire coding regions of SCO2080 and SCO2081 (without stop codons) were amplified from the S. coelicolor M145 chromosome using primer pairs ylmE_F + 1 and ylmE_R + 717 and ylmD_F + 1 and ylmD_R + 726, respectively. The PCR products were digested with StuI/BamHI, and inserted downstream of the native ftsZ promoter region and immediately upstream of egfp in pHJL401. The latter is a highly stable vector with low copy number that generally results in wild-type transcription levels and is well suited for among others complementation experiments40. Thus constructs pGWS757 and pGWS758 were generated that express YlmE-eGFP and YlmD-eGFP, respectively, from the native S. coelicolor ftsZ promoter region. Plasmid pKF41 expresses FtsZ-eGFP from its own promoter region42. Constructs pGWS757 and pGWS758 were also used to complement ylmE and ylmD null mutants, respectively.
eGFP detection using western blot analysis
Strains were grown on SFM agar plates containing the appropriate antibiotics overlaid with cellophane disks. Biomass (mycelium and spores) was collected after 6 days of growth at 30 °C and suspended in urea lysis buffer (8 M urea, 2 M thiourea, 4% CHAPS, 1 mM PMSF and 65 mM DTT), followed by cell disruption via sonication. Western blots were performed as described59. Briefly, cell lysates were obtained after centrifugation at 30,000 × g for 30 min at 4 °C. Proteins were separated on a 12.5% SDS-PAGE gel, and then transferred to PVDF membrane using Mini Trans-Blot (Bio-Rad). As primary antibody a 1:2000 dilution of GFP antibody (ThermoFisher, REF A11122) was used and incubated for 16 h at 4 °C in the presence of 3% BSA. After washing, the membrane was incubated with alkaline phosphatase conjugated anti-rabbit IgG (Sigma) as secondary antibody for 1 h at room temperature. Visualization was done using BCIP/NBT color development substrate.
Microscopy
Fluorescence and light microscopy were performed as described previously60. For peptidoglycan staining we used FITC-labeled wheat germ agglutinin (FITC-WGA) or Oregon Green 488 conjugated WGA (Oregon-WGA); for membrane staining, we used FM5-95 (all obtained from Molecular Probes). All images were background-corrected, setting the signal outside the hyphae to 0 to obtain a sufficiently dark background. These corrections were made using Adobe Photoshop CS4. Cryo-scanning electron microscopy (cryo-SEM) and transmission electron microscopy (TEM) were performed as described61. For quantification of fluorescence intensity of YlmD-eGFP and YlmE-eGFP, the arbitrary average intensity values of manually selected areas of interest were determined as relative fluorescence intensity. All green fluorescent images were made with equal exposure time.
Data analysis
For the Kolmogorov–Smirnov (KS) test, the critical value D crit was calculated for the number of measured spores (n) and significance level (α) of 0.05: \({D}_{crit}=\frac{\sqrt{-0.5\ast \,\mathrm{ln}(\frac{{\rm{\alpha }}}{2})}}{\sqrt{{\rm{n}}}}\). The difference (D n ) between the observed cumulative distribution function (F obs ) and the expected cumulative distribution function (F exp ) was calculated as: D n = |F exp (x) − F obs (x)|. The maximal value was identified as D max . The null hypothesis that the data comes from the specified distribution was rejected if D max > Dcrit. The Mann-Whitney U test was done using the SPSS software release 24.0.
Computer analysis
DNA and protein database searches were completed by using StrepDB (http://strepdb.streptomyces.org.uk/). Phylogenetic analysis was done using the STRING engine at EMBL (www.string.EMBL).
Data availability
All strains, materials and data will be made available upon request.
References
Bi, E. F. & Lutkenhaus, J. FtsZ ring structure associated with division in Escherichia coli. Nature 354, 161–164 (1991).
Goehring, N. W. & Beckwith, J. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr Biol 15, R514–526 (2005).
Adams, D. W. & Errington, J. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat Rev Microbiol 7, 642–653 (2009).
Hale, C. A. & de Boer, P. A. J. Direct Binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88, 175–185 (1997).
RayChaudhuri, D. ZipA is a MAP-Tau homolog and is essential for structural integrity of the cytokinetic FtsZ ring during bacterial cell division. EMBO J 18, 2372–2383 (1999).
Sekine, H., Mimura, J., Yamamoto, M. & Fujii-Kuriyama, Y. Unique and overlapping transcriptional roles of arylhydrocarbon receptor nuclear translocator (Arnt) and Arnt2 in xenobiotic and hypoxic responses. J Biol Chem 281, 37507–37516 (2006).
Gueiros-Filho, F. J. & Losick, R. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev 16, 2544–2556 (2002).
Hamoen, L. W., Meile, J. C., de Jong, W., Noirot, P. & Errington, J. SepF, a novel FtsZ-interacting protein required for a late step in cell division. Mol Microbiol 59, 989–999 (2006).
Ishikawa, S., Kawai, Y., Hiramatsu, K., Kuwano, M. & Ogasawara, N. A new FtsZ-interacting protein, YlmF, complements the activity of FtsA during progression of cell division in Bacillus subtilis. Mol Microbiol 60, 1364–1380 (2006).
Romberg, L. & Levin, P. A. Assembly dynamics of the bacterial cell division protein FTSZ: poised at the edge of stability. Annu Rev Microbiol 57, 125–154 (2003).
Barka, E. A. et al. Taxonomy, physiology, and natural products of the Actinobacteria. Microbiol Mol Biol Rev 80, 1–43 (2016).
Hopwood, D. A. Streptomyces in nature and medicine: the antibiotic makers. (Oxford University Press, 2007).
Wildermuth, H. & Hopwood, D. Septation during sporulation in Streptomyces coelicolor. J Gen Microbiol 60, 51–59 (1970).
Claessen, D., Rozen, D. E., Kuipers, O. P., Sogaard-Andersen, L. & van Wezel, G. P. Bacterial solutions to multicellularity: a tale of biofilms, filaments and fruiting bodies. Nat Rev Microbiol 12, 115–124 (2014).
Flärdh, K. & Buttner, M. J. Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat Rev Microbiol 7, 36–49 (2009).
McCormick, J. R. Cell division is dispensable but not irrelevant in Streptomyces. Curr Opin Microbiol 12, 689–698 (2009).
Celler, K., Koning, R. I., Willemse, J., Koster, A. J. & van Wezel, G. P. Cross-membranes orchestrate compartmentalization and morphogenesis in Streptomyces. Nat Comms 7, 11836 (2016).
Yague, P. et al. Subcompartmentalization by cross-membranes during early growth of Streptomyces hyphae. Nat Comms 7, 12467 (2016).
Holmes, N. A. et al. Coiled-coil protein Scy is a key component of a multiprotein assembly controlling polarized growth in Streptomyces. Proc Natl Acad Sci USA 110, E397–406 (2013).
Celler, K., Koning, R. I., Koster, A. J. & van Wezel, G. P. Multidimensional view of the bacterial cytoskeleton. J Bacteriol 195, 1627–1636 (2013).
Jakimowicz, D. & van Wezel, G. P. Cell division and DNA segregation in Streptomyces: how to build a septum in the middle of nowhere? Mol Microbiol 85, 393–404 (2012).
Schwedock, J., McCormick, J. R., Angert, E. R., Nodwell, J. R. & Losick, R. Assembly of the cell division protein FtsZ into ladder like structures in the aerial hyphae of Streptomyces coelicolor. Mol Microbiol 25, 847–858 (1997).
McCormick, J. R., Su, E. P., Driks, A. & Losick, R. Growth and viability of Streptomyces coelicolor mutant for the cell division gene ftsZ. Mol Microbiol 14, 243–254 (1994).
McCormick, J. R. & Losick, R. Cell division gene ftsQ is required for efficient sporulation but not growth and viability in Streptomyces coelicolor A3(2). J Bacteriol 178, 5295–5301 (1996).
Traag, B. A. & van Wezel, G. P. The SsgA-like proteins in actinomycetes: small proteins up to a big task. Antonie Van Leeuwenhoek 94, 85–97 (2008).
Girard, G. et al. A novel taxonomic marker that discriminates between morphologically complex actinomycetes. Open Biol 3, 130073 (2013).
Willemse, J., Borst, J. W., de Waal, E., Bisseling, T. & van Wezel, G. P. Positive control of cell division: FtsZ is recruited by SsgB during sporulation of Streptomyces. Genes Dev 25, 89–99 (2011).
Keijser, B. J., Noens, E. E., Kraal, B., Koerten, H. K. & van Wezel, G. P. The Streptomyces coelicolor ssgB gene is required for early stages of sporulation. FEMS Microbiol Lett 225, 59–67 (2003).
van Wezel, G. P. et al. ssgA is essential for sporulation of Streptomyces coelicolor A3(2) and affects hyphal development by stimulating septum formation. J Bacteriol 182, 5653–5662 (2000).
Zhang, L., Willemse, J., Claessen, D. & van Wezel, G. P. SepG coordinates sporulation-specific cell division and nucleoid organization in Streptomyces coelicolor. Open Biol 6, 150164 (2016).
Mingorance, J., Tamames, J. & Vicente, M. Genomic channeling in bacterial cell division. J Mol Recognit 17, 481–487 (2004).
Santos-Beneit, F., Gu, J. Y., Stimming, U. & Errington, J. ylmD and ylmE genes are dispensable for growth, cross-wall formation and sporulation in Streptomyces venezuelae. Heliyon 3, e00459 (2017).
Romero, D. A. et al. A comparison of key aspects of gene regulation in Streptomyces coelicolor and Escherichia coli using nucleotide-resolution transcription maps produced in parallel by global and differential RNA sequencing. Mol Microbiol 94, 963–987 (2014).
Swiatek, M. A. et al. The ROK family regulator Rok7B7 pleiotropically affects xylose utilization, carbon catabolite repression, and antibiotic production in Streptomyces coelicolor. J Bacteriol 195, 1236–1248 (2013).
Szklarczyk, D. et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39, D561–D568 (2011).
Claus, H. Laccases and their occurrence in prokaryotes. Arch Microbiol 179, 145–150 (2003).
Eswaramoorthy, S. et al. Structure of a yeast hypothetical protein selected by a structural genomics approach. Acta Crystallogr D Biol Crystallogr 59, 127–135 (2002).
Percudani, R. & Peracchi, A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO R 4, 850–854 (2003).
Tassoni, R., van der Aart, L. T., Ubbink, M., van Wezel, G. P. & Pannu, N. S. Structural and functional characterization of the alanine racemase from Streptomyces coelicolor A3(2). Biochem Biophys Res Comm 483, 122–128 (2017).
van Wezel, G. P., White, J., Hoogvliet, G. & Bibb, M. J. Application of redD, the transcriptional activator gene of the undecylprodigiosin biosynthetic pathway, as a reporter for transcriptional activity in Streptomyces coelicolor A3(2) and Streptomyces lividans. J Mol Microbiol Biotechnol 2, 551–556 (2000).
Zacchetti, B. et al. Aggregation of germlings is a major contributing factor towards mycelial heterogeneity of Streptomyces. Sci Rep 6, 27045 (2016).
Grantcharova, N., Lustig, U. & Flärdh, K. Dynamics of FtsZ assembly during sporulation in Streptomyces coelicolor A3(2). J Bacteriol 187, 3227–3237 (2005).
Yamaguchi, Y., Park, J. H. & Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Ann Rev Genet 45, 61–79 (2011).
Ainsa, J. A., Bird, N., Ryding, N. J., Findlay, K. C. & Chater, K. F. The complex whiJ locus mediates environmentally sensitive repression of development of Streptomyces coelicolor A3(2). Antonie Van Leeuwenhoek 98, 225–236 (2010).
van Dissel, D., Claessen, D. & Van Wezel, G. P. Morphogenesis of Streptomyces in submerged cultures. Adv Appl Microbiol 89, 1–45 (2014).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. Clustal-W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22, 4673–4680 (1994).
Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4, 406–425 (1987).
Tamura, K., Dudley, J., Nei, M. & Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24, 1596–1599 (2007).
MacNeil, D. J. et al. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111, 61–68 (1992).
Bentley, S. D. et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147 (2002).
Cruz-Morales, P. et al. The genome sequence of Streptomyces lividans 66 reveals a novel tRNA-dependent peptide biosynthetic system within a metal-related genomic island. Genome Biol Evol 5, 1165–1175 (2013).
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F. & Hopwood, D. A. Practical Streptomyces genetics. (John Innes Foundation, 2000).
Colson, S. et al. Conserved cis-acting elements upstream of genes composing the chitinolytic system of streptomycetes are DasR-responsive elements. J Mol Microbiol Biotechnol 12, 60–66 (2007).
Vara, J., Lewandowska-Skarbek, M., Wang, Y. G., Donadio, S. & Hutchinson, C. R. Cloning of genes governing the deoxysugar portion of the erythromycin biosynthesis pathway in Saccharopolyspora erythraea (Streptomyces erythreus). J Bacteriol 171, 5872–5881 (1989).
Świątek, M. A., Tenconi, E., Rigali, S. & van Wezel, G. P. Functional analysis of the N-acetylglucosamine metabolic genes of Streptomyces coelicolor and role in control of development and antibiotic production. J Bacteriol 194, 1136–1144 (2012).
Fedoryshyn, M., Welle, E., Bechthold, A. & Luzhetskyy, A. Functional expression of the Cre recombinase in actinomycetes. App Microbiol Biotechnol 78, 1065–1070 (2008).
Khodakaramian, G. et al. Expression of Cre recombinase during transient phage infection permits efficient marker removal in Streptomyces. Nucleic Acids Res 34, e20 (2006).
Sun, J., Kelemen, G. H., Fernandez-Abalos, J. M. & Bibb, M. J. Green fluorescent protein as a reporter for spatial and temporal gene expression in Streptomyces coelicolor A3(2). Microbiology 145, 2221–2227 (1999).
Vijgenboom, E. et al. Three tuf-like genes in the kirromycin producer Streptomyces ramocissimus. Microbiology 140, 983–998 (1994).
Willemse, J. & van Wezel, G. P. Imaging of Streptomyces coelicolor A3(2) with Reduced Autofluorescence Reveals a Novel Stage of FtsZ Localization. PLoS ONE 4, e4242 (2009).
Piette, A. et al. From dormant to germinating spores of Streptomyces coelicolor A3(2): new perspectives from the crp null mutant. J Proteome Res 4, 1699–1708 (2005).
Acknowledgements
This work was supported by the Netherlands Organization for Scientific Research (NWO), via VICI grant 10379 to GPvW.
Author information
Authors and Affiliations
Contributions
L.Z. performed the molecular biology experiments and created the mutants and expression constructs, J.W. performed the imaging and P.A.H. the phylogenetic analysis. G.P.v.W. conceived the experiments together with the other authors. All authors wrote and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Willemse, J., Hoskisson, P.A. et al. Sporulation-specific cell division defects in ylmE mutants of Streptomyces coelicolor are rescued by additional deletion of ylmD. Sci Rep 8, 7328 (2018). https://doi.org/10.1038/s41598-018-25782-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-25782-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
