
Abstract
Tunicates are marine invertebrates that compose the closest phylogenetic group to the vertebrates. These chordates present a particularly diverse range of regenerative abilities and life-history strategies. Consequently, tunicates provide an extraordinary perspective into the emergence and diversity of these traits. Here we describe the genome sequencing, annotation and analysis of the Stolidobranchian Botrylloides leachii. We have produced a high-quality 159 Mb assembly, 82% of the predicted 194 Mb genome. Analysing genome size, gene number, repetitive elements, orthologs clustering and gene ontology terms show that B. leachii has a genomic architecture similar to that of most solitary tunicates, while other recently sequenced colonial ascidians have undergone genome expansion. In addition, ortholog clustering has identified groups of candidate genes for the study of colonialism and whole-body regeneration. By analysing the structure and composition of conserved gene linkages, we observed examples of cluster breaks and gene dispersions, suggesting that several lineage-specific genome rearrangements occurred during tunicate evolution. We also found lineage-specific gene gain and loss within conserved cell-signalling pathways. Such examples of genetic changes within conserved cell-signalling pathways commonly associated with regeneration and development that may underlie some of the diverse regenerative abilities observed in tunicates. Overall, these results provide a novel resource for the study of tunicates and of colonial ascidians.
Similar content being viewed by others


Introduction
Tunicates are a group of worldwide marine invertebrates, the majority of which are subtidal suspension-feeding hermaphrodites. This subphylum is part of the Chordata phylum, phylogenetically positioned between the more basal Cephalochordata and the higher Vertebrata, of which they are considered the closest relatives1 (Fig. 1A). These organisms include a wide range of reproductive methods, regenerative abilities, developmental strategies and life cycles2. Importantly, and despite a drastically different body plan during their adult life cycle, tunicates have a tissue complexity related to that of vertebrates (Fig. 1A), including a heart, a notochord, an endostyle and a vascular system3. In addition, this group of animals is undergoing rapid genomic evolution, with a greater nucleotide substitution rate observed in both their nuclear and mitochondrial genomes, when compared to vertebrates4,5,6,7. Therefore, this chordate subphylum provides an excellent opportunity to study the origin of vertebrates, the emergence of clade specific traits and the function of conserved molecular mechanisms. Biological features that can be investigated in tunicates include, among others, the evolution of colonialism, sessileness, and budding. Moreover, some compound tunicates can undergo whole-body regeneration (WBR), whereby a fully functional adult can be restored from a portion of vascular tissue8. The presence of such an extensive regenerative capacity, in the closest relatives of the vertebrates, renders the study of tunicates particularly well suited for comparative research. In particular, identifying and investigating the shared regulatory mechanisms and signalling pathways required for successful regeneration is of interest to regenerative medicine and ageing research9,10,11,12. However, there are currently only eight Tunicata genomes publicly available7,13,14,15, of which four have been well annotated.

B. leachii phylogenetic position and life cycle. (A) Schematic showing phylogeny of tunicates with respect to the chordate clade (consensus based on4,146,147). (B) Life cycle of B. leachii. The colony expands and grows by asexual reproduction (right loop). During favourable conditions such as warmer water temperatures, members of the colonies start sexual reproduction (left loop). The embryo develops viviparously within the colony in brood pouches until hatching. Motile larvae attach to nearby substrates and begin metamorphosis into oozooids. Abbreviations: zooid (z), system (y), tunic (c), vascular system (v), terminal ampullae (a), buccal siphon (pb), atrial siphon (pa), fertilized oocyte (o), notochord (n), larval tadpole (l), oozooid (zo), bud (b), budlet (bt), regressing zooid (r).
Tunicates are separated into seven orders contained in three classes (Fig. 1A): Appendicularia (order Copelata), Thaliacea (orders Pyrosomida, Salpida and Doliolida) and Ascidiacea (orders Aplousobranchia, Phlebobranchia and Stolidobranchia). Appendicularia is a class of planktonic free-swimming organisms that possess chordate traits common to most tunicate larvae including a notochord, neural tube and pharyngeal slits. These social animals form communities where each individual is enclosed inside a special external mucous structure, termed house, which concentrates and funnels their food. Oikopleura dioica is the sole example of the Appendicularian to have its genome sequenced, showing exceptional compaction (70 Mb)13.
Thaliacea is a class of planktonic pelagic animals forming free-floating compound colonies16. These organisms can reproduce both sexually to initiate novel colonies, as well as asexually, through stolonial budding, to increase the size of the colony. Owing to their peculiar life cycle and habitat, these tunicates have been studied less thoroughly in comparison to other ascidians, and whether they can undergo regeneration remains unknown. A single species, Salpa thompsoni, has been sequenced7 and has a large and repetitive genome (602 Mb, 33% is repetitive sequences).
Ascidiacea consists of both solitary and colonial sessile benthic organisms. Solitary ascidians (Phlebobranchian and some families among the Stolidobranchian) reproduce sexually, releasing eggs through their atrial siphon for external fertilization, hence producing a motile larva. These larvae will explore their environment, attach to a submersed substrate and undergo metamorphosis into a sessile filter-feeding adult. These ascidians can regenerate some organs, including their oral siphon17,18 although regeneration capability reduces as they age19. Ascidiacean genomes represent the majority of the sequenced tunicate genomes, with five published genomes (Ciona robusta [formerly known as C. intestinalis type A], Ciona savigny, Molgula oculata, Molgula occulta, Molgula occidentalis14,20,21,22,23), two yet unpublished species (Phallusia mamilata, Phallusia fumigata22) and two currently being assembled (Halocynthia rorezi, Halocynthia aurantium22). These published genomes are estimated to be between 160–200 Mb (Table 1).
Colonial sessile tunicates (species found in the Aplousobranchia and Stolidobranchia orders) are capable of both sexual and asexual reproduction, through a wide range of budding types (palleal, vascular, stolonial, pyloric and strobilation24), as well as WBR. Colonial ascidians are emerging as increasingly popular model organisms for a variety of studies including immunobiology, allorecognition, angiogenesis and WBR25,26,27,28,29,30,31,32. Only a single colonial Stolidobranchia genome, Botryllus schlosseri, is publicly available, which revealed a considerable expansion of genome size (725 Mb, almost three fold) when compared to the other published ascidian genomes15. A second partially assembled, yet unpublished, genome of colonial ascidian appears to reflect a similar genome expansion (Didemnum vexillum, >542 Mb33). To provide a resource for further studies on the genetics and evolution of this subphylum, as well as research on colonialism and WBR, we have assembled and analysed the genome sequence of Botrylloides leachii (class Ascidiacea, order Stolidobranchia34).
The viviparous colonial ascidian B. leachii (Fig. 1B) lives in colonies composed of genetically identical adults (termed zooids) organized in ladder-like systems and embedded in gelatinous matrix (tunic). While each adult has its own heart, they all share a common vascular system embedded within the tunic. In the presence of sufficient food supply, the size of the colony doubles weekly through synchronized asexual reproduction, known as palleal budding35. During this process, each adult produces two daughter zooids that ultimately replace the mother, which is then resorbed by the colony (Fig. 1B). B. leachii can also reproduce sexually through a tadpole stage that allows the settlement of a new colony onto a substrate (Fig. 1B). Following removal or loss of all zooids from the colony, B. leachii can undergo WBR and restore a single fully-functional adult in as little as 10 days from a small piece of its vascular system26. Furthermore, when facing unfavourable environmental conditions, these colonial tunicates can enter into hibernation, whereby all zooids undergo regression and are resorbed by the remaining vascular system. When a favourable environment is restored, mature adults will develop to re-establish the colony36.
We have assembled and annotated the first de novo draft genome of B. leachii by taking advantage of our recently published transcriptomes37. Using this genome, we have then undertaken a large-scale comparison of the four best-annotated tunicate genomes (B. schlosseri, C. robusta, M. oculata and O. dioica) to gain insights into some of the diverse biological abilities that have evolved within the Tunicata.
Results
Genome assembly and annotation
To minimize contamination from marine algae and bacteria typically present in the pharyngeal basket of feeding B. leachii, we isolated genomic DNA from embryos of a single wild B. leachii colony. Genomic DNA was used to produce two libraries: one short-range consisting of 19,090,212 fragments (300 bp) of which 100 bp were paired-end sequenced, important to obtain high coverage, and a second long-range mate pair with 31,780,788 fragments (1.5–15 kb size range, median ~3 kb) of which 250 bp were paired-end sequenced, to aid scaffold assembly. Following quality checks, low quality reads were removed and sequencing adaptors were trimmed, thus resulting in a high-quality dataset of 86,644,308 paired-end and 12,112,004 single-end sequences (100% with a mean Phred score >=30, <1% with an adapter sequence, Fig. S1A).
We then followed a reference-free genome characterization38 to estimate three properties of the B. leachii genome; provided with statistics from the human, fish (Maylandia zebra39), bird (Melopsittacus undulatus39) and oyster (Crassostrea gigas40) genomes for comparison. Firstly, the SGA-PreQC package38 was used to estimate the genome size to be 194 Mb (194,153,277 bp). This size is similar to that of the solitary C. robusta, C. savigny and M. occidentalis, M. oculata (160 Mb, 190 Mb, 160 Mb and 160 Mb, respectively14,20,23), larger than the compacted 70 Mb genome of O. dioica13 but appreciably smaller than the predicted 725 Mb genome of the closely related colonial ascidian B. schlosseri, of which 580 Mb have been sequenced15. Secondly, by quantifying the structure of the de Brujin graph obtained using the k-mer counts (k = 31), the computational complexity of the assembly was estimated (sequencing errors 1/213; allelic differences 1/233; genomic repeats 1/2,439). With a cumulative occurrence of 1/106, the B. leachii genome is similar to that of bird, more variable than those of fish and human, but still quite less complex than the notably difficult oyster genome38 (Fig. S1B). Lastly, sequence coverage was estimated using the distribution of 51-mers counts, showing a well-separated bimodal distribution with a true-genomic k-mers maximum at 31 × coverage, similar to the human genome but higher than both the fish and the bird. Overall, these metrics suggest that the raw sequencing data was suitable for de novo assembly.
De novo assembly using Metassembler41 produced a genome of 159,132,706 bp (estimated percentage of genome assembled is 82%), with an average sequencing coverage of 66x (after adaptor trimming). The assembly is composed of 1,778 scaffolds, with a N50 scaffold length of 209,776 and a L50 scaffold count of 223. The 7,783 contigs, with a N50 length of 48,085, and a L50 count of 781, represent a total size of 146,061,259 (92%, Table 2). To evaluate the completeness of our assembly, we used the Benchmarking Universal Single-Copy Orthologs (BUSCO42). This tool provides a quantitative measure of genome completeness by verifying the presence of a set of manually curated and highly conserved genes. Out of the 978 orthologs selected in metazoans, 866 (89%) were found in our assembly of the B. leachii genome (File S1), a relatively high score when compared to the BUSCO score of frequently used genome assemblies such as Homo sapiens (89%, GCA_000001405.1542). In addition, we took advantage of our previous assembly of the B. leachii transcriptome37 to further assess the quality of our genome. Using BLAT43, we were able to map 93% of transcript sequences (48,510/52,004) onto our assembly. Overall, these results indicate that the B. leachii de novo genome assembly was largely complete and suitable for annotation.
Ab initio genome annotation was performed using MAKER244 and predicted 15,839 coding genes, of which 13,507 could be classified using InterProScan45. Comparing these predictions with our mapping of the transcriptome, we found out that 83% of our aligned cDNA (40,188/48,510) mapped to a predicted gene locus thus spanning 78% of the annotated genes (12,395/15,839). In addition, a total of 4,213 non-coding sequences were predicted using Infernal46, Snoscan47 and tRNAscan-SE48. Finally, repetitive elements were annotated using RepeatMasker49 and a species-specific library created using the RepeatModeler module50. Ninteen percent of the genome was identified as containing repetitive elements (Table 2, File S2), a majority (17% out of 19%) of these being interspersed repeats.
To further characterize the genome of B. leachii, we compared it to four available Tunicata genomes. The proportion of repetitive elements in B. leachii is similar to other tunicates (File S2) including C. robusta (25%), M. oculata (24%) and O. dioica (15%), while being much lower than B. schlosseri (60%). In particular, there are at least two additional families in the B. schlosseri hAT transposon superfamily and counts for some hAT elements differ dramatically (e.g. hAT-Charlie 366 in B. leachii vs 46,661 in B. schlosseri; File S2). We then quantified the number of sequences from each proteome that mapped onto our assembly using tBLASTn51: C. robusta 71% (10,507/14,740), M. oculata 77% (12,788/16,616), B. schlosseri 71% (32,944/46,519) but only 30% for O. dioica (9,009/29,572).
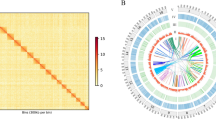
Next, we performed an all-to-all search for protein orthologs between the tunicate genomes using the OrthoMCL clustering approach52 (Fig. 2A). Clustering the combined protein set from all five genomes resulted in 17,710 orthologous groups of annotated genes. By classifying each group based on which tunicate genome(s) they were present within, we identified five orthologous sets of genes: those shared by all species (17% of all groups, 2,972 groups), those shared by all sessile tunicates (11%, 1,927), those between two colonial species (11%, 1,946) and two groups unique to B. schlosseri and O. dioica (15% and 12%, 2,716 and 2,160, respectively; Fig. 2A). Lastly, these proteins specific to a single genome were removed from the corresponding proteome, and a new mapping onto our assembly was performed. Mapping of these two filtered proteomes reached 93% for B. schlosseri and 45% for O. dioica. Altogether, these results indicate that our de novo assembly is highly compatible with that of other tunicates and thus amenable for comparisons with their genomes.

Comparison of tunicate genomes. (A) Clustering of orthologous protein sequences. Indicated are the number of cluster groups, each of which contains at least two proteins. (B) TreeMap representation of the overrepresented GO Biological Processes terms within the ortholog groups shared between B. leachii and B. schlosseri genomes but not with C. robusta, O. dioica and M. oculata. Each rectangle size is proportional to the GOrilla minimum hypergeometric p-value of each GO term. (C) Distribution of the three classes of GO terms for each species. The colour-codes (left) are common for the entire row.
To gain insights into the potential biological function underlying these ortholog groups, we analysed the distribution of Gene Ontology (GO) terms for each cluster and visualized these using REVIGO/Treemap (Figs 2B and S2). Given that the proteins identified by OrthoMCL clustering are potentially novel to colonial ascidians, a cross-species approach for GO enrichment was performed using the Human GO database as background53 (Fig. 2B). The overrepresented genes (Fig. 2B, File S4) function in biological processes such as circulation (GO:0003018, GO:0003013, GO:0050880), wound healing (GO:0072378) and cell communication (GO:0007154); as well as the regulation of immune cell differentiation (GO:0033081, GO:0033089) and immune system processes (GO:0002376, GO:0032608). These biological functions are concordant with that of proteins predicted to be required for the life cycle of colonial ascidian.
Finally, we compared the overall composition of GO terms for all five tunicates (Fig. 2C). Despite B. schlosseri having a larger predicted gene number compared to the other analysed tunicates, the overall proportion of GO group terms were distributed similarly between all genomes (Fig. 2C), indicating no expansion of one particular functional group in B. schlosseri.
Overall, the above analyses showed that our assembly and annotation are consistent with the other tunicate genomes and will provide additional insights into the Tunicata subphylum.
Ancient gene linkages are fragmented in tunicate genomes
To gain further insights in the evolution of the Tunicate genomes, we investigated the organisation of three ancient gene clusters, representing highly conserved sets of genes that are typically located adjacent to each other within a genome54. These clusters arose in a common ancestor and were preserved because of shared regulatory mechanisms. The homeobox-containing Hox gene family55, typically composed of 13 members in vertebrates56, is among the best-studied examples of such an ancient gene cluster, and is critical for embryonic development57. The linear genomic arrangement of genes within the Hox cluster reflects their spatial expression along the anterior-posterior body axis, which establishes regional identity across this axis57.
The basal cephalochordate Branchiostoma floridae genome has all 13 Hox genes located in a single cluster, along with two additional Hox genes (Fig. 3), suggesting that the chordate ancestor also had an intact cluster58. However, in tunicates, this clustering appears to be lost59,60,61,62 (Fig. 3). In C. robusta, the nine identified Hox genes are distributed across five scaffolds, with linkages preserved only between Hox2, Hox3 and Hox4; Hox5 and Hox6; Hox12 and Hox1359,60 (Fig. 3). In O. dioica, the total number of Hox genes is further reduced to eight, split between 6 scaffolds, including a duplication of Hox961,62 (Fig. 3). In M. oculata we could identify only six Hox genes, divided between 4 scaffolds, with clustering retained for the Hox10, Hox11 and Hox12 genes (Fig. 3). In Botryllidae genomes, the same seven Hox genes are conserved (Fig. 3), with a preserved linkage between Hox10, Hox12 and Hox13 in B. leachii and three copies of Hox5 present in B. schlosseri. Of the seven B. leachii Hox genes, transcripts for four are present in our reference transcriptome37 (Hox1, Hox4, Hox10 and Hox12; File S5), indicating that they may still be functional. Two of the Hox genes (Hox2 and Hox5) were not predicted by AUGUSTUS, nor were they present in the transcriptome (File S5); this may represent either partial (non-functional) genes, or a lack of expression in the tissues used to assemble the transcriptome. Altogether, the separation of the tunicate Hox cluster genes supports the hypothesis that reduction and separation of this ancient gene linkage occurred at the base of the tunicate lineage61. In addition, there is no particular pattern to the complement of retained Hox genes, with only Hox1, Hox10 and Hox12 being found in all five examined tunicate genomes (Fig. 3).

Hox genes are dispersed and reduced in number within tunicate genomes. Schematic depicting Hox gene linkages retained in five tunicate genomes in comparison to the ancestral Hox complex, which included thirteen genes. Orthologous genes are indicated by common colours. Chromosome (chr) or scaffold number (S) is shown, along with gene ID when available for newly annotated genomes. For B. floridae and H. sapiens, the length of each Hox gene cluster is given in brackets, and for B. leachii, the total scaffold length is shown. If a gene ID is not available (for unannotated genes), the co-ordinates of the BLAST hit (for the homeobox protein domain) is either given in File S5 or shown in the figure under the putative gene. Transcript IDs for B. leachii Hox genes identified in our transcriptome data37 are also provided in File S5.
A second ancient linkage that we investigated is the pharyngeal cluster, a gene group present in hemichordates, echinoderms and vertebrate genomes that is considered to be Deuterosome specific63. The cluster groups foxhead domain protein (FoxA), Nkx2 (Nkx2.2 and Nkx2.1), Pax1/9, mitochondrial solute carrier family 25 member 21 (slc25A21), mirror-image polydactyly 1 protein (mipol1), egl nine homolog 3 (egln3) and dehydrogenase/reductase member 7 (dhrs7). Among these, slc25a21, Pax1/9, mipol1 and FoxA pairs are also found in protostomes suggesting an even more ancient origin63. The pharyngeal cluster is thought to have arisen due to the location of the regulatory elements of Pax1/9 and FoxA within the introns of slc25A21 and mipol164,65, compelling these genes to remain physically located near each other in a genome.
In the B. floridae genome, the entire cluster is located on the same scaffold, with the exception of the Nkx2.1 and Nk2.2 gene pair located on a separate scaffold, with an average intergenic distance of 14 kb (Fig. 4). In C. robusta, orthologs of FoxA2, slc25a29, Pax1 and dhrs7 are located on the same chromosome (chr 11, Fig. 4), with only Pax1/9 and slc25A29 located in close proximity to each other (~3 kb, Fig. 4). In O. dioica, orthologs of FoxA, Pax1/9 and Nkx2.2 genes were found on different scaffolds, with only one linkage (~10 kb), between Pax1/9 and Nkx2.1 genes, preserved. For both B. schlosseri and M. oculata there was no evidence of clustering between genes (Fig. 4). However, some M. oculata scaffolds are too small (5–13 kb) to make definite conclusions, given that the observed gene dispersal may be an artefact of a fragmented assembly. In the B. leachii genome, mipol1 is the sole missing gene from this cluster and only the pairing of Pax1/9 and slc25A21 remains (Fig. 4).

Ancestral gene linkages remain between a few pharyngeal cluster genes in tunicate genomes. Schematic depicting the organization of the pharyngeal cluster genes among the studied chordate genomes. Double-parallel lines indicate >1 Mb distance between genes. Chromosome (chr) or scaffold (S) number is shown, along with gene ID when available for newly annotated genomes. Orthologous genes are indicated by common colours. Transcript IDs for B. leachii genes identified in our transcriptome data37 are provided in File S5.
A third ancient homeobox-containing gene linkage is the NK cluster. This cluster, predicted to be present in the last common ancestor of bilaterians66, consists of Msx, Lbx, Tlx, NKx1, NKx3, NKx4 and NKx5 (Fig. 5). In B. floridae, linkages between Msx, NKx4 and NKx3; as well as between Lbx and Tlx provide evidence of retained ancestral clustering while NKx5 was lost66 (Fig. 5). However, in vertebrates, only the gene linkages between Lbx and Tlx as well as between NKx4 and NKx3 remain55 (Fig. 5). To further clarify the evolution of this ancestral cluster in tunicates, we determined the structure of the NK cluster within five ascidian genomes. In all these species, NKx1 is absent and no evidence of clustering could be found with all identified orthologs located on different scaffolds or chromosomes (Fig. 5). While some of the assembled scaffolds are small (especially for M. oculata), even those tunicate genomes with assembled chromosome sequences and scaffolds larger than >1 Mb show no evidence of cluster retention, suggesting that most of the tunicates did not conserve the structure of this ancient linkage. In M. oculata only four members of this cluster were identified in the current assembly, with the loss of NKx5 as well as Lbx (Fig. 5). In the colonial tunicates B. leachii and B. schlosseri, Tbx, Lbx and NKx3 are all present. In B. schlosseri, Msx is absent and NKx4 duplicated. In the B. leachii genome, NKx1 is the only ancestral cluster member to be missing and NKx5 has been duplicated (Fig. 5). These results suggest that there has been a loss of NKx5 in cephalochordates, one of NKx1 in tunicates and that the retention of both Lbx and Tbx may be specific to colonial ascidians. However, only Msx and NKx4 were identified in our transcriptome (File S5), therefore we cannot be certain if NKx3, Lbx1, Tlx and NKx5 genes are expressed and functional in B. leachii.

NK homeobox cluster genes are fragmented within tunicate genomes. Schematic depicting the organization of the NK homeobox cluster genes among the studied chordate genomes. Double-parallel lines indicate >1 Mb distance between genes. Chromosome (chr) or scaffold (S) number is shown, along with gene ID when available for newly annotated genomes. Orthologous genes are indicated by common colours. Transcript IDs for B. leachii genes identified in our transcriptome data37 are provided in File S5.
Taken together, these three results suggest that most of the tunicates did not conserve the structure of some ancient gene linkages. Further studies are needed to determine the consequences to both gene expression and function following loss of gene clustering.
Lineage-specific changes to cell-signalling pathways in Botryllidae genomes
To examine the evolution of colonial ascidians more directly, we examined the genomes of B. leachii and B. schlosseri, looking for key components of signalling pathways required for metazoan development and regeneration. Of particular interest, we focused on the Wingless-related integration site (Wnt), Notch and Retinoic acid (RA) signalling pathways. All three of these pathways have been implicated in WBR and asexual reproduction in colonial tunicates25,37,67.
Wnt pathway
Wnt ligands are secreted glycoproteins that have roles in axis patterning, morphogenesis and cell specification68. The ancestral gene family appears to have originated early during multi-cellular evolution and is composed of eleven members69,70. The Wnt gene family expanded to 19 members in the human genome, while independent gene loss has reduced this family to 7 genes in Drosophila melanogaster and Caenorhabditis elegans71. Consequently, we investigated whether the Wnt gene family has either expanded or contracted during Tunicata speciation.
We found an increase in the number of Wnt5a genes among Styelidae genomes (Fig. 6A). In B. schlosseri, we identified 15 Wnt members, including seven Wnt5a genes on multiple scaffolds (Fig. 6A). In the B. leachii genome, fourteen Wnt ligand genes were identified, including four Wnt5a genes located on the same scaffold near Wnt4 (Fig. S3). M. oculata has only 7 Wnt ligand genes, including three Wnt5a genes (Fig. 6A). In comparison, C. robusta has a total of 11 Wnt genes, including a single copy of Wnt5a60 (Fig. 6A). In the compact O. dioica genome, this number is reduced to 6 (Wnts 3, 4, 7, 11 and 16), none of which are Wnt5a orthologs (Fig. 6A). The various orthologs of the duplicated Wnt5a genes show a similar exon-intron structure (Fig. S3), which indicates that they are likely to have arisen through gene duplication72. Overall, our data suggests that an expansion, possibility through gene duplication, of the Wnt5a family occurred during tunicate evolution, but was lost in some lineages.

Duplication of components of the Wnt signalling pathway in tunicate genomes. Schematic showing the organization of (A) the Wnt genes within each indicated genome and (B) of the downstream effectors. Note that no Wnt5 ortholog is present in the O. dioica genome. Genome browser images for the Wnt5 genes are shown in Fig. S4.
To assess the functionality of the Wnt pathway in tunicates, we evaluated whether its downstream effectors are also present in the available genomic data. The downstream pathways activated by Wnt ligands are divided into canonical, non-canonical calcium and non-canonical planar cell polarity. The Wnt5a ligand is associated with both of the non-canonical pathways through binding of membrane receptors that include frizzled (Fzd4), receptor tyrosine kinase-like orphan receptor 1/2 (Ror1/2) and atypical tyrosine kinase receptor (Ryk)68. Further downstream, dishevelled (Dvl), β-catenin (Cnntb), Axin, low-density lipoprotein receptor-related protein 5/6 (LRP5/6) and nuclear factor of activated T-cells (NFAT) are proteins essential for triggering intracellular responses to Wnt signalling73. We identified orthologs for each of these signalling transduction molecules in all Tunicata genomes (Fig. 6B), with no evidence of further gene duplication events. Importantly, 90% of the genes identified in B. leachii (35/39) have a corresponding transcript in the transcriptome37 (File S5). This supports the interpretation that signalling through the Wnt pathway is functional in tunicates.
Notch pathway
Notch receptors are transmembrane proteins that are involved in cell-cell signalling during development, morphogenesis and regeneration74. Following activation through the binding of the Delta or Jagged/Serrate ligands, the intracellular domain of Notch is cleaved and induces the expression of downstream target genes including the hairy and enhancer of split (hes) gene family members75. The presence of both Notch and the Delta/Serrate/lag-2 (DSL) proteins in most metazoan genomes suggests that their last common ancestor had a single copy of each gene76. To establish how this pathway has evolved in tunicates, we screened these genomes for the Notch receptor using the conserved Lin12/Notch Repeat (LNR) domain, as well as for genes encoding probable Notch ligands.
In all examined genomes, only a single Notch receptor gene was identified while the number of ligand genes varied (Fig. S4A). The C. robusta genome contains two DSL genes, while O. dioica, M. oculata and B. schlosseri possess only a single DSL gene. By contrast, we found three DSL genes in B. leachii (Fig. S4A). To determine the relationships between these identified tunicate DSL-like genes, a phylogeny was constructed with other chordate DSL proteins. All three B. leachii genes are Delta orthologs, two of them related to the B. schlosseri and Cionidae proteins; the third one closer to the M. oculata and H. roretzi variants. The mouse, human and zebrafish delta and delta-like (DLL) proteins form a discrete clade loosely related to the genes found in cephalochordates and tunicates (Fig. 7, red box). Jagged proteins form a separate clade where no subphylum-specific clustering is observed (Fig. 7, blue box). The tunicate DSL-like proteins show long phylogenetic branches, suggestive of greater diversity, also observed in the protein alignment (Fig. S5A). This suggests that the tunicate DSL proteins are diverging rapidly from each other, indicative of lineage specific evolution of DSL-like genes.

Tunicate Delta proteins. Bayesian phylogenetic tree depicting the relationship between tunicate and vertebrate DSL proteins, using Drosophila Delta to root the tree. Tunicate proteins are shown in bold and shaded areas correspond to Delta and Jagged groupings. Branch support values (probabilities) are indicated.
Retinoic acid signalling
Retinoic acid (RA) is an extracellular metabolite that is essential for chordate embryonic development. RA is synthesized from retinol (vitamin A) by two successive oxidation steps. In the first step, retinol dehydrogenase (Rdh) transforms retinol into retinal. Then RA is produced by aldehyde dehydrogenase (Aldh), a superfamily of enzymes with essential roles in detoxification and metabolism77. RA influences the expression of downstream target genes by binding to the RA receptors, RAR and RXR78 (Fig. 8A). Finally, RA is metabolized by the cytochrome P450 family 26 (Cyp26) enzyme, which absence of expression can restrict RA-induced responses to specific tissues or cell types79,80. Components of this pathway have been found in non-chordate animals, suggesting a more ancient origin80. This pathway is required for B. leachii WBR25 and Ciona development, yet several genes essential for RA signalling appear to be missing in O. dioica80,81.

Evolution of the RA pathway in tunicates. (A) Overview of the RA synthesis and degradation pathway. In bold are the major proteins that contribute to RA signalling during animal development. Indicated below these are changes to the number of copies present in examined genomes. (B) Maximum likelihood phylogenetic tree depicting the relationship between invertebrate and vertebrate CYP26 proteins using CYP4 and CYP51 proteins as an out-group. Tunicate proteins are shown in bold. No Cyp26 gene has been identified in the O. dioica genome81. Values for the approximate likelihood-ratio test (aLRT) are indicated.
Rdh10 is the major dehydrogenase associated with the first steps of RA production, although the Rdh16 and RdhE2 enzymes can also substitute this function82,83,84. The O. dioica genome has no orthologs for either Rdh10 or Rdh16 but it does have four genes that encode for RdhE2 proteins81. O. dioica also lacks both an Aldh1-type gene, as well as a Cyp26 gene, but has one single RXR-ortholog81 (Fig. S5B). In contrast, the C. robusta genome contains single copies of Rdh10, Rdh16 and RdhE2 genes and a total of four Aldh1 genes, located on two chromosomes80. Consistent with C. robusta, M. oculata, B. leachii and B. schlosseri genomes all have single copies of Rdh10, Rdh16 and RdhE2 genes, as well as three Aldh1 genes on separate scaffolds (Fig. S5B).
Three retinoic acid receptor genes were identified within the B. leachii genome, one of which had been previously cloned25 (g03013 in our assembly; File S5). All three were also found in C. robusta, M. oculata and B. schlosseri genomes (File. S5B). While there is only one potential Cyp26 gene in M. oculata, four paralogs were identified in B. leachii and B. schlosseri. A phylogenetic analysis, using related chordate Cyp proteins as an outgroup, showed that these paralogs cluster with Cyp26 proteins (Figs 8B and S5B). Altogether, these results show a loss of key RA-pathway genes in O. dioica (Rdh10, Rdh16, Cyp26 and Aldh1) while, in non-larvacean tunicates, the copy numbers of some genes has increased, suggesting that RA signalling pathway is still functional.
Discussion
Genomic diversity within the Stolidobranchia
The diversity of changes to developmental pathways observed between the B. leachii genome and that of closely related ascidians, along with previous genomic analyses of other ascidian species, supports the widely held view that ascidian genomes are diverse and rapidly evolving, which is particularly evident in the Stolidobranchia group5,13,14,15,85,86,87,88. Nevertheless, Styelidae were sufficiently similar in external appearance and morphology for early researchers to suggest that Botrylloides could be a subgenus of Botryllus89,90. Strikingly however, the B. schlosseri genome differs from that of B. leachii, as well as from other sequenced tunicate genomes (Table 1). The main genomic differences between B. leachii and B. schlosseri are in their genome sizes (194 Mb vs 725 Mb), their fraction of repetitive sequences (19% vs 60%15) and the number of predicted genes (15,839 vs 27,46315). The genome size of B. schlosseri resembles more that of the Thaliacean S. thompsoni7 than that of B. leachii (S. thompsoni genome size: 602 Mb, repetitive sequences: 60–70%, genes: 26,415; Table 1). Altogether, these comparisons indicate that genome expansion is not necessary for coloniality in ascidians, and that the B. schlosseri genome has an architecture divergent from that of B. leachii by having undergone a significant increase in its genomic content, including repetitive element expansion (File S2).
Rapid genome evolution, and active transposable elements in particular, are proposed to aid adaptation to new environments for invasive species91. Differences have been noted in the range of tolerable environmental conditions, such as salinity or temperature, which permits the colonization of a given habitat by tunicates, not only between B. leachii and B. schlosseri92,93,94, but even within the B. schlosseri cryptic species complex87,90. It is possible that such plasticity in genome characteristics, like transposon diversity, genome size and gene number, assists the observed invasive success of tunicate species95.
Ancient homeobox genes clusters whose structure has been retained over millions of years of evolution in many organisms appear fragmented in the available tunicate genomes. Because the expression of each Hox gene across the anterior-posterior axis relates to their genomic location within the Hox gene cluster57, cluster breaks are predicted to have consequences for patterning processes. However, an adult body plan with correct spatial orientation of its body axes is also established during sexual and asexual development, including WBR, in ascidians. Embryonic patterning events in tunicate species have only been well characterized during Ciona sexual reproduction. Early stages of development (prior to gastrulation) follow a mosaic pattern of developmental axis formation, where inheritance of maternally provided factors establishes the body axes96. Hox gene knockdown experiments in C. robusta revealed that Hox genes have very limited roles, with defects only observed in the development of the neurons and tails of the larvae97. Therefore, it appears that embryonic patterning events in C. robusta are not dependent upon Hox genes function to establish regional identity. However, Hox genes do play a role later on, during metamorphosis where knockdown of Hox1, Hox10 and Hox12 causes tissue malformation and adult death97,98, while no functions have been attributed to the other Hox genes98. These three posterior Hox genes are the ones present in all studies tunicate genomes (Fig. 3). Thus, it will be of interest to determine the consequences of Hox cluster dispersion and gene loss to the formation of adult organs during sexual and asexual reproduction in colonial ascidians. In animals many mechanisms, in addition to molecular factors, act to establish regional tissue patterning99. In B. schlosseri, the entry point of the connective test vessel into the developing bud determines the posterior end of the new zooid100. Therefore, we hypothesize that physical and/or environmental cues could help compensate for the loss of Hox gene function in determining regional identity during asexual development. A wider analysis comprising multiple tunicate species will be necessary to investigate the exact consequences of homeobox cluster dispersion and compensatory mechanisms.
These three examples highlight the genomic diversity which exists among tunicates, and within the Stolidobranchia in particular. These organisms provide a unique opportunity to study the functional impact of such genomic variations by comparing closely related species.
Gene orthology analysis and innovations
As a first step towards investigating the genetics underlying tunicate biology, we have performed an all-to-all search for protein orthologs between five tunicate species (File S3). Among the tunicate orthologous clusters that we obtained, we identified several groups of genes that are not shared by all the tunicate genomes (Fig. 2A). Of particular interest are genes found only in the B. schlosseri and B. leachii genomes, as these may function in biological processes unique to colonial tunicates. Many of these genes have orthologs not only in vertebrates, but also in more evolutionarily distant animals such as C. elegans (File S4). This suggests that these genes have a more ancient origin, which was retained specifically in Botryllidae genomes. The overrepresented biological processes include circulation, wound healing and cell communication; as well as regulation of immune cell differentiation and immune system processes. Unlike solitary tunicate species, colonial ascidians share a complex system of single cell-lined vessels, used to transport haemocytes and facilitate communication between zooids within the colony, that is the exclusive site of WBR following zooid loss101. In addition, immune response is known to have roles in wound healing, vasculogenesis, allorecognition and regeneration102,103,104. Therefore, it is possible that these genes, found only in Botryllus and Botrylloides, contribute to biological pathways and cellular processes that have important roles in colonialism. Expansion of ortholog analysis to include additional genomes from other newly sequenced tunicates will further refine the set of candidate genes belonging to these processes. For instance, including the Thaliacean S. thompsoni (which has both colonial and solitary life stages) would be of interest for studying colonialism, while incorporating regenerating Phlebobranchian species such as Perophora viridis105,106 would help identify genes involved in regeneration.
Both O. dioica and B. schlosseri had a high number (2160 and 2716 respectively) of clusters unique to their genomes (Fig. 2A). While the O. dioica genome has undergone considerable loss of ancestral genes13,107, the total number of genes in this species is similar to that of other tunicates (Table 2). Taken together, these observations suggest that there has been a duplication of the retained genes such as Otx (3 copies in O. dioica, one in Ciona108). The B. schlosseri genome has an ~10,000 higher predicted gene number compared to other tunicates (Table 2), suggesting partial genome duplication. Further analysis will be required to determine whether these are novel or duplicated genes, hence providing important insights in the evolution of Tunicata genomes.
Lineage-specific changes to evolutionarily conserved cell communication pathways
Cell signalling pathways are critical for morphogenesis, development and adult physiology. We have focused our analysis on three highly conserved pathways: Wnt, Notch and RA signalling. Representatives of all twelve Wnt gene subfamilies are found in metazoans, suggesting that they evolved before the emergence of the bilaterians109. We identified members of each Wnt subfamily among the studied tunicate genomes, along with numerous examples of lineage-specific gene loss and/or duplication. The most striking was an increase in Wnt5a gene copy number in B. leachii, B. schlosseri and M. oculata. Indeed, most invertebrate genomes, including the basal chordate B. floridae, contain a single Wnt5a gene while most vertebrate genomes have two Wnt5a paralogs, believed to be a result of whole genome duplication110. Potentially, these additional genes have been co-opted into novel roles and were retained during tunicate evolution. Wnt5a ligands have numerous biological roles, including a suppressive one during zebrafish regeneration111 and a promotive one during amphioxus regeneration112. Components of both Wnt signaling pathways are differentially expressed during B. leachii WBR37, it is possible that Wnt5a gene number has expanded in colonial tunicates to sustain WBR. The functional characterization of Wnt5a genes during B. leachii WBR will be explored in future studies.
All components of the Notch pathway are present in the genomes we investigated. Of particular interest, the DSL Notch ligand appears to be rapidly evolving in tunicates. This indicates that tunicate DSL proteins are under less pressure to conserve their sequence than their vertebrate orthologs. Given that the interaction between the DSL domain and the Notch receptor is central to signalling pathway activation113, it will be interesting to assess whether the functional ligand-receptor interactions between tunicate DSL proteins and tunicate Notch proteins have adapted accordingly.
Components of the RA signalling pathway have also been identified in all the tunicate genomes. However, Oikopleura has seemingly lost a functional RA synthesis pathway, while still forming a functional body plan. This suggests that O. dioica utilizes an alternative synthesis approach, that the RA signalling function has been replaced or, and rather uniquely, that RA is not involved in the development of this species. Conversely, lineage specific increases in RA pathway gene numbers have been observed in C. robusta114 (Aldh1) and Botrylloides (Cyp26 genes, Fig. 8), suggestive of a functional role at some stage of their development.
RA, Notch and Wnt pathways play roles in regeneration and development in many species, including Stolidobranchian tunicates25,37,67 and Cionidae19,74. The involvement of such conserved signalling pathways opens a number of interesting hypothesis. While the regenerative potential of O. dioica has not been characterized, the observed loss of RA signalling genes may implicate a reduced regeneration ability compared to the other tunicates. Given the unique WBR potential developed by colonial tunicates, the selective pressure on their genomes to retain these pathways might be higher than that of other chordates. Additionally, because of the morphological similarities between WBR and colony reactivation following hibernation, it appears plausible that these three pathways play a similar role in these processes.
Among tunicates, even between closely related species, there exist significant differences in life cycle, reproduction and regeneration ability, which likely reflect an underlying diversity in genomic content. For instance, differences in both asexual and sexual reproduction have been observed within the Botryllidae family19,35,92,93. Furthermore, B. schlosseri can only undergo WBR during a short time frame of its asexual reproductive cycle when the adults are reabsorbed by the colony8,115 while B. leachii can undergo WBR throughout its adult life116. Overall, this indicates that despite a generally similar appearance, the rapid evolution of the Tunicata subphylum has provided diversity and innovations within its species. In future studies, it will be interesting to investigate how such genomic plasticity balances between adaptation to new challenges and constraint, preserving common morphological features.
Summary
In conclusion, our assembly of the B. leachii genome provides an essential resource for the study of this colonial ascidian as well as a crucial point of comparison to gain further insights into the remarkable genetic diversity among tunicate species. In addition, the genome of B. leachii will be most useful for dissecting WBR in chordates; particularly through comparison with B. schlosseri for understanding how the initiation of WBR can be blocked during specific periods of its life cycle. Furthermore, given the key phylogenetic position of tunicates with respect to vertebrates, the analysis of their genomes will provide important insights in the emergence of chordate traits and the origin of vertebrates.
Methods
Sampling, library preparation and sequencing
B. leachii colonies were collected from Nelson harbour (latitude 41.26°S, longitude 173.28°E) in New Zealand. To reduce the likelihood of contamination, embryos were dissected out of a colony and pooled before carrying out DNA extraction using E.Z.N.A SP Plant DNA Mini Kit (Omega Biotek). A total of 4 µg DNA was sent to New Zealand Genomics Limited (University of Otago, NZ) for two runs of library preparation and sequencing. Short read sequencing of Illumina TruSeq libraries in a HiSeq. 2500 generated 19,090,212 paired-end reads of 100 bp (average fragment size: 450 bp, adaptor length: 120 bp). A second sequencing (Illumina Nextera MiSeq Mate Pair), without size-selection, generated 31,780,788 paired-end sequences of 250 bp (fragment size: 1.5–15 kb, median size: ~3 kb, adaptor length: 38 bp).
Pre-quality check report was generated using the String Graph Assembler software package38 and quality metrics before assembly with both FastQC117 as well as MultiQC118. These analyses revealed that 91% of sequences had a mean Phred quality score >=30, 96% a mean Phred quality score >=30 and 39% of sequences had an adapter sequence (either Illumina or Nextera, Fig. S1). Adaptor trimming was performed with NxTrim119 for the mate pair library, followed by Trimmomatic120 with the following options: MINLEN:40 ILLUMINACLIP:2:30:12:1:true LEADING:3 TRAILING:3 MAXINFO:40:0.4 MINLEN:40 for both libraries. After trimming, 86,644,308 paired-end (85%) and 12,112,004 (12%) single-end sequences remained (100% with a mean Phred quality score >=30, <1% with an adapter sequence, Fig. S1).
Genome assembly
De novo assembly was performed in three consecutive iterations following a Meta-assembly approach (Table S5). First, both libraries were assembled together in parallel, using a k-mer size of 63 (when available) following the results from KmerGenie121, by five assemblers: ABySS122, Velvet123, SOAPdenovo2124, ALLPATHS-LG125, MaSuRCA126. The MaSuRCA assembler was run twice, once running the adapter filtering function (here termed “MaSuRCA-filtered”), the other without it (termed simply “MaSuRCA”). Their respective quality was then estimated using three different metrics: the N50 length, the BUSCO core-genes completion42 and the number of genes predicted by Glimmer127. Second, these drafts were combined by following each ranking using Metassembler41, hence producing three new assemblies (limiting the maximum insert size at 15 kb). Third, the B. leachii transcriptome37 was aligned to each meta-assembly using STAR128, and their alignment percentage was used as ranking in a third run using Metassembler, limiting the maximum insert size at either 3 kb, 8 kb or 15 kb. Finally, the quality of the meta-meta-assemblies was estimated using the BUSCO score and the best one (Table S5) selected as the reference de novo assembly.
Data access
All data was retrieved from the indicated sources in January 2016. Note that Ciona intestinalis type A14 has recently been recognized as a distinct species (Ciona robusta129).
B. schlosseri, C. robusta, M. oculata: Ascidian Network for In Situ Expression and Embryonic Data (ANISEED, https://www.aniseed.cnrs.fr/aniseed/).
O. dioica: Oikopleura Genome Browser (http://www.genoscope.cns.fr/externe/GenomeBrowser/Oikopleura/).
B. floridae, H. sapiens: Joint Genome Institute (JGI, http://genome.jgi.doe.gov).
Quality assessment comparison data for Homo sapiens, Maylandia zebra, Melopsittacus undulatus, Crassostrea gigas: String Graph Assembler (https://github.com/jts/sga/tree/master/src/examples/preqc).
The data of the B. leachii genome is available from the following sources:
Raw sequence reads: National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra) with the accession number SRP127769.
Assembled and annotated genome, predicted transcriptome, predicted proteome, species-specific AUGUSTUS and SNAP models: ANISEED (https://www.aniseed.cnrs.fr/aniseed/).
Repeat region analysis
A de novo repeat library was built for each tunicate genome using RepeatModeler50. This utilizes the RECON tandem repeats finder from the RepeatScout packages to identify species-specific repeats in a genome assembly. RepeatMasker49 was then used to mask those repeats. This repeat library is available on ANISEED (https://www.aniseed.cnrs.fr/aniseed/).
Gene annotation
Ab initio genome annotation was performed using MAKER244 with AUGUSTUS130 and SNAP131 for gene prediction. In addition, we used our previously published transcriptome37 and a concatenation of UniProtKB132, C. robusta and B. schlosseri proteins into a custom proteome as evidence of gene product. Using the predicted genes, AUGUSTUS and SNAP were then trained to the specificity of B. leachii genome. A second round of predictions was then performed, followed by a second round of training. The final annotation of the genome was obtained after running a third round of predictions, and the provided trained AUGUSTUS and SNAP configurations after a third round of training. Non-coding RNA sequences were annotated using Infernal46 with the Rfam library 12.0133, tRNAscan-SE48 and snoRNA47. Finally, the identified sequences were characterized by InterProScan45.
Analysis of Gene Ontology terms
Distribution of Gene Ontology (GO) terms were computed for each species as follows. GO terms were extracted from the genome annotation and the number of occurrence for each term determined using a custom Python script. The resulting list of frequencies was then simplified using REVIGO134 (similarity factor “small” of 0.5) and the TreeMap output retrieved. The hierarchy of every GO term present was reconstructed following the schema defined by the core gene ontology (go.obo135) using a custom Python script selecting the shortest path to the root of the tree, favouring smaller GO terms identification number in case of multiple paths. Finally, frequencies were displayed using the sunburstR function of the Data-Driven Documents library136 (D3).
Predicted amino acid sequences for all species were retrieved and clustered into 17,710 groups by OrthoMCL52. Protein sequences within each group were then aligned into a Multiple Sequence Alignment by Clustal-Omega137, and the corresponding consensus sequence inferred by cons (EMBOSS138). Consensus sequences were matched to the Swiss-Prot curated database using BLASTp51 (e-value cut-off of 10−5), and the GO terms corresponding to the best match retrieved. GO terms frequencies were analysed as described above and displayed using REVIGO and TreeMap. Results from OrthoMCL (groups of clustered genes), Clustal-Omega (multiple sequence alignments), EMBOSS (consensus sequences) and BLASTp (top hit) are provided in File S3.
The overrepresentation analysis was performed using GOrilla139 with Homo sapiens as the organism background, using a p-value threshold of 10−3 and REVIGO TreeMap (similarity factor “medium” of 0.7) for visualization. Results are provided in File S4.
Analysis of specific gene families
Genes and transcripts for each examined genome were identified by a tBLASTn search (e-value cut-off of 10−5) using the SequencerServer software140. This was followed by a reciprocal BLAST using SmartBLAST141, to confirm their identity.
Conserved protein domains used to identify the corresponding orthologous proteins within tunicate genomes are found in Table S2.
Phylogenetics
Sequences were aligned with ClustalX142 before using ProtTest 3143 to determine the best-fit model of evolution. The best-fit models for the DSL and CYP26 phylogeny were WAG + I + G and LG + I + G, respectively.
Bayesian inference (BI) phylogenies were constructed using MrBayes144 with a mixed model for 100,000 generations and summarized using a Sump burn-in of 200. Maximum Likelihood (ML) phylogenies were generated by PhyML145, using the estimated amino acid frequencies.
Accession numbers are provided in File S5 and sequence alignments are provided in Fig. S5. Analyses carried out with BI and ML produced identical tree topologies. Trees were displayed using FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
References
Delsuc, F., Brinkmann, H., Chourrout, D. & Philippe, H. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 439, 965–968 (2006).
Lemaire, P., Smith, W. C. & Nishida, H. Ascidians and the plasticity of the chordate developmental program. Curr Biol 18, R620–631 (2008).
Millar, R. H. The biology of ascidians. Advanced Marine Biology 9, 1–100 (1971).
Tsagkogeorga, G. et al. An updated 18S rRNA phylogeny of tunicates based on mixture and secondary structure models. BMC evolutionary biology 9 (2009).
Rubinstein, N. D. et al. Deep Sequencing of Mixed Total DNA without Barcodes Allows Efficient Assembly of Highly Plastic Ascidian Mitochondrial Genomes. Genome biology and evolution 5, 1185–1199 (2013).
Berna, L. & Alvarez-Valin, F. Evolutionary genomics of fast evolving tunicates. Genome biology and evolution 6, 1724–1738 (2014).
Jue, N. K. et al. Rapid Evolutionary Rates and Unique Genomic Signatures Discovered in the First Reference Genome for the Southern Ocean Salp, Salpa thompsoni (Urochordata, Thaliacea). Genome biology and evolution 8, 3171–3186 (2016).
Kurn, U., Rendulic, S., Tiozzo, S. & Lauzon, R. J. Asexual propagation and regeneration in colonial ascidians. The Biological bulletin 221, 43–61 (2011).
Goessling, W. & North, T. E. Repairing quite swimmingly: advances in regenerative medicine using zebrafish. Disease models & mechanisms 7, 769–776 (2014).
Karami, A., Tebyanian, H., Goodarzi, V. & Shiri, S. Planarians: an In Vivo Model for Regenerative Medicine. Int J Stem Cells 8, 128–133 (2015).
Jones, D. L. & Rando, T. A. Emerging models and paradigms for stem cell ageing. Nat Cell Biol 13, 506–512 (2011).
Voskoboynik, A. & Weissman, I. L. Botryllus schlosseri, an emerging model for the study of aging, stem cells, and mechanisms of regeneration. Invertebr Reprod Dev 59, 33–38 (2015).
Seo, H. C. et al. Miniature genome in the marine chordate Oikopleura dioica. Science 294, 2506 (2001).
Dehal, P. et al. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 298, 2157–2167 (2002).
Voskoboynik, A. et al. The genome sequence of the colonial chordate. Botryllus schlosseri. eLife 2, e00569 (2013).
Piette, J. & Lemaire, P. Thaliaceans, the Neglected Pelagic Relatives of Ascidians: A Developmental and Evolutionary Enigma. Q Rev Biol 90, 117–145 (2015).
Auger, H., Sasakura, Y., Joly, J. S. & Jeffery, W. R. Regeneration of oral siphon pigment organs in the ascidian Ciona intestinalis. Developmental biology 339, 374–389 (2010).
Dahlberg, C. et al. Refining the Ciona intestinalis model of central nervous system regeneration. PloS one 4, e4458 (2009).
Jeffery, W. R. Regeneration, Stem Cells, and Aging in the Tunicate Ciona: Insights from the Oral Siphon. Int Rev Cell Mol Biol 319, 255–282 (2015).
Small, K. S., Brudno, M., Hill, M. M. & Sidow, A. A haplome alignment and reference sequence of the highly polymorphic Ciona savignyi genome. Genome biology 8, R41 (2007).
Vinson, J. P. et al. Assembly of polymorphic genomes: algorithms and application to Ciona savignyi. Genome research 15, 1127–1135 (2005).
Brozovic, M. et al. ANISEED 2015: a digital framework for the comparative developmental biology of ascidians. Nucleic acids research 44, D808–818 (2016).
Stolfi, A. et al. Divergent mechanisms regulate conserved cardiopharyngeal development and gene expression in distantly related ascidians. eLife 3, e03728 (2014).
Brown, F. D. & Swalla, B. J. Evolution and development of budding by stem cells: ascidian coloniality as a case study. Developmental biology 369, 151–162 (2012).
Rinkevich, Y., Paz, G., Rinkevich, B. & Reshef, R. Systemic bud induction and retinoic acid signaling underlie whole body regeneration in the urochordate Botrylloides leachi. PLoS biology 5, e71 (2007).
Rinkevich, B., Shlemberg, Z. & Fishelson, L. Whole-body protochordate regeneration from totipotent blood cells. Proceedings of the National Academy of Sciences of the United States of America 92, 7695–7699 (1995).
Ballarin, L., Franchini, A., Ottaviani, E. & Sabbadin, A. Morula cells as the major immunomodulatory hemocytes in ascidians: evidences from the colonial species Botryllus schlosseri. The Biological bulletin 201, 59–64 (2001).
Manni, L., Zaniolo, G., Cima, F., Burighel, P. & Ballarin, L. Botryllus schlosseri: a model ascidian for the study of asexual reproduction. Developmental dynamics: an official publication of the American Association of Anatomists 236, 335–352 (2007).
Gasparini, F., Burighel, P., Manni, L. & Zaniolo, G. Vascular regeneration and angiogenic-like sprouting mechanism in a compound ascidian is similar to vertebrates. Evolution & development 10, 591–605 (2008).
Lauzon, R. J., Brown, C., Kerr, L. & Tiozzo, S. Phagocyte dynamics in a highly regenerative urochordate: Insights into development and host defense. Developmental biology (2012).
Rinkevich, Y. et al. Repeated, long-term Cycling of Putative Stem cells between Niches in a Basal Chordate. Developmental Cell 24, 76–88 (2013).
Franchi, N. et al. Immune roles of a rhamnose-binding lectin in the colonial ascidian Botryllus schlosseri. Immunobiology 216, 725–736 (2011).
Velandia-Huerto, C. A., Gittenberger, A. A., Brown, F. D., Stadler, P. F. & Bermudez-Santana, C. I. Automated detection of ncRNAs in the draft genome sequence of a colonial tunicate: the carpet sea squirt Didemnum vexillum. BMC genomics 17, 691 (2016).
Savigny, J.-C. Mémoires sur les animaux sans vertèbres (A Paris: Chez Deterville: Chez Treuttel et Würtz; à Londres; à Strasbourg, 1816).
Berrill, N. J. The developmental cycle of Botrylloides. Q J Microsc Sci 88, 393–407 (1947).
Burighel, P., Brunetti, R. & Zaniolo, G. Hibernation of the Colonial Ascidian Botrylloides Leachi (Savigny): Histological Observations. Bolletino di zoologia 43, 293–301 (1976).
Zondag, L. E., Rutherford, K., Gemmell, N. J. & Wilson, M. J. Uncovering the pathways underlying whole body regeneration in a chordate model, Botrylloides leachi using de novo transcriptome analysis. BMC genomics 17, 114 (2016).
Simpson, J. T. Exploring genome characteristics and sequence quality without a reference. Bioinformatics 30, 1228–1235 (2014).
Bradnam, K. R. et al. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience 2, 10 (2013).
Zhang, G. et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54 (2012).
Wences, A. H. & Schatz, M. C. Metassembler: merging and optimizing de novo genome assemblies. Genome biology 16, 207 (2015).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome research 12, 656–664 (2002).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC bioinformatics 12, 491 (2011).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Lowe, T. M. & Eddy, S. R. A computational screen for methylation guide snoRNAs in yeast. Science 283, 1168–1171 (1999).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25, 955–964 (1997).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0, 2013–2015).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0., http://www.repeatmasker.org (2008–2015).
Camacho, C. et al. BLAST + : architecture and applications. BMC bioinformatics 10, 421 (2009).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome research 13, 2178–2189 (2003).
Primmer, C. R., Papakostas, S., Leder, E. H., Davis, M. J. & Ragan, M. A. Annotated genes and nonannotated genomes: cross-species use of Gene Ontology in ecology and evolution research. Mol Ecol 22, 3216–3241 (2013).
Graham, G. J. Tandem genes and clustered genes. J Theor Biol 175, 71–87 (1995).
Garcia-Fernandez, J. The genesis and evolution of homeobox gene clusters. Nature reviews. Genetics 6, 881–892 (2005).
Hoegg, S. & Meyer, A. Hox clusters as models for vertebrate genome evolution. Trends in genetics: TIG 21, 421–424 (2005).
Pascual-Anaya, J., D’Aniello, S., Kuratani, S. & Garcia-Fernandez, J. Evolution of Hox gene clusters in deuterostomes. BMC developmental biology 13, 26 (2013).
Takatori, N. et al. Comprehensive survey and classification of homeobox genes in the genome of amphioxus. Branchiostoma floridae. Development genes and evolution 218, 579–590 (2008).
Spagnuolo, A. et al. Unusual number and genomic organization of Hox genes in the tunicate Ciona intestinalis. Gene 309, 71–79 (2003).
Wada, S. et al. A genomewide survey of developmentally relevant genes in Ciona intestinalis. II. Genes for homeobox transcription factors. Development genes and evolution 213, 222–234 (2003).
Edvardsen, R. B. et al. Remodelling of the homeobox gene complement in the tunicate Oikopleura dioica. Curr Biol 15, R12–13 (2005).
Seo, H. C. et al. Hox cluster disintegration with persistent anteroposterior order of expression in Oikopleura dioica. Nature 431, 67–71 (2004).
Simakov, O. et al. Hemichordate genomes and deuterostome origins. Nature 527, 459–465 (2015).
Santagati, F. et al. Identification of Cis-regulatory elements in the mouse Pax9/Nkx2-9 genomic region: implication for evolutionary conserved synteny. Genetics 165, 235–242 (2003).
Wang, W., Zhong, J., Su, B., Zhou, Y. & Wang, Y. Q. Comparison of Pax1/9 locus reveals 500-Myr-old syntenic block and evolutionary conserved noncoding regions. Molecular biology and evolution 24, 784–791 (2007).
Luke, G. N. et al. Dispersal of NK homeobox gene clusters in amphioxus and humans. Proceedings of the National Academy of Sciences of the United States of America 100, 5292–5295 (2003).
Rinkevich, Y., Rinkevich, B. & Reshef, R. Cell signaling and transcription factor genes expressed during whole body regeneration in a colonial chordate. BMC developmental biology 8, 100 (2008).
Loh, K. M., van Amerongen, R. & Nusse, R. Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev Cell 38, 643–655 (2016).
Kusserow, A. et al. Unexpected complexity of the Wnt gene family in a sea anemone. Nature 433, 156–160 (2005).
Guder, C. et al. The Wnt code: cnidarians signal the way. Oncogene 25, 7450–7460 (2006).
Prud’homme, B., Lartillot, N., Balavoine, G., Adoutte, A. & Vervoort, M. Phylogenetic analysis of the Wnt gene family. Insights from lophotrochozoan members. Curr Biol 12, 1395 (2002).
Holland, P. W., Marletaz, F., Maeso, I., Dunwell, T. L. & Paps, J. New genes from old: asymmetric divergence of gene duplicates and the evolution of development. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 372 (2017).
MacDonald, B. T., Tamai, K. & He, X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17, 9–26 (2009).
Hamada, M., Goricki, S., Byerly, M. S., Satoh, N. & Jeffery, W. R. Evolution of the chordate regeneration blastema: Differential gene expression and conserved role of notch signaling during siphon regeneration in the ascidian Ciona. Developmental biology 405, 304–315 (2015).
Guruharsha, K. G., Kankel, M. W. & Artavanis-Tsakonas, S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nature reviews. Genetics 13, 654–666 (2012).
Gazave, E. et al. Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC evolutionary biology 9, 249 (2009).
Jackson, B. et al. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum Genomics 5, 283–303 (2011).
Cunningham, T. J. & Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nature reviews. Molecular cell biology 16, 110–123 (2015).
Ross, A. C. & Zolfaghari, R. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu Rev Nutr 31, 65–87 (2011).
Canestro, C., Postlethwait, J. H., Gonzalez-Duarte, R. & Albalat, R. Is retinoic acid genetic machinery a chordate innovation? Evolution & development 8, 394–406 (2006).
Marti-Solans, J. et al. Coelimination and Survival in Gene Network Evolution: Dismantling the RA-Signaling in a Chordate. Molecular biology and evolution 33, 2401–2416 (2016).
Belyaeva, O. V., Chang, C., Berlett, M. C. & Kedishvili, N. Y. Evolutionary origins of retinoid active short-chain dehydrogenases/reductases of SDR16C family. Chem Biol Interact 234, 135–143 (2015).
Belyaeva, O. V., Lee, S. A., Kolupaev, O. V. & Kedishvili, N. Y. Identification and characterization of retinoid-active short-chain dehydrogenases/reductases in Drosophila melanogaster. Biochimica et biophysica acta 1790, 1266–1273 (2009).
Lee, S. A., Belyaeva, O. V. & Kedishvili, N. Y. Biochemical characterization of human epidermal retinol dehydrogenase 2. Chem Biol Interact 178, 182–187 (2009).
Tsagkogeorga, G., Turon, X., Galtier, N., Douzery, E. J. P. & Delsuc, F. Accelerated Evolutionary Rate of Housekeeping Genes in Tunicates. Journal of Molecular Evolution 71, 153–167 (2010).
Tsagkogeorga, G., Cahais, V. & Galtier, N. The Population Genomics of a Fast Evolver: High Levels of Diversity, Functional Constraint, and Molecular Adaptation in the Tunicate Ciona intestinalis. Genome biology and evolution 4, 852–861 (2012).
Bock, D. G., MacIsaac, H. J. & Cristescu, M. E. Multilocus genetic analyses differentiate between widespread and spatially restricted cryptic species in a model ascidian. P Roy Soc B-Biol Sci 279, 2377–2385 (2012).
Griggio, F. et al. Ascidian Mitogenomics: Comparison of Evolutionary Rates in Closely Related Taxa Provides Evidence of Ongoing Speciation Events (vol 6, pg 591, 2014). Genome biology and evolution 6, 931–931 (2014).
Saito, Y., Shirae, M., Okuyama, M. & Cohen, S. Phylogeny of Botryllid ascidians. Biology of Ascidians 315–320 (2001).
Nydam, M. L., Giesbrecht, K. B. & Stephenson, E. E. Origin and Dispersal History of Two Colonial Ascidian Clades in the Botryllus schlosseri Species Complex. PloS one 12 (2017).
Stapley, J., Santure, A. W. & Dennis, S. R. Transposable elements as agents of rapid adaptation may explain the genetic paradox of invasive species. Mol Ecol 24, 2241–2252 (2015).
Brunetti, R. Observations on the Life Cycle of Botryllus schlosseri (Pallas) (Ascidiacea) in the Venetian Lagoon. Bolletino di zoologia 41, 225–251 (1974).
Brunetti, R. Biological Cycle of Botrylloides leachi (Savigny) (Ascidiacea) in Venetian Lagoon. Vie Milieu a Biol Ma26, 105-& (1976).
Brunetti, R., Beghi, L., Bressan, M. & Marin, M. G. Combined Effects of Temperature and Salinity on Colonies of Botryllus schlosseri and Botrylloides leachi (Ascidiacea) from the Venetian Lagoon. Mar Ecol Prog Ser 2, 303–314 (1980).
Zhan, A. B., Briski, E., Bock, D. G., Ghabooli, S. & MacIsaac, H. J. Ascidians as models for studying invasion success. Mar Biol 162, 2449–2470 (2015).
Nishida, H. Specification of embryonic axis and mosaic development in ascidians. Developmental dynamics: an official publication of the American Association of Anatomists 233, 1177–1193 (2005).
Ikuta, T., Satoh, N. & Saiga, H. Limited functions of Hox genes in the larval development of the ascidian Ciona intestinalis. Development 137, 1505–1513 (2010).
Sasakura, Y. & Hozumi, A. Formation of adult organs through metamorphosis in ascidians. Wiley Interdiscip Rev Dev Biol (2017).
Benazeraf, B. & Pourquie, O. Formation and segmentation of the vertebrate body axis. Annu Rev Cell Dev Biol 29, 1–26 (2013).
Sabbadin, A., Zaniolo, G. & Majone, F. Determination of Polarity and Bilateral Asymmetry in Palleal and Vascular Buds of Ascidian Botryllus schlosseri. Developmental biology 46, 79–87 (1975).
Sabbadin, A., Zaniolo, G. & Majone, F. Determination of polarity and bilateral asymmetry in palleal and vascular buds of the ascidian Botryllus schlosseri. Developmental biology 46, 79–87 (1975).
Voskoboynik, A. et al. Identification of a colonial chordate histocompatibility gene. Science 341, 384–387 (2013).
Taketa, D. A. et al. Molecular evolution and in vitro characterization of Botryllus histocompatibility factor. Immunogenetics 67, 605–623 (2015).
Gutierrez, S. & Brown, F. D. Vascular budding in Symplegma brakenhielmi and the evolution of coloniality in styelid ascidians. Developmental biology 423, 152–169 (2017).
Freeman, G. The role of blood cells in the process of asexual reproduction in the tunicate Perophora viridis. Journal of Experimental Zoology 156, 157–183 (1964).
Goldin, A. Regeneration in Perophora viridis. The Biological bulletin 94, 184–193 (1948).
Albalat, R. & Canestro, C. Evolution by gene loss. Nature reviews. Genetics 17, 379–391 (2016).
Canestro, C., Bassham, S. & Postlethwait, J. Development of the central nervous system in the larvacean Oikopleura dioica and the evolution of the chordate brain. Developmental biology 285, 298–315 (2005).
Janssen, R. et al. Conservation, loss, and redeployment of Wnt ligands in protostomes: implications for understanding the evolution of segment formation. BMC evolutionary biology 10, 374 (2010).
Martin, A., Maher, S., Summerhurst, K., Davidson, D. & Murphy, P. Differential deployment of paralogous Wnt genes in the mouse and chick embryo during development. Evolution & development 14, 178–195 (2012).
Stoick-Cooper, C. L. et al. Distinct Wnt signaling pathways have opposing roles in appendage regeneration. Development 134, 479–489 (2007).
Somorjai, I. M., Somorjai, R. L., Garcia-Fernandez, J. & Escriva, H. Vertebrate-like regeneration in the invertebrate chordate amphioxus. Proceedings of the National Academy of Sciences of the United States of America 109, 517–522 (2012).
Chillakuri, C. R., Sheppard, D., Lea, S. M. & Handford, P. A. Notch receptor-ligand binding and activation: insights from molecular studies. Semin Cell Dev Biol 23, 421–428 (2012).
Sobreira, T. J. et al. Structural shifts of aldehyde dehydrogenase enzymes were instrumental for the early evolution of retinoid-dependent axial patterning in metazoans. Proceedings of the National Academy of Sciences of the United States of America 108, 226–231 (2011).
Voskoboynik, A. et al. Striving for normality: whole body regeneration through a series of abnormal generations. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 21, 1335–1344 (2007).
Rinkevich, Y., Douek, J., Haber, O., Rinkevich, B. & Reshef, R. Urochordate whole body regeneration inaugurates a diverse innate immune signaling profile. Developmental biology 312, 131–146 (2007).
Andrews, S. (http://www.bioinformatics.babraham.ac.uk/projects/fastqc, 2010).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics (2016).
O’Connell, J. et al. NxTrim: optimized trimming of Illumina mate pair reads. Bioinformatics 31, 2035–2037 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Chikhi, R. & Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 30, 31–37 (2014).
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome research 19, 1117–1123 (2009).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research 18, 821–829 (2008).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proceedings of the National Academy of Sciences of the United States of America 108, 1513–1518 (2011).
Zimin, A. V. et al. The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677 (2013).
Delcher, A. L., Harmon, D., Kasif, S., White, O. & Salzberg, S. L. Improved microbial gene identification with GLIMMER. Nucleic acids research 27, 4636–4641 (1999).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Brunetti, R. et al. Morphological evidence that the molecularly determined Ciona intestinalis type A and type B are different species: Ciona robusta and Ciona intestinalis. J Zoolog Syst Evol Res 53, 183–193 (2015).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl 2), ii215–225 (2003).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 59 (2004).
UniProt, C. UniProt: a hub for protein information. Nucleic acids research 43, D204–212 (2015).
Nawrocki, E. P. et al. Rfam 12.0: updates to the RNA families database. Nucleic acids research 43, D130–137 (2015).
Supek, F., Bosnjak, M., Skunca, N. & Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS one 6, e21800 (2011).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29 (2000).
Bostock, M., Ogievetsky, V. & Heer, J. D(3): Data-Driven Documents. IEEE Trans Vis Comput Graph 17, 2301–2309 (2011).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539 (2011).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends in genetics: TIG 16, 276–277 (2000).
Eden, E., Navon, R., Steinfeld, I., Lipson, D. & Yakhini, Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC bioinformatics 10, 48 (2009).
Priyam, A. et al. Sequenceserver: a modern graphical user interface for custom BLAST databases. Biorxix (2015).
Coordinators, N. R. Database resources of the National Center for Biotechnology Information. Nucleic acids research 44, D7–19 (2016).
Jeanmougin, F., Thompson, J. D., Gouy, M., Higgins, D. G. & Gibson, T. J. Multiple sequence alignment with Clustal X. Trends in biochemical sciences 23, 403–405 (1998).
Abascal, F., Zardoya, R. & Posada, D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105 (2005).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic biology 61, 539–542 (2012).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic biology 59, 307–321 (2010).
Stach, T. & Turbeville, J. M. Phylogeny of Tunicata inferred from molecular and morphological characters. Mol Phylogenet Evol 25, 408–428 (2002).
Rubinstein, N. D. et al. Deep sequencing of mixed total DNA without barcodes allows efficient assembly of highly plastic ascidian mitochondrial genomes. Genome biology and evolution 5, 1185–1199 (2013).
Acknowledgements
Funding support was provided to M.J.W. by the Otago BMS Deans Bequest and Department of Anatomy. S.B. was supported by the Swiss National Science Foundation (SNSF) grant numbers P2ELP3_158873 and PZ00P3_173981. We would like to thank Peter Maxwell and the New Zealand eScience Infrastructure (NeSI); Christelle Dantec and ANISEED for help and advice during the annotation process, as well as for the accompanying B. leachii genome browser; Aude Blanchoud and James Smith for proofreading the manuscript.
Author information
Authors and Affiliations
Contributions
S.B. and K.R. conducted the genome assembly. L.Z. collected and performed DNA extraction for B. leachii. M.W. and S.B. carried out data analysis, and drafted the article. M.W. and N.G. designed the project. All authors were involved in the revision of the manuscript before publication.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Blanchoud, S., Rutherford, K., Zondag, L. et al. De novo draft assembly of the Botrylloides leachii genome provides further insight into tunicate evolution. Sci Rep 8, 5518 (2018). https://doi.org/10.1038/s41598-018-23749-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23749-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
