
Abstract
In this study, we report transcription of genes involved in aerobic and anaerobic benzene degradation pathways in a benzene-degrading denitrifying continuous culture. Transcripts associated with the family Peptococcaceae dominated all samples (21–36% relative abundance) indicating their key role in the community. We found a highly transcribed gene cluster encoding a presumed anaerobic benzene carboxylase (AbcA and AbcD) and a benzoate-coenzyme A ligase (BzlA). Predicted gene products showed >96% amino acid identity and similar gene order to the corresponding benzene degradation gene cluster described previously, providing further evidence for anaerobic benzene activation via carboxylation. For subsequent benzoyl-CoA dearomatization, bam-like genes analogous to the ones found in other strict anaerobes were transcribed, whereas gene transcripts involved in downstream benzoyl-CoA degradation were mostly analogous to the ones described in facultative anaerobes. The concurrent transcription of genes encoding enzymes involved in oxygenase-mediated aerobic benzene degradation suggested oxygen presence in the culture, possibly formed via a recently identified nitric oxide dismutase (Nod). Although we were unable to detect transcription of Nod-encoding genes, addition of nitrite and formate to the continuous culture showed indication for oxygen production. Such an oxygen production would enable aerobic microbes to thrive in oxygen-depleted and nitrate-containing subsurface environments contaminated with hydrocarbons.
Similar content being viewed by others


Introduction
Benzene is an important component of petroleum. It easily dissolves in water, but is one of the least reactive aromatic hydrocarbons and a potential human carcinogen1. Benzene can be readily degraded aerobically, however, anaerobic benzene degradation is challenging2. Lacking potentially destabilizing or reactive substituents, the benzene molecule is thermodynamically very stable especially under anoxic conditions3. Both aerobic and anaerobic degradation pathways include benzene activation and channeling towards key intermediates (catechol in aerobic and benzoyl-CoA in anaerobic pathways), the upper pathway for dearomatization and ring cleavage and the lower pathway for generation of tricarboxylic acid cycle intermediates (reviewed in2,4,5,6). The genes and enzymes involved in anaerobic benzene activation are not well-studied7. Three putative reactions have been proposed for anaerobic benzene activation: hydroxylation to phenol8,9,10,11, direct carboxylation to benzoate8,12,13,14,15 and methylation to toluene16.
In contrast to many aerobic benzene-degrading pure cultures, only few anaerobic benzene-degrading axenic cultures have been described. The hyperthermophilic archaeon Ferroglobus placidus was proposed to employ a putative UbiD-related carboxylase in anaerobic benzene activation17, and anaerobic benzene oxidation in Geobacter metallireducens was shown to proceed via hydroxylation to phenol18,19. In contrast to these strictly anaerobic iron-reducers that employ oxygen-independent activation routes, the chlorate-reducing Alicycliphilus denitrificans strain BC20 was shown to degrade benzene via an oxygenase-mediated pathway21. Such ‘intra-aerobic’ anaerobes apparently derive oxygen species from inorganic oxo-compounds such as nitrate or chlorate for classical aerobic degradation of hydrocarbons22,23,24,25. The nitrate-reducing facultatively anaerobic Dechloromonas26 may recruit enzymes of a yet unknown pathway for initial benzene activation27. This hypothesis is based on the finding that the genome of Dechloromonas aromatica strain RCB lacks the genes involved in anaerobic degradation of monoaromatic compounds whereas it contains genes for their aerobic activation, including several mono- and dioxygenases28. Moreover, the oxygen incorporated into benzene to produce phenol by this bacterium does not originate from water9 whereas the oxygen source for anaerobic metabolism of benzene to phenol is water11. The benzene degradation pathways of the nitrate-reducing Azoarcus strains29 have not been investigated in details.
Due to the limited availability of anaerobic benzene-degrading isolates, mixed microbial communities were predominantly studied to reveal the physiology and phylogeny of anaerobic benzene degraders and potential anaerobic benzene activation genes and mechanisms12,15,29,30,31,32,33,34,35,36,37,38,39,40,41,42. Among different microbial communities involved in anaerobic benzene degradation, members of the strictly anaerobic Peptococcaceae (Clostridiales) were prevalently found in enrichment cultures with different electron acceptors and proposed as the key players in the initial steps of benzene degradation12,30,31,32,36,38,40,41,42. Among these studies, two cultures were suggested to activate benzene via carboxylation41,42. A proteogenomic analysis using a benzene-degrading iron-reducing enrichment culture identified a putative benzene degradation gene cluster41. The products of the putative benzene carboxylase genes (AbcAD) were specifically detected in cultures growing on benzene but not in those growing on phenol or benzoate, suggestive for their role in initial benzene carboxylation41. A metatranscriptomic analysis using nitrate-reducing enrichment cultures showed high levels of transcripts of the proposed benzene carboxylation genes (abcAD, bzlA)42. Also in this case, these high levels were seen only in benzene-amended cultures but not in benzoate-fed cultures42.
In this study our aim was to elucidate anaerobic benzene degradation using a nitrate-reducing continuous enrichment culture growing for more than 15 years. A former DNA-stable isotope probing (SIP) study with 13C-labeled-benzene identified Peptococcaceae as the predominant members involved in initial benzene degradation38. Efforts to isolate benzene-degrading members of the Peptococcaceae have failed, likely because they require syntrophic interactions with partner species. Recent microbial community analysis using Illumina MiSeq next generation technology sequencing (NGS) and quantitative PCR (qPCR) showed high (relative) abundance of the Peptococcaceae 16S ribosomal RNA (rRNA) gene and abcA gene, further supporting the role of Peptococcaceae in benzene degradation via initial carboxylation40. Here, we performed a metatranscriptomic study using the same enrichment culture. Our results are in line with the former studies on benzene carboxylation by Peptococcaceae41,42 corroborating the concept that carboxylation initiates benzene degradation in the absence of oxygen. The observed downstream pathway involved in further breakdown of the benzoate mostly resembled that of facultative anaerobes. Interestingly, transcripts of genes involved in oxygenase-mediated aerobic benzene degradation were also identified.
Results and Discussion
In the present study, we aimed to elucidate benzene degradation pathways in an anaerobic continuous biofilm culture that was initially inoculated with soil from a benzene-polluted industrial location and enriched for years with benzene as substrate and nitrate as the electron acceptor. The culture was shown to be dominated by Gram-positive Peptococcaceae-related microorganisms38,40. We here conducted a metatranscriptomic analysis of this microbial consortium to track transcripts involved in anaerobic benzene degradation. We analyzed six samples in our transcriptomic study obtained from two types of biofilm morphologies growing in the reactor: four samples containing white biofilm (samples 1–4) and two samples containing brown biofilm (samples 5–6) (Table S1). After rRNA depletion, cDNA synthesis and sequencing using the Illumina HiSeq platform, a total of 83,662,373 reads was initially obtained with rRNA reads ranging between 0.3–6.9% (Table S2).
Active community members
Diverse microbial groups were found in the transcriptome dataset even though the continuous culture was running for more than 15 years (Fig. 1). This could be due to the presence of scavengers growing on dead biomass and cheaters that do not directly contribute to benzene degradation36. The transcripts associated with strictly anaerobic Firmicutes dominated all samples with 36–59% relative abundance (Fig. 1). Among these were high levels of transcripts assigned to members of the Peptococcaceae (21–36% relative abundance). In line with former reports, this suggests a key role of Peptococcaceae in anaerobic benzene degradation12,30,31,32,36,38,40,41,42. The transcripts assigned to Candidatus Kuenenia (Planctomycetes) were found at a higher relative abundance in samples 4–5 (Table S1). In our previous microbial biofilm community analysis using DNA-SIP with 13C-labeled benzene and 16S rRNA gene clone libraries, members of the phyla Firmicutes (37% of clones) and Planctomycetes (28% of clones) dominated the libraries38. In contrast, Planctomycetes were not among the most predominant community members in our recent phylogenetic analysis at DNA-level using MiSeq sequencing of PCR-amplified partial 16S rRNA genes40. In turn, members of the families Anaerolineaceae, Rhodocyclaceae, Comamonadaceae and SJA-28 were identified as predominant community members40, but not in the metatranscriptomic analysis described here. Discrepancy between abundance and activity of microbes has been described previously43,44,45.

Taxonomic comparison of active microbial communities at mRNA level. Samples 1–4 are from white biofilms and sample 5–6 are from the brown biofilms.
Transcription of genes involved in anaerobic benzene degradation
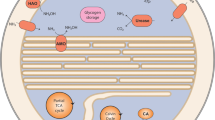
As described in more detail in the following sections, we found transcription of genes potentially involved in anaerobic benzene activation and subsequent pathways for further degradation of the initially formed benzoyl-CoA (Fig. 2A,C, Table 1).

Gene transcripts identified in reactor samples corresponding to known or hypothesized enzymes involved in anaerobic (A) and aerobic (B) benzene degradation in different microbes and their relative abundances (%) (C). Gene transcripts that could not be distinguished due to overlapping assignment with similar genes in the pathway are shown with question marks (full list is given in Table 1). Note that only the substrate and products of each enzymatic reaction are given for clarity. The bar showing the number of relative abundance was log scaled and 0 values were removed.
Benzene activation mechanisms
We did not find transcripts indicating methylation of benzene to toluene (the proposed pathway is shown in Figure S1). The bssA gene encoding the α-subunit of the key enzyme benzylsuccinate synthase was also not detected by qPCR in the co-extracted DNA samples40. In line with our results, genes of the toluene activation pathway were absent in a metatranscriptomic study conducted using another benzene-degrading nitrate-reducing culture42, although benzene methylation mechanism was proposed for this culture in the past16. To date, known benzene-degrading anaerobes do not seem to employ activation by methylation as (i) no proteins mediating benzene methylation were found in a proteogenomic analysis of a benzene-degrading culture that used iron as the electron acceptor41, (ii) none of the investigated benzene-degrading pure cultures seems to employ a methylation step for benzene activation, (iii) no intermediates such as the key product of anaerobic toluene activation, benzylsuccinate, have been detected, and (iv) some anaerobic benzene-degrading enrichment cultures failed to degrade toluene12,15,35.
We found transcripts potentially involved in benzene hydroxylation to phenol (Fig. 2A,C). Among these was a polycistronic transcript that contained genes for the synthesis of UbiD and UbiX, along with a hydroxylase candidate (contig-100_193). The hydroxylase candidate showed low identity (58% at the amino acid level) to a NUDIX family hydrolase from the deltaproteobacterial strain NaphS246 that is not reported to be involved in anaerobic benzene activation. In addition, we found transcripts of phenylphosphate synthase genes (ppsABC) and phenylphosphate carboxylase genes (ppcC). We also detected transcripts of a gene similar to the hcrL gene encoding 4-hydroxybenzoate-CoA ligase, however, it is not possible to differentiate between hcrL and the benzoate-CoA ligase gene (bzlA) (Table 1). Generally, the specificity of the CoA ligases for 4-hydroxybenzoate and benzoate is difficult to predict solely on the basis of sequence similarity47,48. The transcript of a 4-hydroxybenzoyl-CoA reductase gene (hcrA) was also identified. Taken together, these findings might indicate hydroxylation of benzene to phenol in this consortium. Anaerobic benzene oxidation via phenol was documented for G. metallireducens18,19. However, besides lack of an identifiable hydroxylase, we did not find a full set of transcripts encoding all subunits of the essential enzymes for this pathway in our study. Likewise, the reconstructed genome of the Pelotomaculum candidate BPL did not show a full repertoire of genes involved in anaerobic phenol degradation49.
The high level of transcripts involved in the proposed anaerobic benzene carboxylase pathway (abcA in contiq_100_0_8 and contig-100_751_1, and abcD in contiq_100_0_9) and a benzoate-CoA ligase gene (bzlA contiq_100_0_7)41 as revealed in this study (Fig. 2A,C, Table 1) corroborates that benzene carboxylation to benzoate is the main initial benzene degradation pathway in our culture. In line with our results, genes encoding UbiD/UbiX-related carboxylases were also highly transcribed in yet another benzene-degrading nitrate-reducing enrichment, suggesting benzene carboxylation to benzoate as the main mechanism of anaerobic benzene activation42. Similarly, the hyperthermophilic archaeon F. placidus was proposed to employ a benzene-induced UbiD-related benzene carboxylase (Frep_1630) for anaerobic benzene oxidation17. Although biochemical data to demonstrate benzene carboxylation is pending, the compiling evidence on carboxylation of benzene17,41,42 and naphthalene46,50,51,52 indicates carboxylation as an important initial reaction involved in the anaerobic degradation of non-substituted aromatic hydrocarbons7,53. Most recently, a novel UbiD-related decarboxylase was shown to mediate anaerobic phthalate degradation by decarboxylation of phthaloyl-CoA to benzoyl-CoA, further reinforcing the importance of UbiD-related (de)carboxylases in anaerobic degradation of aromatic compounds54.
Co-localization of the putative genes involved in benzene carboxylation
The putative benzene carboxylation genes transcribed in this study showed high similarity (>96% at the amino acid level) and gene synteny to a cluster previously proposed to encode putative enzymes for benzene carboxylation to benzoate41 (Figure S2). Similar observations were made with another benzene-degrading nitrate-reducing enrichment culture indicating a highly conserved set of genes involved in benzene carboxylation in these types of enrichments42. Noteworthy, the three enrichments in which these gene clusters were identified were obtained from geographically distinct locations in Poland32,41, Canada37,42 and the Netherlands38,40, and operated under iron-reducing32,41 or nitrate-reducing conditions37,38,40,42. Similarly, gene clusters encoding enzymes involved in carboxylation reactions in the anaerobic degradation of naphthalene are co-localized in the genomes of the sulfate-reducing cultures N4752 and NaphS246. The genes for the degradation of aromatic compounds are usually clustered at a single genomic locus55. Furthermore, the co-localization and co-transcription of genes encoding a transcriptional regulator, MarR, and multidrug resistance protein MRP homologue (Figure S2) suggest a functional relationship between these genes and the abcAD and bzlA genes. As such, in the genome of the facultatively anaerobic benzoate-degrading Thauera aromatica and the phototrophic bacterium Rhodopseudomonas palustris, marR is co-localized with benzoate degradation genes and proposed to regulate their transcription56,57,58. In contrast, the proposed gene encoding a UbiD-related carboxylase in F. placidus (Frep_1630) is not co-localized with genes coding for carboxylase proteins, benzoate-CoA ligase proteins, or any other proteins involved in the metabolism of aromatic compounds17, even though most of the other genes involved in anaerobic aromatic degradation in F. placidus are localized within gene clusters17,59. Genes homologous to abcA were also present in the genome of Pelotomaculum candidate BPL (single copy with 33% amino acid sequence identity)49 and in the metagenomes of hydrocarbon-degrading enrichment cultures60,61. However, genes homologous to abcD49,60,61 or bzlA49,60 were absent.
Another interesting finding in our study was transcripts of genes for phage-related proteins and transposable elements some of which were located within the same contig that contained putative aromatic-degrading genes (e.g. contig-100_0; Table S3). This implies potential distribution of xenobiotic degradation genes by horizontal gene transfer62.
Upper pathway
Dearomatization
Reductive dearomatization of the benzene ring by benzoyl-CoA reductase (BCR) is the key step in anaerobic degradation of benzoyl-CoA, and BCR is the only oxygen-sensitive enzyme within the benzoyl-CoA pathway55. There are two types of BCRs: class I are ATP-dependent FeS enzymes composed of four different subunits63 whereas class II are ATP-independent enzymes that contain eight subunits and harbour a tungsten-containing cofactor in the active site64. All known monoaromatic-degrading strict anaerobes apply class II BCRs with the exception of the benzene-degrading archaeon F. placidus that lacks the genes coding for the class II BCRs59 and employs an ATP-dependent Azoarcus-type BCR65. We found transcription of bam-like genes (bamBCDEI, from strict anaerobes55) and at much lower relative abundance genes analogous to one subunit of class I BRC (bzdQ and its homologs bcrA/badF, from facultative anaerobes55) in our enrichment culture (Fig. 2A,C, Table 1). This finding indicates that class II BCRs are recruited similar to strictly anaerobic microorganisms. In accordance with our results, Bam-like proteins were detected in a proteogenomic analysis of a benzene-degrading and iron-reducing enrichment culture, indicating that benzoyl-CoA reduction steps are analogous to the activities of class II BCRs41. Genomic and proteomic evidence also proposed benzoate-CoA degradation via Bam-like BCR by Pelotomaculum candidate BPL49. Similarly, bam-like genes were almost exclusively transcribed in a nitrate-reducing enrichment culture growing on benzene but not when it was growing on benzoate42.
Modified β-oxidation
Modified β-oxidation of the dearomatized diene product (cyclohexadienoyl-CoA) by specific hydratases, dehydrogenases and hydrolases results in ring cleavage and diene conversion to an aliphatic C7-dicarboxyl-CoA (Fig. 2A,C). The β-oxidation reactions are similar in facultative and strict anaerobes55. We found transcription of Azoarcus-type bzdXYW genes66 (Fig. 2A,C, Table 1) indicating that the modified β-oxidation reactions in our culture are related to those of denitrifying bacteria. The bzd genes are located in a catabolic operon (bzdNOPQMSTUVWXYZA) in Azoarcus sp. strain CIB66. The bzdXYW gene transcripts identified in our dataset were similarly co-located (contig100_24_4 to contig100_24_6, Table 1) implying a functional relationship. Transcripts of bzdXYW-like genes from Azoarcus were also identified in two other benzene-degrading enrichment cultures41,42.
Lower pathway
The C7-dicarboxyl-CoA is degraded to three acetyl-CoAs and CO2 through a series of reactions that involve a dicarboxylic acid β-oxidation pathway (leading to glutaryl-CoA), a glutaryl-CoA dehydrogenase (leading to crotonyl-CoA), and a short-chain fatty acid β-oxidation pathway (leading to two acetyl-CoAs) (Fig. 2A,C)55. We found transcription of the pimE and pimB genes encoding 3-hydroxypimeloyl-CoA dehydrogenase and acetyl-CoA acyltransferase, respectively (Fig. 2, Table 1). These enzymes which link pimeloyl-CoA to the central metabolism via glutaryl-CoA, are best described for R. palustris, in which they are encoded by the pim operon67. The subsequent decarboxylation of glutaryl-CoA to crotonyl-CoA is the second reaction in the benzoyl-CoA degradation pathway (the first being the dearomatization of benzoyl-CoA, see above), catalyzed by different enzymes in obligate and facultative anaerobes7,68. Facultative anaerobes employ a decarboxylating glutaryl-CoA dehydrogenase with crotonyl-CoA as the product67. Obligate anaerobes on the other hand employ a non-decarboxylating glutaryl-CoA dehydrogenase (that forms glutaconyl-CoA as an intermediate) in combination with a glutaconyl-CoA decarboxylase. The latter is sodium-dependent and will allow ATP synthesis by coupling the subsequent decarboxylation of its product (glutaconyl-CoA) with a translocation of sodium ions across the membrane69. We found transcription of a non-decarboxylating glutaryl-CoA dehydrogenase encoding gene (acd) accompanied by genes that code for a sodium-translocating glutaconyl-CoA decarboxylase (gcdBC) on the same contig (contig-100_40) (Fig. 2A,C, Table 1). This implies that energy-conserving mechanisms were employed by our culture, similar to strict anaerobes degrading aromatic compounds e.g. Syntrophus aciditrophicus70 and Desulfococcus multivorans68. We also found transcription of a decarboxylating glutaryl-CoA dehydrogenase gene (gcdH) in our enrichment culture (Fig. 2A,C, Table 1). However, the assembled transcripts observed here only encoded a rather short fragment of 66 amino acids compared to a usual decarboxylating glutaryl-CoA dehydrogenase of around 400 amino acids in length. Hence, the actual function could not be unambiguously predicted due to the truncated nature of the assembly.
Transcription of genes involved in aerobic benzene degradation
A striking finding was the identification of transcripts from a full set of genes involved in aerobic benzene degradation (Fig. 2B,C, Table 1). Both toluene monooxygenase (tmoABCDEF)71 and phenol hydroxylase (dmoKLMNOP)21 were shown to oxidise benzene to catechol. The catechol 2,3-dioxygenase encoded by dmpB mediates oxidative ring cleavage of catechol, which is then further converted to pyruvate and acetyl-CoA by enzymes of the lower pathway encoded by dmpCDEFGHI72. The dmp genes were characterized from the phenol-catabolizing plasmid pVI150 of Pseudomonas sp. CF60072 and are homologous to phe genes from the phenol-utilizing strain Bacillus thermoglucosidasius A773, tdn genes from the aniline-catabolizing plasmid pTDN1 of P. putida UCC2274, nah genes from the naphthalene-catabolizing plasmid NAH7 of P. putida G775 and nag genes from the naphthalene-utilizing strain Ralstonia sp. U276.
Oxygen production in the anaerobic benzene degrading culture
Possible explanations for the observation of transcripts for enzymes involved in aerobic metabolism under nitrate-reducing conditions might be oxygen influx or production in the enrichment culture. It has been shown that oxygen can be produced by a selected set of species that employ a nitric oxide dismutase (Nod) during nitrate reduction22. The resulting low concentrations of oxygen can be effectively scavenged in biofilms by the activities of monooxygenases and respiratory enzymes, such that strict anaerobes are protected from oxygen toxicity77. As such, biofilms can provide the necessary barrier for spatial separation of anaerobic and aerobic microbes.
To test the possibility of internal oxygen production, we added 0.5 mM nitrite to the continuous culture, but no oxygen production was detected within 2.5 hours. However, after addition of 1 mM formate to stimulate nitrite reduction, an oxygen concentration of up to 2.1% (5.25 µM) was detected by the oxygen electrode in the liquid phase of the continuous culture after 1.5 hours and by headspace oxygen analysis using GC-TCD (Figure S3). Nitrite was depleted after 12 days, and subsequently a second spike of 0.5 mM nitrite and 1 mM formate was added to the continuous culture. This time, no oxygen was detected (oxygen detection limit <0.1%, 0.25 µM). It is tempting to speculate that the aerobic organisms enriched during the first nitrite/formate spike effectively scavenged the oxygen formed during the second spike.
Typical Nod have a tandem histidine, one to ligate the low spin haem, the other to ligate the high spin reaction center haem78. This second histidine is absent from the Nod sequences, and therefore a characteristic discriminator between nitric oxide reductases and dismutases78. A search for the conserved Nod motifs did not reveal any matches in our dataset, however, this does not rule out an intermediate role for oxygen in the activation of benzene during denitrification. For example, D. aromatica strain RCB lacks genes encoding enzymes for anaerobic aromatic degradation and for the key enzyme Nod28, yet it was reported to degrade benzene under denitrifying conditions26. Moreover, the anaerobic methanotroph Candidatus Methylomirabilis oxyfera contains the entire pathway for aerobic methane oxidation but lacks key genes for anaerobic methane and hydrocarbon degradation, and activates methane in the presence of nitrite with oxygen and nitrogen formation22. Likewise, the alkane-degrading facultative denitrifying γ-proteobacterium strain HdN1 lacks genes for anaerobic alkane degradation but contains genes encoding monooxygenases25. However, in contrast to D. aromatica strain RCB, both Candidatus M. oxyfera and γ-proteobacterium strain HdN1 contain highly identical putative Nod78. These findings suggest a yet unknown pathway for oxygen formation from nitrate/nitrite that can be used for aerobic hydrocarbon degradation under anoxic conditions.
Transcripts for oxygenases associated with oxidation of monoaromatic compounds, particularly genes of benzoyl-CoA oxygenases (box genes), were also reported during growth on benzene and benzoate in a nitrate-reducing enrichment culture42. The box genes expressed under anoxic conditions in benzoate-degrading Azoarcus cultures were proposed to constitute an alternative oxygen-scavenging mechanism79 and may assist in a strategy to rapidly shift to aerobic degradation if oxygen levels become higher4,79.
Compound specific isotope analysis (CSIA) might help to further elucidate benzene biodegradation mechanisms. Interestingly, a recent combined carbon (C) and hydrogen (H) CSIA showed that isotope enrichment in a benzene-degrading nitrate-reducing enrichment culture (ΛC/H = 12 ± 3)80 was distinct from the same culture grown under sulfate-reducing condition (ΛC/H = 28 ± 3)81. In turn, it was similar to the isotope fractionation patterns of aerobic benzene degraders employing monooxygenase i.e. Cupriavidus necator ATCC 17697 (ΛC/H = 11 ± 6) and Alicycliphilus denitrificans strain BC (ΛC/H = 10 ± 4)81. This suggests involvement of monooxygenase-mediated degradation under nitrate-reducing condition80. Unfortunately, we were unable to grow our continuous culture in batch cultures for CSIA even when biofilm material was used as inoculum.
Transcription of genes involved in nitrate metabolism
We found transcripts from a number of genes involved in nitrate reduction (narGHI/nrxAB, nirK, norB, nosZ, nrfAH), including both denitrification and dissimilatory nitrate reduction to ammonium (DNRA). Interestingly, among the genes necessary for stepwise denitrification, transcription of the narGHI and nirK genes (mediating reduction of nitrate - > nitrite - > nitric oxide) was higher than that of the downstream norB and nosZ genes (mediating reduction of nitric oxide - > nitrous oxide - > dinitrogen) (Figure S4, Table S4). This suggests that nitrous oxide is not likely the main product of nitric oxide reduction. We also identified transcripts for assimilatory nitrate reduction (nsaA, narB), nitrogen fixation (nifDH) and nitrification (amoAB) (Figure S4, Table S4). The latter might also indicate oxygen presence in the culture.
In summary, our metatranscriptomic study of a benzene-degrading nitrate-reducing continuous culture provides insights into benzene degradation mechanisms. This culture appears to activate benzene predominantly via carboxylation, and employs ATP-independent BCR similar to what has been reported for strict anaerobes. The downstream pathway resembles that found in facultative anaerobes except for a non-decarboxylating glutaryl-CoA dehydrogenase that might enable energy conservation similar to strict anaerobes.
The likelihood of oxygen production from nitrate reduction proposed in our study and elsewhere80 is in agreement with field data. For example, a recent study showed unexpected diversity and high abundance of putative nod genes in BTEX-contaminated aquifers82. Interestingly, ample nod sequences were retrieved from the highly reduced core of an anoxic BTEX plume82 for which high abundance of tmoA genes had previously been revealed83. Likewise, a metagenomic study of anoxic hydrocarbon resource environments that had been subjected to nitrate injection showed high proportions of genes for enzymes involved in aerobic hydrocarbon metabolism84. Oxygenic denitrifiers may offer ecological advantages by enabling the aerobic microbes to thrive in hydrocarbon-contaminated anoxic subsurface environments.
Methods
Enrichment culture
A chemostat (Applikon, Schiedam, the Netherlands) culture that originated from soil samples obtained from a benzene-contaminated site located in the northern part of the Netherlands has been maintained with benzene as electron donor and nitrate as electron acceptor for more than 15 years38. Details of media composition and culture conditions were described previously40.
Sampling, RNA extraction and sequencing
Biofilms grown on the glass wall of the reactor had different morphologies40. Four suspended biofilm samples were taken from the areas with white biofilm: three on 31st October 2014 and one on 3rd November 2014. Moreover, two suspended biofilm samples were taken from the areas with brown biofilm on 3rd November 2014 (Table S1). Defined areas of biofilm attached to the glass wall were scraped off under a constant N2/CO2 (80/20%) flow. The liquid phase in the vessel was stirred for 5 minutes at 200 rpm to dislodge the biofilm aggregates followed by liquid phase sampling as described previously40. The samples were immediately stored at −80 °C. DNA and RNA co-extraction and purification was done as described previously85. The DNA samples were used for community analysis using MiSeq sequencing and quantification of key benzene degradation genes as described elsewhere40. The RNA samples were used for metatranscriptomic analysis in this study. Removal of rRNA, synthesis of cDNA and adding indices for Illumina library preperation were performed using the ScriptSeqTM Complete Kit (Bacteria) (Epicentre) following the manufacturer’s protocol. Single read sequencing was done with an Illumina HiSeq. 2500 (GATC-Biotech, Konstanz, Germany) generating reads between 6.02 to 46.4 M per sample. The read length was 150 bp.
Data quality assessment and filtering
SortMeRNA v1.986 was used to remove rRNA reads. Trueseq adapters were trimmed with cutadapt v1.2.187 with the –b settings. Quality trimming was performed with PRINSEQ Lite v0.20.288, with a minimum sequence length of 40 bp and a minimum quality of 30 on both ends of the read and as mean quality. All reads with non-IUPAC characters were discarded as well as reads containing more than three Ns.
Assembly and annotation
All reads which passed the quality assessment were pooled and cross-assembled with IDBA_UD version 1.1.1 with standard parameters89. All contigs, which contained more than 90% of a single base, more than 90% GC or AT, or which contained 50 or more bases of the same type in a row, were removed from further processing. On the assembled meta-transcriptome, Prodigal v2.5 was used for prediction of protein coding DNA sequences (CDS) with the option for meta samples90. Reads were mapped to the meta-transcriptome with Bowtie2 v2.0.691 using default settings. BAM files were converted with SAMtools v0.1.1892, and gene coverage was calculated with subread version 1.4.693.
The proteins were annotated with KAAS94, with SBH and ghostX as settings and with InterProScan 5.6-48.095. The annotation was further enhanced by adding EC numbers via PRIAM version March 06, 201396. Further EC numbers were derived by text mining and matching all InterproScan derived domain names against the BRENDA database (download 13.06.13)97. This text mining was done as outlined in supporting information. All EC and KO numbers were mapped with custom scripts onto the KEGG database98 and visualized using Python Scipy version 1.6.1 and NumPy version 0.9.099.
Taxonomic assignments
All assembled contigs were analysed with Blast 2.2.29100 against the NCBI NT database (download 22.01.2014) with standard parameters besides an e-value of 0.0001, the human microbiome (download 08.05.2014), the NCBI bacterial draft genomes (download 23.01.2014), the NCBI protozoa genomes (download 08.05.2014), and the human genome (download 30.12.2013, release 08.08.2013, NCBI Homo sapiens annotation release 105). Taxonomy was estimated with a custom version of the LCA algorithm as implemented in MEGAN101, but with changed default parameters. Only hits exceeding a bitscore of 50 were considered and of these only hits with a length of more than 100 nucleotides and that did not deviate more than 10% from the longest hit were used. All contigs, for which this did not result in any assignment, were again analysed with Blast using all the above mentioned databases, but with the –blastn option and the taxonomic assignment was calculated as mentioned.
Testing oxygen production
The oxygen production experiment was performed at a dilution rate of 0.1 day−1 at 25 °C with an influent benzene concentration of 100 µM as previously described40. The influent medium was similar to the medium used for metatranscriptomic analyses except that the vitamins were excluded and (NH4)2SO4 was replaced with 1.9 mM Na2SO4. The oxygen production was measured using an oxygen electrode submerged in the liquid phase of the continuous culture (AppliSens, Applikon). The oxygen electrode was calibrated by sparging nitrogen gas (0% O2) or air (100% O2) through demineralized water at 20 °C corresponding to 0 µM or 250 µM dissolved oxygen, respectively. The oxygen detection limit was 0.1% (0.25 µM). Headspace oxygen concentrations were measured with a Varian 3800 gas chromatographic (GC) system equipped with a thermal conductivity detector (TCD) and a tandem column (Molsieve 5 A/Porabond Q, Agilent, CA, USA). The TCD detector was set at 80 °C and the filament temperature was 160 °C. The oven temperature was constant at 45 °C for 8 min with helium as carrier gas. Headspace samples of 250 µl were taken from the continuous culture with a 250 µl Pressure-Lock gas syringe and a 0.6 × 25 mm sterile needle (Henke Sass Wolf, Tuttlingen, Germany) followed by 50 µl injection into the GC-TCD. Nitrite was added from a 1 M anoxic stock solution (NaNO2) to a final concentration of 0.5 mM. Formate was added from a 2 M anoxic stock solution (HCOONa) to a final concentration of 1 mM. Benzene was measured in 0.5 ml headspace samples of the reactor on a GC-FID system, as described previously40.
Sequence Data
All sequence data from this study were deposited at the European Bioinformatics Institute under the accession numbers ERS1670018 to ERS1670023. Further, all assigned genes, taxonomy, function, sequences of contigs, genes and proteins can be found in Table S3.
References
Dean, B. J. Recent findings on the genetic toxicology of benzene, toluene, xylenes and phenols. Mutat. Res/Rev. Gen. Toxicol 154, 153–181 (1985).
Vogt, C., Kleinsteuber, S. & Richnow, H. H. Anaerobic benzene degradation by bacteria. Microb. Biotechnol. 4, 710–724 (2011).
Langenhoff, A. A., Zehnder, A. J. & Schraa, G. Behaviour of toluene, benzene and naphthalene under anaerobic conditions in sediment columns. Biodegradation 7, 267–274 (1996).
Díaz, E., Jiménez, J. I. & Nogales, J. Aerobic degradation of aromatic compounds. Curr. Opin. Biotechnol. 24, 431–442 (2013).
Fuchs, G., Boll, M. & Heider, J. Microbial degradation of aromatic compounds- From one strategy to four. Nat. Rev. Microbiol. 9, 803–816 (2011).
Weelink, S. A. B., van Eekert, M. H. A. & Stams, A. J. M. Degradation of BTEX by anaerobic bacteria: physiology and application. Rev. Environ. Sci. Biotechnol. 9, 359–385 (2010).
Meckenstock, R. U. et al. Anaerobic degradation of benzene and polycyclic aromatic hydrocarbons. J. Mol. Microbiol. Biotechnol. 26, 92–118 (2016).
Caldwell, M. E. & Suflita, J. M. Detection of phenol and benzoate as intermediates of anaerobic benzene biodegradation under different terminal electron-accepting conditions. Environ. Sci. Technol. 34, 1216–1220 (2000).
Chakraborty, R. & Coates, J. D. Hydroxylation and carboxylation - Two crucial steps of anaerobic benzene degradation by Dechloromonas strain RCB. Appl. Environ. Microbiol. 71, 5427–5432 (2005).
Grbic-Galic, D. & Vogel, T. M. Transformation of toluene and benzene by mixed methanogenic cultures. Appl. Environ. Microbiol. 53, 254–260 (1987).
Vogel, T. M. & Grbic-Galic, D. Incorporation of oxygen from water into toluene and benzene during anaerobic fermentative transformation. Appl. Environ. Microbiol. 52, 200–202 (1986).
Abu Laban, N., Selesi, D., Jobelius, C. & Meckenstock, R. U. Anaerobic benzene degradation by Gram-positive sulfate-reducing bacteria. FEMS Microbiol. Ecol. 68, 300–311 (2009).
Kunapuli, U., Griebler, C., Beller, H. R. & Meckenstock, R. U. Identification of intermediates formed during anaerobic benzene degradation by an iron-reducing enrichment culture. Environ. Microbiol. 10, 1703–1712 (2008).
Phelps, C. D., Zhang, X. & Young, L. Y. Use of stable isotopes to identify benzoate as a metabolite of benzene degradation in a sulphidogenic consortium. Environ. Microbiol. 3, 600–603 (2001).
Musat, F. & Widdel, F. Anaerobic degradation of benzene by a marine sulfate-reducing enrichment culture, and cell hybridization of the dominant phylotype. Environ. Microbiol. 10, 10–19 (2008).
Ulrich, A. C., Beller, H. R. & Edwards, E. A. Metabolites detected during biodegradation of 13C6-benzene in nitrate-reducing and methanogenic enrichment cultures. Environ. Sci. Technol. 39, 6681–6691 (2005).
Holmes, D. E., Risso, C., Smith, J. A. & Lovley, D. R. Anaerobic oxidation of benzene by the hyperthermophilic archaeon Ferroglobus placidus. Appl. Environ. Microbiol. 77, 5926–5933 (2011).
Zhang, T. et al. Anaerobic benzene oxidation via phenol in Geobacter metallireducens. Appl. Environ. Microbiol. 79, 7800–7806 (2013).
Zhang, T. et al. Identification of genes specifically required for the anaerobic metabolism of benzene in Geobacter metallireducens. Front. Microbiol. 5, 245 (2014).
Weelink, S. A. B. et al. Isolation and characterization of Alicycliphilus denitrificans strain BC, which grows on benzene with chlorate as the electron acceptor. Appl. Environ. Microbiol. 74, 6672–6681 (2008).
Oosterkamp, M. J. et al. Genome Analysis and Physiological Comparison of Alicycliphilus denitrificans Strains BC and K601T. PLoS ONE 8, e66971 (2013).
Ettwig, K. F. et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548 (2010).
Mehboob, F. et al. Genome and proteome analysis of Pseudomonas chloritidismutans AW‐1T that grows on n‐decane with chlorate or oxygen as electron acceptor. Environ. Microbiol. 18, 3247–3257 (2016).
Peng, P. et al. Concurrent haloalkanoate degradation and chlorate reduction by Pseudomonas chloritidismutans AW-1T. Appl. Environ. Microbiol. 83, e00325–00317 (2017).
Zedelius, J. et al. Alkane degradation under anoxic conditions by a nitrate-reducing bacterium with possible involvement of the electron acceptor in substrate activation. Env. Microbiol. Rep. 3, 125–135 (2011).
Coates, J. B. et al. Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two strains of Dechloromonas. Nature 411, 1039–1043 (2001).
Meckenstock, R. U. & Mouttaki, H. Anaerobic degradation of non-substituted aromatic hydrocarbons. Curr. Opin. Biotechnol. 22, 406–414 (2011).
Salinero, K. K. et al. Metabolic analysis of the soil microbe Dechloromonas aromatica str. RCB: Indications of a surprisingly complex life-style and cryptic anaerobic pathways for aromatic degradation. BMC Genomics 10, 351 (2009).
Kasai, Y., Takahata, Y., Manefield, M. & Watanabe, K. RNA-based stable isotope probing and isolation of anaerobic benzene-degrading bacteria from gasoline-contaminated groundwater. Appl. Environ. Microbiol. 72, 3586–3592 (2006).
Herrmann, S. et al. Functional characterization of an anaerobic benzene-degrading enrichment culture by DNA stable isotope probing. Environ. Microbiol. 12, 401–411 (2010).
Kleinsteuber, S. et al. Molecular characterization of bacterial communities mineralizing benzene under sulfate-reducing conditions. FEMS Microbiol. Ecol. 66, 143–157 (2008).
Kunapuli, U., Lueders, T. & Meckenstock, R. U. The use of stable isotope probing to identify key iron-reducing microorganisms involved in anaerobic benzene degradation. ISME J. 1, 643–653 (2007).
Liou, J. S. C., Derito, C. M. & Madsen, E. L. Field-based and laboratory stable isotope probing surveys of the identities of both aerobic and anaerobic benzene-metabolizing microorganisms in freshwater sediment. Environ. Microbiol. 10, 1964–1977 (2008).
Luo, F., Devine, C. E. & Edwards, E. A. Cultivating microbial dark matter in benzene-degrading methanogenic consortia. Environ. Microbiol. 18, 2923–2936 (2016).
Oka, A. R. et al. Identification of critical members in a sulfidogenic benzene-degrading consortium by DNA stable isotope probing. Appl. Environ. Microbiol. 74, 6476–6480 (2008).
Taubert, M. et al. Protein-SIP enables time-resolved analysis of the carbon flux in a sulfate-reducing, benzene-degrading microbial consortium. ISME J. 6, 2291–2301 (2012).
Ulrich, A. C. & Edwards, E. A. Physiological and molecular characterization of anaerobic benzene-degrading mixed cultures. Environ. Microbiol. 5, 92–102 (2003).
van der Zaan, B. M. et al. Anaerobic benzene degradation under denitrifying conditions: Peptococcaceae as dominant benzene degraders and evidence for a syntrophic process. Environ. Microbiol. 14, 1171–1181 (2012).
Weelink, S. A. B. et al. Physiological and phylogenetic characterization of a stable benzene-degrading, chlorate-reducing microbial community. FEMS Microbiol. Ecol. 60, 312–321 (2007).
van der Waals, M. J. et al. Benzene degradation in a denitrifying biofilm reactor: activity and microbial community composition. Appl. Microbiol. Biotechnol. 101, 5175–5188 (2017).
Abu Laban, N., Selesi, D., Rattei, T., Tischler, P. & Meckenstock, R. U. Identification of enzymes involved in anaerobic benzene degradation by a strictly anaerobic iron-reducing enrichment culture. Environ. Microbiol. 12, 2783–2796 (2010).
Luo, F. et al. Metatranscriptome of an anaerobic benzene-degrading, nitrate-reducing enrichment culture reveals involvement of carboxylation in benzene ring activation. Appl. Environ. Microbiol. 80, 4095–4107 (2014).
Frias-Lopez, J. et al. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. 105, 3805–3810 (2008).
Shi, Y., Tyson, G. W., Eppley, J. M. & Delong, E. F. Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J. 5, 999–1013 (2011).
Chen, L. X. et al. Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J. 9, 1579–1592 (2015).
Didonato, R. J. Jr et al. Genome sequence of the deltaproteobacterial strain NaphS2 and analysis of differential gene expression during anaerobic growth on naphthalene. PLoS ONE 5, e14072 (2010).
Butler, J. E. et al. Genomic and microarray analysis of aromatics degradation in Geobacter metallireducens and comparison to a Geobacter isolate from a contaminated field site. BMC Genomics 8, 180 (2007).
Peters, F., Heintz, D., Johannes, J., Van Dorsselaer, A. & Boll, M. Genes, enzymes, and regulation of para-cresol metabolism in Geobacter metallireducens. J. Bacteriol. 189, 4729–4738 (2007).
Dong, X. et al. Reconstructing metabolic pathways of a member of the genus Pelotomaculum suggesting its potential to oxidize benzene to carbon dioxide with direct reduction of sulfate. FEMS Microbiol. Ecol., fiw254 (2016).
Mouttaki, H., Johannes, J. & Meckenstock, R. U. Identification of naphthalene carboxylase as a prototype for the anaerobic activation of non-substituted aromatic hydrocarbons. Environ. Microbiol. 14, 2770–2774 (2012).
Musat, F. et al. Anaerobic degradation of naphthalene and 2-methylnaphthalene by strains of marine sulfate-reducing bacteria. Environ. Microbiol. 11, 209–219 (2009).
Bergmann, F. et al. Genomic insights into the metabolic potential of the polycyclic aromatic hydrocarbon degrading sulfate-reducing Deltaproteobacterium N47. Environ. Microbiol. 13, 1125–1137 (2011).
Boll, M., Löffler, C., Morris, B. E. L. & Kung, J. W. Anaerobic degradation of homocyclic aromatic compounds via arylcarboxyl-coenzyme A esters: Organisms, strategies and key enzymes. Environ. Microbiol. 16, 612–627 (2014).
Ebenau-Jehle, C. et al. An unusual strategy for the anoxic biodegradation of phthalate. ISME J. 11, 224–236 (2017).
Carmona, M. et al. Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol. Mol. Biol. Rev. 73, 71–133 (2009).
Egland, P. G. & Harwood, C. S. BadR, a new MarR family member, regulates anaerobic benzoate degradation by Rhodopseudomonas palustris in concert with AadR, an Fnr family member. J. Bacteriol. 181, 2102–2109 (1999).
Schühle, K. et al. Benzoate-coenzyme a ligase from Thauera aromatica: An enzyme acting in anaerobic and aerobic pathways. J. Bacteriol. 185, 4920–4929 (2003).
Egland, P. G. & Harwood, C. S. HbaR, a 4-hydroxybenzoate sensor and FNR-CRP superfamily member, regulates anaerobic 4-hydroxybenzoate degradation by Rhodopseudomonas palustris. J. Bacteriol. 182, 100–106 (2000).
Holmes, D. E., Risso, C., Smith, J. A. & Lovley, D. R. Genome-scale analysis of anaerobic benzoate and phenol metabolism in the hyperthermophilic archaeon Ferroglobus placidus. ISME J. 6, 146–157 (2012).
Tan, B., Dong, X., Sensen, C. W. & Foght, J. Metagenomic analysis of an anaerobic alkane-degrading microbial culture: potential hydrocarbon-activating pathways and inferred roles of community members. Genome 56, 599–611 (2013).
Tan, B. et al. Comparative analysis of metagenomes from three methanogenic hydrocarbon-degrading enrichment cultures with 41 environmental samples. ISME J. 9, 2028–2045 (2015).
Top, E. M. & Springael, D. The role of mobile genetic elements in bacterial adaptation to xenobiotic organic compounds. Curr. Opin. Biotechnol. 14, 262–269 (2003).
Boll, M. & Fuchs, G. Benzoyl‐coenzyme A rrductase (Ddaromatizing), a key enzyme of anaerobic aromatic metabolism: ATP dependence of the reaction, purification and some properties of the enzyme from Thauera Aromatica strain K172. Eur. J. Biochem. 234, 921–933 (1995).
Kung, J. W. et al. Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc. Natl. Acad. Sci. 106, 17687–17692 (2009).
Schmid, G., Auerbach, H., Pierik, A. J., Schünemann, V. & Boll, M. ATP-dependent electron activation module of benzoyl-coenzyme A reductase from the hyperthermophilic archaeon Ferroglobus placidus. Biochemistry 55, 5578–5586 (2016).
López Barragán, M. J. et al. The bzd gene cluster, coding for anaerobic benzoate catabolism, in Azoarcus sp. strain CIB. J. Bacteriol. 186, 5762–5774 (2004).
Harrison, F. H. & Harwood, C. S. The pimFABCDE operon from Rhodopseudomonas palustris mediates dicarboxylic acid degradation and participates in anaerobic benzoate degradation. Microbiology 151, 727–736 (2005).
Wischgoll, S. et al. Decarboxylating and nondecarboxylating glutaryl-coenzyme A dehydrogenases in the aromatic metabolism of obligately anaerobic bacteria. J. Bacteriol. 191, 4401–4409 (2009).
Dimroth, P. & Schink, B. Energy conservation in the decarboxylation of dicarboxylic acids by fermenting bacteria. Arch. Microbiol. 170, 69–77 (1998).
McInerney, M. J. et al. The genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. Proc. Natl. Acad. Sci. 104, 7600–7605 (2007).
Tao, Y., Fishman, A., Bentley, W. E. & Wood, T. K. Oxidation of benzene to phenol, catechol, and 1, 2, 3-trihydroxybenzene by toluene 4-monooxygenase of Pseudomonas mendocina KR1 and toluene 3-monooxygenase of Ralstonia pickettii PKO1. Appl. Environ. Microbiol. 70, 3814–3820 (2004).
Shingler, V., Powlowski, J. & Marklund, U. Nucleotide sequence and functional analysis of the complete phenol/3, 4-dimethylphenol catabolic pathway of Pseudomonas sp. strain CF600. J. Bacteriol. 174, 711–724 (1992).
Duffner, F. M., Kirchner, U., Bauer, M. P. & Müller, R. Phenol/cresol degradation by the thermophilic Bacillus thermoglucosidasius A7: cloning and sequence analysis of five genes involved in the pathway. Gene 256, 215–221 (2000).
Fukumori, F. & Saint, C. P. Complete nucleotide sequence of the catechol metabolic region of plasmid pTDN1. J. Gen. Appl. Microbiol. 47, 329–333 (2001).
Sota, M. et al. Genomic and functional analysis of the IncP-9 naphthalene-catabolic plasmid NAH7 and its transposon Tn4655 suggests catabolic gene spread by a tyrosine recombinase. J. Bacteriol. 188, 4057–4067 (2006).
Zhou, N.-Y., Fuenmayor, S. L. & Williams, P. A. nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism. J. Bacteriol. 183, 700–708 (2001).
Atashgahi, S. et al. Geochemical parameters and reductive dechlorination determine aerobic cometabolic vs aerobic metabolic vinyl chloride biodegradation at oxic/anoxic interface of hyporheic zones. Environ. Sci. Technol. 51, 1626–1634 (2017).
Ettwig, K. F. et al. Bacterial oxygen production in the dark. Front. Microbiol. 3, 273 (2012).
Valderrama, J. A. et al. Bacterial degradation of benzoate: cross-regulation between aerobic and anaerobic paathways. J. Biol. Chem. 287, 10494–10508 (2012).
Keller, A. H., Kleinsteuber, S. & Vogt, C. Anaerobic benzene mineralization by nitrate-reducing and sulfate-reducing microbial consortia enriched from the same site: Comparison of community composition and degradation characteristics. Microbiol. Ecol., 1–13 (2017).
Fischer, A. et al. Combined carbon and hydrogen isotope fractionation investigations for elucidating benzene biodegradation pathways. Environ. Sci. Technol. 42, 4356–4363 (2008).
Zhu, B. et al. Unexpected diversity and high abundance of putative nitric oxide dismutase (Nod) genes in contaminated aquifers and wastewater treatment systems. Appl. Environ. Microbiol. 83, e02750–02716 (2017).
Larentis, M., Hoermann, K. & Lueders, T. Fine‐scale degrader community profiling over an aerobic/anaerobic redox gradient in a toluene‐contaminated aquifer. Env. Microbiol. Rep. 5, 225–234 (2013).
An, D. et al. Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common. Environ. Sci. Technol. 47, 10708–10717 (2013).
Rajeev, L. et al. Dynamic cyanobacterial response to hydration and dehydration in a desert biological soil crust. ISME J. 7, 2178–2191 (2013).
Kopylova, E., Noe, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal 17, 10–12 (2011).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011).
Peng, Y., Leung, H. C., Yiu, S. M. & Chin, F. Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC bioinformatics 11, 119 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108 (2013).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35, W182–185 (2007).
Hunter, S. et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40, D306–312 (2012).
Claudel-Renard, C., Chevalet, C., Faraut, T. & Kahn, D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res 31, 6633–6639 (2003).
Chang, A. et al. BRENDA in 2015: exciting developments in its 25th year of existence. Nucleic Acids Res 43, D439–446 (2015).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40, D109–114 (2012).
van der Walt, S., Colbert, C. & Varoquaux, G. The NumPy Array: a structure for efficient numerical computation. Comput. Sci. Engin. 13, 22–30 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Huson, D. H., Mitra, S., Ruscheweyh, H. J., Weber, N. & Schuster, S. C. Integrative analysis of environmental sequences using MEGAN4. Genome Res 21, 1552–1560 (2011).
Acknowledgements
This study was supported by a grant of BE-Basic-FES funds from the Dutch Ministry of Economic Affairs. The research of A.J.M. Stams is supported by an ERC grant (project 323009) and the gravitation grant “Microbes for Health and Environment” (project 024.002.002) of the Netherlands Ministry of Education, Culture and Science. F. Hugenholtz was supported by the same gravitation grant (project 024.002.002). B. Hornung is supported by Wageningen University and the Wageningen Institute for Environment and Climate Research (WIMEK) through the IP/OP program Systems Biology (project KB-17-003.02-023).
Author information
Authors and Affiliations
Contributions
H.S., J.G., A.J.M.S. and R.v.S. planned the research; S.A., M.J.v.d.W., U.N.d.R. and F.H. did the experiments; S.A., B.H., B.N. and D.M. did data analysis; and all authors wrote and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Atashgahi, S., Hornung, B., van der Waals, M.J. et al. A benzene-degrading nitrate-reducing microbial consortium displays aerobic and anaerobic benzene degradation pathways. Sci Rep 8, 4490 (2018). https://doi.org/10.1038/s41598-018-22617-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22617-x
This article is cited by
-
Metatranscriptomic responses and microbial degradation of background polycyclic aromatic hydrocarbons in the coastal Mediterranean and Antarctica
Environmental Science and Pollution Research (2023)
-
Bacteria-plant interactions synergistically enhance biodegradation of diesel fuel hydrocarbons
Communications Earth & Environment (2022)
-
Taxonomic and functional trait-based approaches suggest that aerobic and anaerobic soil microorganisms allow the natural attenuation of oil from natural seeps
Scientific Reports (2022)
-
Lignin intermediates lead to phenyl acid formation and microbial community shifts in meso- and thermophilic batch reactors
Biotechnology for Biofuels (2021)
-
Diversity and metagenome analysis of a hydrocarbon-degrading bacterial consortium from asphalt lakes located in Wietze, Germany
AMB Express (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
