
Abstract
To optimise fecal sampling for reproducible analysis of the gut microbiome, we compared different methods of sample collection and sequencing of 16S rRNA genes at two centers. Samples collected from six individuals on three consecutive days were placed in commercial collection tubes (OMNIgeneGut OMR-200) or in sterile screw-top tubes in a home fridge or home freezer for 6–24 h, before transfer and storage at −80 °C. Replicate samples were shipped to centers in Australia and the USA for DNA extraction and sequencing by their respective PCR protocols, and analysed with the same bioinformatic pipeline. Variation in gut microbiome was dominated by differences between individuals. Minor differences in the abundance of taxa were found between collection-processing methods and day of collection, and between the two centers. We conclude that collection with storage and transport at 4 °C within 24 h is adequate for 16S rRNA analysis of the gut microbiome. Other factors including differences in PCR and sequencing methods account for relatively minor variation compared to differences between individuals.
Similar content being viewed by others

Introduction
The fecal or ‘gut’ microbiome is shaped strongly by diet but also by the host genotype, age, hygiene and antibiotic exposure, and is altered in many pathophysiological states1,2. The composition of gut microbiota differs greatly between individuals3 and therefore maximizing the detection of biological and disease-related changes requires minimization of variation due to methods of sample collection and analysis.
Previous studies have shown that fecal microbial composition overall was not altered when DNA was extracted from a fresh fecal sample compared to a sample that had been immediately frozen and stored at −80 °C for up to 6 months4,5. Storage at different temperatures for varying times has been compared with immediate freezing and storage at −80 °C. One study5 reported a decrease in Bacteroidetes and an increase in Firmicutes phyla after 30 minutes at room temperature, but the majority4,6,7,8,9,10,11 have found that storage at room temperature for at least a day, or at 4, −20 or −80 °C for up to 14 days, had little effect on the relative abundance of taxa. Moreover, microbial composition was not significantly altered in DNA extracted from fecal occult blood test cards that had been at room temperature for three days12. Recent studies have also evaluated the OMNIgene®•GUT (OMR-200) collection and liquid storage tube, which is reported to stabilize DNA at room temperature for 14 days13. Samples immediately collected into these tubes and stored for three days at room temperature showed little difference in microbial composition by 16S rRNA gene sequencing compared with samples immediately frozen at −80 °C7. A similar result was obtained when samples in OMR-200tubes stored for 1–28 days at room temperature were compared with samples from which DNA was freshly extracted14,15. However, the relative abundance of Bacteroides increased after seven days and in infants (who had lower microbial diversity than adults) significant differences from fresh samples were observed after 14 days storage.
As microbiome studies expand into larger populations at multiple sites, stringent quality control remains critical. With this in mind and to optimise analysis of the gut microbiome in a multi-site, longitudinal pregnancy-birth cohort study16, we evaluated the impact of different collection-processing methods on three sequential daily fecal samples from six individuals for 16S rRNA gene sequencing in two centers.
Methods
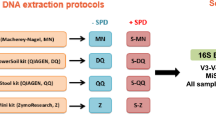
Six healthy adult volunteers, three males and three females aged 35–70, provided fecal samples on three successive days. Multiple aliquots were taken from each bulk sample and those for bacterial microbiome analysis were stored by one of four methods A-D (Fig. 1).

Schematic of the four collection methods for a home-collected fecal sample. Six individuals collected samples over three days. Each sequencing center received three aliquots per person-day-method combination (216 in total).
Collection-processing methods
Method A: individuals placed aliquots of feces into 6× OMNIgene®•GUT (OMR-200)13 tubes as per manufacturer’s protocol. These were stored at room temperature for 6–24 h before delivery to a laboratory for transfer to sterile 5 mL screw cap tubes and storage at −80 °C. Method B: individuals placed aliquots of feces into 6× sterile 5 ml screw cap tubes, which were stored in the home freezer for 6–24 h before delivery to the laboratory in an insulated container for storage at −80 °C. Method C: individuals placed aliquots of feces into 6× sterile 5 m screw cap tubes, which were stored in the home refrigerator for 6–24 h before delivery to the laboratory in an insulated container for storage at −80 °C. Method D: individuals placed a bulk fecal sample into a sterile 70 ml collection jar, which was stored in home refrigerator for 6–24 h before delivery to the laboratory in an insulated container, transfer of aliquots into 6× sterile 5 m screw cap tubes and storage at −80 °C.
In the laboratory, samples were handled under sterile conditions in a Biosafety Level 2 cabinet. This collection-processing procedure yielded a total of 432 samples (24 from each of 6 individuals per day for 3 days) (Fig. 1). After 2–4 weeks at −80 °C, three sample aliquots per person-day-method (n = 216) were transported on dry ice to sequencing centers in Australia (Walter and Eliza Hall Institute of Medical Research, Melbourne, Victoria; WEHI), and the USA (Baylor College of Medicine, Alkek Center for Metagenomics and Microbiome Research, Houston, Texas; BCM), for 16S rRNA gene amplicon sequencing. At both WEHI and BCM samples were stored at −80 °C for 4 weeks before sequencing.
DNA sequencing methods
Samples were thawed on ice and DNA extracted at both WEHI and BCM with the MoBio PowerSoil kit (MoBio Laboratories, Carlsbad, CA), as in the Human Microbiome Project17. At both WEHI and BCM, the V4 hypervariable region of the bacterial 16S rRNA marker gene (16Sv4) was PCR-amplified with primers 515F-OH1 and 806R-OH2. Analogues of these are described, respectively, by the common name U515F, new name S-*-Univ-0515-a-S-19, and the common name 806R, new name S-D-Bact-0787-b-A-20 in18.
At WEHI, these primers (GTGACCTATGAACTCAGGAGTCGGACTACNVGGGTWTCTAAT) and (CTGAGACTTGCACATCGCAGCGTGYCAGCMGCCGCGGTAA) included unique sequences (underlined) to provide a target for the subsequent introduction of Illumina sequencing adaptors and dual index barcodes to the amplicon target for paired-end sequencing on the Illumina MiSeq instrument19. Primary 16S rRNA gene PCR amplification was performed in duplicate with the following conditions: 94 °C for 3 minutes followed by 20 cycles at 94 °C for 45 seconds each, 55 °C for 1 minute, and 72 °C for 1 minute 30 seconds and a final extension step at 72 °C for 10 minutes. Successful amplification was determined by agarose gel electrophoresis. Amplicons from the primary amplification were diluted 1/10 and used as template for the secondary amplification. In the secondary amplification, the overhang sequences were used to introduce Illumina sequencing adaptors and dual index barcodes to the amplicon target. Individual amplicons were identified using 8 base index sequences from the Illumina Nextera design. Sixteen forward index primers and 24 reverse index primers were designed for a 96-well plate format with the potential to generate a maximum of 384 dual index amplicons. PCR conditions were as for the primary amplification, except for an increase to 25 cycles. Reactions were performed in triplicate in separate plates, with extra wells added, up to a maximum of 9 wells, if PCR product appeared low. Amplicon size distribution was determined by agarose gel electrophoresis. One sample failed to yield sufficient product in any well. Reactions from the three replicate plates were pooled and the PCR amplicons purified with 1.0× NGS Beads (Macherey-Nagel). Each dual indexed library plate pool was quantified with the Agilent Tapestation and the Qubit™ DNR BR assay kit for Qubit 3.0® Fluorometer (Life Technologies). The indexed pool was diluted to 12pM and sequenced with the paired end 600-cycle (2 × 311) kit on a MiSeq instrument. After quality filtering, 702 technical replicate samples comprising 215 biological samples from the 6 individuals were obtained.
At BCM, the 16S V4 sequencing methods were adapted from the NIH-Human Microbiome Project17 and the Earth Microbiome Project20,21. The primers used for amplification contained adapters for MiSeq sequencing and single-end barcodes allowing pooling and direct sequencing of PCR products21. PCR-amplified amplicons were normalized by concentration before pooling and sequences were generated in one lane of a MiSeq instrument using the v2 kit (2 × 250 bp paired-end protocol). The 216 samples were analysed in a single pool.
Bioinformatic analysis
Sequences were clustered into operational taxonomic units (OTUs) with 97% similarity using QIIME (Version 1.8.1)22,23,24,25, and taxonomically classified by aligning the representative sequences to the Greengenes 13_08 database26. Paired-end sequences were assembled with PEAR27 with parameters -v 100 -m 600 -n 80, where -v is minimum overlap, -m is maximum assembled length and -n is minimum assembled length. WEHI index sequences were extracted with QIIME script extract_barcodes.py, and bases up to and including the 533F to 805R V4 region amplicon primers were trimmed. BCM sequences were supplied as trimmed sequences with a separate barcode index file.
QIIME’s split_libraries_fastq.py script was used for quality filtering, with Phred quality scores required to be above 29, and 90% of a read’s length required to have consecutive, high-quality base calls. In order to minimize differences between WEHI and BCM sequences, WEHI sequences were further filtered by aligning to the SILVA 123 16S rRNA gene database using MOTHUR v1.38.128,29 with start = 8390 and end = 17068, and by removing sequences that were not in this position.
QIIME’s open-reference OTU-picking was used with the Uclust algorithm22,23 to form clusters at 97% similarity. The representative sequences from each OTU were aligned with gaps to a reference set using QIIME’s implementation of PyNAST, then filtered for chimeric sequences using UChime with default settings30. After making a filtered OTU table with minimum count 3, and assigning taxonomy with Greengenes_13_0826, the data were analyzed in R.
WEHI data were derived from 759 (PCR-well) samples. Those with sequence counts <1000 or with descriptions of PCR products as ‘None’ or ‘Very faint’ were discarded, which included all samples for individual 55, method B, day 3, aliquot 3. OTU counts for the remaining technical replicates were summed to give 215 biological-replicate samples.
Alpha and beta diversity and differential abundance analyses were performed in R using the Phyloseq package31 and DESeq232. Most analyses used 215 or 216 samples with minimum OTU size of 20; alpha diversities were calculated with the minimum-count = 3 OTUs with technical and biological replicates combined, giving 72 samples.
Data availability
The sequences analysed during this study are available in the ENA Short Read Archive, with accession PRJNA393083 (SRP116702), https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA393083. The OTU tables generated are available through Figshare with accession 5401594, https://figshare.com/articles/ENDIA_microbiome_QC_OTUs_and_metadata/5401594.
Results
For the WEHI data set, library sizes after quality filtering, clustering, and combining PCR replicates ranged from 30,000 to 250,000 sequences per sample, with a median of 67,000 (Figure S1A); sequences clustered into 12,652 OTUs of minimum size 20. For the BCM data set, library sizes ranged from 5,000 to 56,000, with a median of 27,000 (Figure S1B); sequences clustered into 3,675 OTUs of minimum size 20. The final number of sequences reflects differing sequencing and filtering protocols, including the use of multiple PCR replicates at WEHI.
Taxonomic overview
Samples were dominated at the phylum level by Bacteroidetes and Firmicutes, as expected. The mean summed proportion of these two phyla was 94%, varying from 71.9% to 99.7% between individual samples. Likewise, a single order from each of these two phyla, Bacteroidales and Clostridiales, was dominant, with three orders from the phylum Proteobacteria contributing another 1–2% overall (Fig. 2A,D; see also Figure S2).

Overview of the fecal bacterial microbiome from sequencing at WEHI (A,B,C) or BCM (D,E,F). (A,D) Dominant bacterial genera in fecal samples, or higher taxa where genus was not available. Bars are colour-coded by phyla: red Bacteroidetes, blue Firmicutes, green Proteobacteria, brown Actinobacteria, yellow Verrucomicrobia. (B,E) Alpha diversity within samples. Two measures are shown: observed number of OTUs per sample, an estimate of richness, and Inverse-Simpson index indicating the evenness of the sample. Samples were sub-sampled to the smallest sample size, and values are the mean of 10 random sub-samples. Boxes show the inter-quartile range for the four methods on three days. (C,F) Beta diversity. NMDS ordination of the UniFrac distance between samples, a representation of phylogenetic similarity.
Alpha (α) diversity is used to characterise the richness of the microbiome and its evenness (heterogeneity) or distribution of proportions. Samples showed a considerable spread of α diversity (Fig. 2B,E). Samples from individual 66 had the lowest observed richness (number of OTUs per sample) and the lowest Inverse-Simpson diversity index, the latter indicating dominance by a smaller number of OTUs. This is reflected in the genera plots (Fig. 2A,D). In contrast, samples from individual 11 had a high observed richness but a comparatively low Inverse-Simpson index, consistent with the presence of a few high-abundance and multiple low-abundance genera.
Analysis of β diversity by non-metric multidimensional scaling (NMDS) ordination of the UniFrac distance showed that samples cluster strongly by individual, with marked separation between individuals (Fig. 2C,F).
Differences between WEHI and BCM data sets
For WEHI and BCM data sets the most abundant phyla were similar, but the proportions of less abundant phyla and higher taxonomic resolution differed. For example, the mean proportion of genus Akkermansia in the order Verrucomicrobiales was greater in BCM (0.7%) than WEHI (0.02%). The proportion of Bacteroides was lower in some individual samples for BCM than WEHI (Fig. 2B; also S2).
The BCM data set yielded fewer OTUs and therefore had lower values for observed richness (Fig. 2B,E). The number of OTUs observed per sample was dependent on sampling depth (Figure S3); values shown are based on the smallest sample sizes for each of the two data sets. Richness was similar between the WEHI and BCM data sets, with samples from individual 66 showing the lowest alpha diversity and those from individual 44 the highest. For the Inverse-Simpson diversity index, which is not dependent on library size at this depth of sequencing, the BCM data set had a greater range of values, and a greater range for samples from some individuals. Both data sets had similar patterns of beta diversity between individuals (Fig. 2C,F), although the BCM data set had several outliers.
Initial analysis was performed separately on the WEHI and BCM datasets. For better comparison of the taxonomies, the bioinformatic pipeline was re-applied to a data set comprising the BCM sequences and one of the three WEHI technical replicates (Fig. 3). The ordination plot shows ‘batch’ effects between the two sequencing centers and greater between-sample differences in the BCM data set.

Beta diversity between samples from two sequencing centers. Ordination plot of Bray-Curtis distances between samples, using Detrended Correspondence Analysis. Points represent samples from BCM and a single technical replicate from WEHI.
DESeq2 was used to make generalized linear models for the counts at phylum, order and OTU levels (Table 1). The model included individual ID, day and collection-processing method as factors. At the phylum level, the largest change was in the Verrucomicrobia. At the OTU level, 3% of OTUs were significantly different (Figure S4, Additional data S1). Most of the differentially abundant OTUs belonged to the orders Clostridiales (63%) and Bacteroidales (31%). The direction of change in OTUs was not consistent, and there were no significant differences in counts for Clostridiales and Bacteroidales between WEHI and BCM data sets.
Effect of collection-processing method on taxonomic analysis
Testing of the WEHI data set for differential abundances between collection-processing methods, using DESeq2 with a design controlling for the effect of person and day, revealed no significant differences in counts by phylum, order or family (Table 2, Fig. 4A). Five OTUs (0.04% of OTUs comprising 0.2% of sequences) were different under collection-processing Method A. With the BCM data set, collection-processing Methods A and B were taxonomically different, with a decrease in Actinobacteria in Method A (Fig. 4D) and an increase in Lentisphaerae, although counts were very low (p < 0.001, Additional Table S1). Lentisphaerae were also increased in Method A compared with Methods C and D (p < 0.05).

Effect of collection-processing method from sequencing at WEHI (A,B,C) or BCM (D,E,F). (A,D) Log of standardised counts (scaled by library size) of the four most abundant phyla. Points show mean and bars standard deviation (sd) for each individual and collection-processing method. Method A has the smallest average sd for Bacteroidetes and Actinobacteria. (B,E) The Inverse-Simpson α diversity index for each sample (compare with Fig. 2). (C,F) Mean log (standardised count) plotted against the mean over the collection-processing methods, and a linear regression applied. Method A has the greatest average deviation from the linear model for the WEHI data set.
Diversity varied within a sample depending on collection-processing method (Fig. 4B,E) but the effect was small and inconsistent. After fitting a linear model with inputs for method and individual, 20–30% of variation was unaccounted for, while collection-processing method accounted for only 2%. Overall, alpha diversity was slightly lower with Method A in the WEHI data set, and higher with Method B in the BCM dataset. (Table 3).
Different methods of collection-processing might also increase the variance between samples, reducing the reproducibility of a result. Two approaches were used to test for this. Greater variance between samples is equivalent to greater distance between samples by some measure. The Bray-Curtis dissimilarity between OTU counts was calculated for pairs of samples from each individual and method, and a Tukey Honest Significant Difference test applied to a linear model of the dissimilarity. There was no evidence that the dissimilarity between samples was different for collection-processing methods (smallest p = 0.1) in the WEHI data set. There were significant differences in Bray-Curtis distances between samples in the BCM data set (p < 0.001), with collection-processing Method A associated with smaller differences between samples from the same individual than Methods B, C and D (Additional Table S2).
In addition, we looked for differences in the variance of the four most abundant phyla. The log transformed standardised counts for Bacteroidetes, Firmicutes, Proteobacteria and Actinobacteria per sample were compared with the mean across collection-processing methods for each individual (Fig. 4C,F). In the WEHI data set Methods B, C and D gave similar results, while method A had lower variance within samples from the same individual but greater deviation from the mean compared with the other methods.
Effect of collection-processing method on library size
Collection-processing methods were compared after quality filtering, barcode extraction and clustering. In the WEHI data set, the number of DNA sequences extracted per sample was not different by collection-processing method; in the BCM data set, collection-processing Method D resulted in fewer sequences than other collection-processing methods, but the difference was small compared with total variation (Figure S5). Batch effects (sequencing run) were more significant (p < 10−5) than collection-processing method, but batch and method together contributed less than 5% of the variation in library size.
Discussion
This was a quality assessment study prior to commencing a multi-site, longitudinal pregnancy-birth cohort study. Our main focus was the comparison of fecal sample collection and storage methods, with samples sequenced at two centers. Not unexpectedly, given between-center differences in DNA extraction and sequencing methodologies, some differences in WEHI and BCM data sets were observed. For example, the proportions of phyla Verrucomicrobia and Actinobacteria, and order Enterobacteriales were increased in the BCM compared to WEHI data set. Although samples were transported and arrived on dry ice at both centers, covert effects of shipping and handling can’t be excluded.
Different collection-processing methods for fecal samples were associated with only minor differences in fecal microbiota derived by 16S rRNA gene sequencing. In the BCM data set Method A using the OMNIgeneGut OMR-200 device exhibited differences in phyla Actinobacteria and Lentisphaerae proportions compared with the methods that involved either immediate freezing or refrigeration. In the WEHI data set, no method was associated with significantly more distance between samples at the OTU level. However, in the BCM data set beta diversity was reduced between Method A samples compared to the other methods, possibly reflecting mitigation of storage and transport effects by the OMR-200 device. In WEHI data set, the log-values of proportions of the four dominant phyla had lower variance within samples from the same individual, but greater deviation from the mean, for samples collected with Method A.
In conclusion, collection-processing methods and day of collection contribute to only minor variation in fecal microbiome extracted sequences, composition and diversity, the major variation being at the level of the individual. Collection with storage and transport at 4 °C within 24 h is adequate for analysis of the fecal microbiome. These findings complement and support the results of several previous studies4,5,6,7,8,9,10,11. They are relevant to the quality control of gut microbiome studies, in particular to population-based multi-site studies, samples from which would ideally be analysed by the same methodology.
Declarations
Ethics approval and consent to participate
Volunteers gave informed consent for self-collection of non-identifiable stool samples and basic, demographic information. In accordance with the Australian NHMRC National Statement on Ethical Conduct in Human Research, the research was considered as ‘negligible risk’ with no foreseeable risk of harm or discomfort to the volunteers and therefore deemed exempt from ethical review. The methods for sample collection have been approved by the Women’s and Children’s Health Network Human Research Ethics Committee under project numbers HREC/16/WCHN/66 and HREC/13/WCHN/29.
Availability of data and materials
Additional Figures S1–S6 and Additional Tables S1 and S2 attached as Additional Figures and Tables. Additional data attached as xlsx file.
R code is available as joint_plots_tables.Rmd from github.com/PapenfussLab/endiaQC/.
References
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227, http://www.nature.com/nature/journal/v486/n7402/abs/nature11053.html-supplementary-information (2012).
De Filippo, C. et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and ruralAfrica. Proceedings of the National Academy of Sciences 107, 14691–14696, https://doi.org/10.1073/pnas.1005963107 (2010).
The Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214, http://www.nature.com/nature/journal/v486/n7402/abs/nature11234.html-supplementary-information (2012).
Carroll, I. M., Ringel-Kulka, T., Siddle, J. P., Klaenhammer, T. R. & Ringel, Y. Characterization of the fecal microbiota using high-throughput sequencing reveals a stable microbial community during storage. PloS one 7, e46953, https://doi.org/10.1371/journal.pone.0046953 (2012).
Gorzelak, M. A. et al. Methods for Improving Human Gut Microbiome Data by Reducing Variability through Sample Processing and Storage of Stool. PloS one 10, e0134802, https://doi.org/10.1371/journal.pone.0134802 (2015).
Cardona, S. et al. Storage conditions of intestinal microbiota matter in metagenomic analysis. BMC microbiology 12, 158, https://doi.org/10.1186/1471-2180-12-158 (2012).
Choo, J. M., Leong, L. E. X. & Rogers, G. B. Sample storage conditions significantly influence faecal microbiome profiles. Scientific Reports 5, 16350, https://doi.org/10.1038/srep16350 (2015).
Lauber, C. L., Zhou, N., Gordon, J. I., Knight, R. & Fierer, N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS microbiology letters 307, 80–86, https://doi.org/10.1111/j.1574-6968.2010.01965.x (2010).
Roesch, L. F. et al. Influence of fecal sample storage on bacterial community diversity. The open microbiology journal 3, 40–46, https://doi.org/10.2174/1874285800903010040 (2009).
Tedjo, D. I. et al. The effect of sampling and storage on the fecal microbiota composition in healthy and diseased subjects. PloS one 10, e0126685, https://doi.org/10.1371/journal.pone.0126685 (2015).
Wu, G. D. et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC microbiology 10, 206, https://doi.org/10.1186/1471-2180-10-206 (2010).
Dominianni, C., Wu, J., Hayes, R. B. & Ahn, J. Comparison of methods for fecal microbiome biospecimen collection. BMC microbiology 14, 103, https://doi.org/10.1186/1471-2180-14-103 (2014).
Doukhanine, E. B. A., Merino, C., Pozza L. OMNIgene®•GUTenables reliable collection of high quality fecal samples for gut microbiome studies. DNA Genotek 2015–16 (2014).
Anderson, E. L. et al. A robust ambient temperature collection and stabilization strategy: Enabling worldwide functional studies of the human microbiome. Sci Rep 6, 31731, https://doi.org/10.1038/srep31731 (2016).
Hill, C. J. et al. Effect of room temperature transport vials on DNA quality and phylogenetic composition of faecal microbiota of elderly adults and infants. Microbiome 4, 19, https://doi.org/10.1186/s40168-016-0164-3 (2016).
Penno, M. A. et al. Environmental determinants of islet autoimmunity (ENDIA): a pregnancy to early life cohort study in children at-risk of type 1 diabetes. BMC pediatrics 13, 124, https://doi.org/10.1186/1471-2431-13-124 (2013).
The Human Microbiome Project, C. A framework for human microbiome research. Nature 486, 215–221, http://www.nature.com/nature/journal/v486/n7402/abs/nature11209.html-supplementary-information (2012).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic acids research 41, e1, https://doi.org/10.1093/nar/gks808 (2013).
Aubrey, B. J. et al. An Inducible Lentiviral Guide RNA Platform Enables the Identification of Tumor-Essential Genes and Tumor-Promoting Mutations In-Vivo. Cell Reports 10, 1422–1432, https://doi.org/10.1016/j.celrep.2015.02.002 (2015).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America 108, 4516–4522, https://doi.org/10.1073/pnas.1000080107 (2011).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME Journal 6, 1621–1624, https://doi.org/10.1038/ismej.2012.8 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods, https://doi.org/10.1038/nmeth.f.303 (2010).
Edgar, R. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one 5, e9490, https://doi.org/10.1371/journal.pone.0009490 (2010).
Caporaso, J. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267 (2010).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME journal 6, 610–618, https://doi.org/10.1038/ismej.2011.139 (2012).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620 (2014).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids research 41, D590–D596, https://doi.org/10.1093/nar/gks1219 (2013).
Schloss, P. D. et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Applied and environmental microbiology 75, 7537–7541, https://doi.org/10.1128/aem.01541-09 (2009).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200, https://doi.org/10.1093/bioinformatics/btr381 (2011).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PloS one 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 1–21, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Acknowledgements
We acknowledge Tulin Ayvaz, Ginger Metcalf, Donna Muzny and Richard Gibbs and the Human Genome Sequencing Center at Baylor College of Medicine (BCM) for assistance with sample processing and data generation for the BCM dataset. This research (ANZCTR registration number ACTRN1261300794707) was supported by the Juvenile Diabetes Research Foundation (JDRF) Australia, the Australian Research Council Special Research Initiative in Type 1 Juvenile Diabetes, The Helmsley Charitable Trust, JDRF International, and the National Health and Medical Research Council (NHMRC) of Australia (Program Grants 1037321 and 1054618). L.C.H. is the recipient of a NHMRC Senior Principal Research Fellowship (1080887). A.T.P. is the recipient of a NHMRC Senior Research Fellowship (1116955). The work was made possible through Victorian State Government Operational Infrastructure Support and NHMRC Research Institute Infrastructure Support Scheme. The funding sources had no role in the design of the study and collection, analysis, and interpretation of data, or in writing the manuscript.
Author information
Authors and Affiliations
Author notes
A comprehensive list of consortium members appears at the end of the paper
Consortia
Contributions
L.C.H., M.A.S.P. and J.J.C. conceived the study. M.A.S.P., L.C.H., E.B.-S., J.M.W., S.C.B. and C.Y.B. collected and processed samples. K.M.N., S.A.W. and N.J.A. performed DNA extraction and sequencing. J.S.P., A.J.R.-S., A.T.P., N.J.A. and J.F.P. performed the bioinformatics analyses. J.S.P., A.T.P. and L.C.H. wrote the initial draft of the paper, which was reviewed by all authors.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Penington, J.S., Penno, M.A.S., Ngui, K.M. et al. Influence of fecal collection conditions and 16S rRNA gene sequencing at two centers on human gut microbiota analysis. Sci Rep 8, 4386 (2018). https://doi.org/10.1038/s41598-018-22491-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22491-7
This article is cited by
-
Multicenter evaluation of gut microbiome profiling by next-generation sequencing reveals major biases in partial-length metabarcoding approach
Scientific Reports (2023)
-
The gut microbiome of obese postpartum women with and without previous gestational diabetes mellitus and the gut microbiota of their babies
Diabetology & Metabolic Syndrome (2022)
-
Gestational weight gain and visceral adiposity in adult offspring: Is there a link with the fecal abundance of Acidaminococcus genus?
European Journal of Clinical Nutrition (2022)
-
Detection of cell-free microbial DNA using a contaminant-controlled analysis framework
Genome Biology (2021)
-
ddPCR allows 16S rRNA gene amplicon sequencing of very small DNA amounts from low-biomass samples
BMC Microbiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
