
Abstract
LytR-cpsA-Psr (LCP) domain containing proteins fulfil important functions in bacterial cell wall synthesis. In Mycobacterium tuberculosis complex (Mtbc) strains, the causative agents of tuberculosis (TB), the genes Rv3484 and Rv3267 encode for LCP proteins which are putatively involved in arabinogalactan transfer to peptidoglycan. To evaluate the significance of Rv3484 for Mtbc virulence, we generated a deletion mutant in the Mtbc strain H37Rv and studied its survival in mice upon aerosol infection. The deletion mutant failed to establish infection demonstrating that Rv3484 is essential for growth in mice. Following an initial phase of marginal replication in the lungs until day 21, the Rv3484 deletion mutant was almost eliminated by day 180 post-infectionem. Interestingly, the mutant also showed higher levels of resistance to meropenem/clavulanate and lysozyme, both targeting peptidoglycan structure. We conclude that Rv3484 is essential for Mtbc virulence in vivo where its loss of function cannot be compensated by Rv3267.
Similar content being viewed by others

Introduction
Tuberculosis (TB) is, in conjunction with HIV infection, the leading cause of mortality and morbidity due to a single infectious agent worldwide1. Increasing numbers of cases with multidrug (MDR) and extensively drug (XDR) resistant Mycobacterium tuberculosis complex (Mtbc) strains complicate TB control and potentially lead to an era in which current anti-TB drugs are no longer effective1,2. This underscores the urgent need to identify new drug targets to design new and more effective treatment regimens. An ideal drug target is essential for bacterial survival under in vivo infection conditions. Several of the current antibiotics target cell wall synthesis and thereby interfere with an essential structure for bacterial cell integrity3.
Accordingly, we were interested in genes putatively involved in mycobacterial cell wall homeostasis. Based on the previously defined core transcriptome of 17 Mtbc strains responding to intracellular conditions in murine bone marrow derived macrophages (mBMDM)4, we selected Rv3484, which contains besides the LytR-cpsA-psr (E-value 3.49e-51) (LCP) domain conserved Csp2a (E-value 1.48e-75), PRK09379 (E-value 2.30e-16) and LytR_C (E-value 3.44e-11) domains as detected by NCBI CDD search5. Proteins containing these domains have been associated with bacterial cell wall metabolism and regulatory functions even though the exact mechanism remained elusive6,7,8,9,10,11,12,13. Phosphotransferase activity was described for LCP proteins of Gram-positive bacteria, where the transfer of anionic cell wall polymers, such as teichoic acids or capsular polysaccharides, to peptidoglycan (PGN) was attributed to LCP proteins14,15. Recently, in vitro reconstitution studies revealed ligase activity for the Staphylococcus aureus LCP proteins, LcpA, LcpB and LcpC. These proteins transferred wall teichoic acid precursor molecules to PGN oligomers16. Additional results further indicated that LCP proteins catalyze the transfer of diverse substrates to different acceptor moieties such as the transfer of the lipid-linked ManNAc-GlcNAc-GlcNAc of Bacillus anthracis. In B. anthracis, the LCP homologues, lcpB2, lcpC and lcpD were also able to complement for the lack of wall teichoic acid attachment to PGN in S. aureus LCP mutants strains17. In addition glycoprotein glycosylation in Actinomyces oris has been ascribed to an LCP protein18.
The cell wall core of mycobacteria and corynebacteria consists of PGN, which is covalently bound to arabinogalactan (AG) and mycolic acids. In Mtbc this structure builds up the scaffold for the asymmetric outer lipid bilayer, which in turn, serves as a matrix for virulence associated lipids such as phtiocerol dimycocerosates (PDIM), phenolic glycolipids (PGL) and trehalose dimycolate (TDM), as well as e.g. mannosylated lipoarabinomannan (manLAM). The cell wall in its complexity is responsible for many of the properties of the Mtbc, e.g. its rigidity and resistance to common disinfectants or its low permeability for antibiotics; it provides immune modulatory compounds and ligands for host cell interaction and preserves bacterial cell integrity19,20,21. Homologues of the LCP proteins have been identified in the genomes of Corynebacterium glutamicum, M. marinum and Mtbc22,23. In M. marinum, orthologues of all LCP proteins of Mtbc are present (MMAR_4858, MMAR_1274, MMAR_4966, and MMAR_5392; Rv0822c, Rv3267, Rv3484, and Rv3840, respectively). A M. marinum transposon mutant defective in MMAR_4966 revealed altered colony morphology, cell surface properties, lower growth rate in vitro, enhanced susceptibility to erythromycin, vancomycin and penicillin and altered cell wall permeability for hydrophobic compounds. Comparing cell wall compositions between wild-type and mutant M. marinum cells showed an imbalance in the AG/PGN-ratio. Furthermore the mutant was less virulent in the murine macrophage cell line RAW 264.7 and the zebrafish model22.
Grzegorzewicz et al. analyzed heterologously expressed Rv3484, Rv3267 and Rv0822c which showed pyrophosphatase activity, corroborating their proposed function as LCP proteins. They obtained single mutants for Rv3484 and Rv3267 in the virulent Mtb CDC1551 strain and on the background of an avirulent auxotroph H37Rv mutant, whereas a double mutant could only be generated for Rv0822c/Rv3267 but not for the other combinations. It was concluded that Rv3484 and Rv3267 substitute for each other’s function, however, together are essential for viability in vitro whereas Rv0822c plays a redundant role. Analyzing the rhamnose/GlcNAc ratio as marker for AG and PGN corroborated this hypothesis, as no significant differences were observed between both mutants and the parental wild-type strain. Furthermore, total lipids, mycolic acids and cell wall polysaccharides did not differ between mutants and wild-type mycobacteria. Notably, the Rv3267 deficient mutant exhibited enhanced sensitivity to antibiotics, which was speculated to be due to synergistic effects between Rv3267 deficiency and cell wall targeting antibiotics such as vancomycin or β-lactams. A predominant role of Rv3267 in AG attachment was suggested23. Catalyzing the transfer of labelled galactose containing cell wall material to peptidoglycan in vitro was also described for Rv326724.
To further characterize the role of Rv3484 for virulence, we generated an unmarked in-frame deletion mutant of Rv3484 in Mtb H37Rv and investigated its survival in vivo. Our findings show that inactivation of Rv3484 alone is sufficient to abolish long term growth of Mtb upon aerosol infection in C57BL/6 mice.
Results
Deletion of Rv3484 of Mtb H37Rv
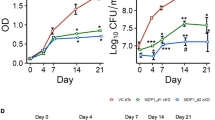
Rv3484 is a gene of 1539 bp encoding a 512 amino acids long protein. The majority (1233 bp) of Rv3484 was deleted in-frame using a two-step homologous recombination procedure resulting in a marker free mutant strain. Figure 1a shows the genomic locus of Rv3484 in Mtb H37Rv, the strain in which the plasmid providing the homologous recombination substrate had co-integrated into the chromosome, as well as Mtb H37RvΔRv3484, including restriction endonuclease sites used for Southern blotting, localization of the probe and expected fragment sizes. Inactivation of Rv3484 was confirmed by Southern blotting (Fig. 1b), PCR (Fig. 1c) and sequencing of the Rv3484 region (data not shown). Rv3484 is not essential in vitro as mutant clones could readily be obtained after the second intrachromosomal recombination event. Mtb H37RvΔRv3484 showed normal growth kinetics in vitro in 7H9 medium (Fig. 2a) and retained its acid-fast properties as shown by Ziehl-Neelsen staining (Fig. 2b).

Genomic locus and deletion of Rv3484 from the genome of Mtb H37Rv. (a) Genomic organization of the Rv3484 locus including restriction sites and localization of the probe for Southern analysis and localization of primers for PCR analysis. The in frame deletion comprises 1233 bp. Southern blot of the Mtb H37Rv Rv3484 deletion mutant. (b) Southern analysis using StuI treated genomic DNA resulting in signals at 3878 bp for the mutant strain, two signals at 3878 bp and 4264 bp for the strain which underwent a single crossover resulting in cointegration of a deleted copy of Rv3484 and 5111 bp for the wild type. pSvM2 cut with PacI served as a control and resulted in a fragment of 2266 bp. PCR analysis of the Mtb H37Rv Rv3484 deletion mutant. (c) Agarose gels showing Mtb H37Rv wild type (wt), the strain carrying a deletion in Rv3484 (ΔRv3484) besides the complemented mutant strain (ΔRv3484::Rv3484), the mutant strain carrying the empty vector used for complementation (ΔRv3484::pMV306) and the plasmid used for transformation of the deleted copy of the gene (pSvM2) and a PCR negative control ((−)) as controls. Primers #172 and #185: wt 1481 bp, ΔRv3484::Rv3484 1481 bp. Primers #184 and #190: wt 2913 bp, ΔRv3484, ΔRv3484::Rv3484, ΔRv3484::pMV306, pSvM2: 1680 bp.

Growth kinetics of the Rv3484 deletion mutant strain in 7H9 medium and acid fast properties. (a) Bacteria were grown in fully supplemented 7H9 medium and OD600 measured at indicated time points. Means ± SD of biological triplicates performed in technical duplicates are shown. (b) Acid fast Ziehl-Neelsen staining of Mtb H37Rv, Mtb H37RvΔRv3484, Mtb H37RvΔRv3484::Rv3484 as well as H37RvΔRv3484::pMV306 was performed according to standard procedures (1000× magnification, scale ≙ 10 µm).
Resistance of Mtb H37RvΔRv3484 to in vitro stress conditions and hydrophobic properties
First, we addressed the question whether deletion of Rv3484 affects the resistance of Mtb H37RvΔRv3484 to certain stresses in vitro. To test the resistance to lysozyme, an enzyme cleaving the β-1,4-glycosidic bonds of the peptidoglycan backbone, we used the resazurin (7-hydroxy-10-oxidophenoxazin-10-ium-3-one) microplate assay. Redox dyes such as resazurin have been widely used to assess metabolic activity of viable cell lines25 as well as efficacy of antibiotics to bacterial pathogens including the Mtbc26,27. This assay measures the metabolic activity of the cells by means of resazurin reduction, yielding the fluorescent product resorufin. Results of this approach have been shown to correspond well with those obtained from traditional drug susceptibility testing28,29,30, and is recommended by the WHO for non-commercial drug susceptibility testing31. A lack of metabolic activity is regarded as indicative for an inhibitory activity of the compound tested but not necessarily its bactericidal activity. The resazurin microplate assay indicated that the deletion of Rv3484 resulted in an increased resistance to lysozyme (Fig. 3a). In the presence of 0.25 mg/ml lysozyme, we observed a significant decrease in fluorescent units of the Mtb H37Rv as compared to Mtb H37RvΔRv3484; in addition, differences were significant between the complemented mutant (Mtb H37RvΔRv3484::Rv3484) compared to Mtb H37RvΔRv3484 or the mutant strain transformed with the empty vector pMV306 (Mtb H37RvΔRv3484::pMV306).

Resistance of the Mtb H37Rv Rv3484 deletion mutant against lysozyme. (a) Mtb wild type (H37Rv), Mtb H37RvΔRv3484, Mtb H37RvΔRv3484::Rv3484 as well as the mutant strain transformed with the empty vector pMV306 were exposed to lysozyme for 7 days and metabolic activity assessed using resazurin. Fluorescence was read at 590 nm in a microplate reader and the means and SEM of fluorescence intensities corrected by subtracting background fluorescence from wells containing dH2O and resazurin alone were plotted. Two-way ANOVA and Bonferroni Post-hoc-test were used for statistical evaluation of the data. P-values ≤ 0.01 (**) and ≤0.0001 (****) were considered as significant. (b) Resistance of the Mtb H37Rv Rv3484 deletion mutant strain against meropenem/clavulanate. Mtb wild type (H37Rv), Mtb H37RvΔRv3484, Mtb H37RvΔRv3484::Rv3484 as well as the mutant strain transformed with the empty vector pMV306 were exposed to 0, 0.04, 0.08, 0.16, 0.32, 0.63, 1.25, 2.5, 5 µg/ml meropenem (each sample supplemented with 2.5 µg/ml clavulanate) and metabolic activity of the bacteria was assessed after 1 week by the addition of resazurin. Data are expressed as normalized fluorescence intensities after subtraction of background fluorescence emitted by wells containing resazurin in 7H9 medium. Comparisons were based on two-way ANOVA and the Bonferroni Post-hoc-test. P-values ≤ 0.01 (**) or ≤0.001 (***) were considered as significant. Means with error bars representing the SEM are shown.
In order to validate the experimental approach, we determined the OD600 of the samples prior to adding resazurin. Lysozyme was used as a stressor. The OD600 reads showed relatively high variation and less resolution than the fluorescence reads (Supplementary Fig. 1). Overall, the OD600 corresponded to the results obtained from the fluorescence reads as Mtb H37Rv and Mtb H37RvΔRv3484::Rv3484 tended to have the least OD600 reads when compared to Mtb H37RvΔRv3484 and Mtb H37RvΔRv3484::pMV306. Another way to read resazurin assays is to determine visually the concentration at which no metabolic activity could be detected making use of the change in color as a result of resazurin reduction. We detected a clear difference of one two-fold dilution between the pairs Mtb H37RvΔRv3484, Mtb H37RvΔRv3484::pMV306 and Mtb H37Rv, Mtb H37RvΔRv3484::Rv3484, respectively. We conclude that analysis of the fluorescence reads from the resazurin microplate assays is a suitable approach and allows for statistical analysis and assessment of observed effects of independent replicates.
Moreover, we analyzed whether osmotic stress (0.5 M NaCl) as an indicator for compromised cell wall homeostasis impacts Mtb H37RvΔRv3484 differently from Mtb H37Rv. However, we could not detect significant differences between mutant and the Mtb H37Rv regarding resistance against osmotic stress (Supplementary Fig. 2).
In addition, we tested the hydrophobicity of the bacteria by means of bacterial affinity to the hydrocarbon hexadecane in a biphasic system, but did not find differences between Mtb H37RvΔRv3484, Mtb H37Rv and Mtb H37RvΔRv3484::Rv3484 (Supplementary Fig. 2).
Resistance of Mtb H37RvΔRv3484 to antibiotics
Since the mycobacterial cell wall is an important factor for the Mtbc to withstand antibiotic intervention, we tested the sensitivity of Mtb H37RvΔRv3484 to antibiotics. Bacteria of the Mtbc encode for a β-lactamase32; therefore, we added 2.5 µg/ml of the β-lactamase inhibitor clavulanate proven to render β-lactam antibiotics such as meropenem more effective33. Results obtained from the resazurin assay suggested that Mtb H37RvΔRv3484 is more resistant to meropenem/clavulanate than Mtb H37Rv or Mtb H37RvΔRv3484::Rv3484 (Fig. 3b).
For rifampicin we achieved ambiguous results (Supplementary Fig. 3). In addition, we did not observe differences in the susceptibilities to isoniazid, ethambutol, streptomycin, kanamycin, hygromycin and vancomycin between Mtb H37RvΔRv3484 and Mtb H37Rv (Supplementary Fig. 4). As expected, the control strains bearing the vector used for complementation or the empty vector were resistant to hygromycin as they carry a hygromycin resistance cassette as selection marker whereas no difference could be observed for Mtb H37RvΔRv3484 and Mtb H37Rv.
Mtb H37RvΔRv3484 is severely attenuated in the mouse model of tuberculosis
In order to examine the role of Rv3484 as a virulence determinant, we infected C57BL/6 mice with an aerosol containing Mtb H37Rv, Mtb H37RvΔRv3484 and the complemented mutant strain Mtb H37RvΔRv3484::Rv3484 (infection dose approximately 100 CFU). Infections were performed twice with 5 mice per bacterial strain and time point. Mtb H37RvΔRv3484 was unable to establish an infection as compared to Mtb H37Rv and Mtb H37RvΔRv3484::Rv3484 (Fig. 4). The pulmonary bacterial loads of mice infected with Mtb H37RvΔRv3484 were similar or only slightly increased at day 21 to the infection dose assessed at day 1 indicating only weak, if any, replication in the early phase of infection (Fig. 4). Subsequently, the bacterial load declined and no bacteria could be detected 180 days post infectionem (p.i.). In comparison, both, the Mtb H37Rv and the complemented mutant strain grew with bacterial loads reaching 6–7 log units by day 21 p.i., which remained at a plateau until day 180 p.i. Despite the fact of extremely low bacterial loads in lungs of mice infected with Mtb H37RvΔRv3484, some bacteria were able to disseminate to liver and spleen. However, there was no evidence for replication. Single bacteria were detectable in some mice up to day 90. At day 180 Mtb H37RvΔRv3484 was cleared from all organs examined.

In vivo characterization of the Rv3484 deletion mutant of Mtb H37Rv. C57BL/6 mice were infected in two replicate experiments by aerosol containing either Mtb H37Rv ((a) 177 (b) 88 CFU/lung), Mtb H37Rv ΔRv3484 ((a) 98 (b) 210 CFU/lung) or Mtb H37Rv ΔRv3484::Rv3484 ((a) 274 (b) 171 CFU/lung) (infection dose determined with n = 2–3). At indicated time points the bacterial load of lung, spleen and liver was determined by enumerating bacteria after plating organ homogenates of 7H10 agar plates (n = 4–5 in each replicate experiment). A detection limit was calculated for samples for which at least for one animal no bacterial load could be detected by plating 100 µl of the organ homogenates. In this case raw CFU counts were set to 0.99, the theoretical CFU/organ calculated and defined as detection limit. Data points for organs of single mice as well as means ± SEM are shown.
To assess whether the mutant strain was totally eliminated we plated 1 ml undiluted homogenate from mice infected for 180 days. Only very few colonies were recovered after 6 weeks of incubation (1, 1, 2, 5 and 10 CFU [5 animals out of 10; 3× lung, 1× liver, 1× spleen]). Four out of these five mice came from the replicate experiment, in which the infection dose for Mtb H37RvΔRv3484 was twice the one of Mtb H37Rv (210 versus 88 CFU/lung), which may be the reason for the slight delay in bacterial clearance. Strong attenuation of Mtb H37RvΔRv3484 is also reflected by the absence of inflammatory infiltrates in lungs from mice infected with Mtb H37RvΔRv3484 at all time points p.i. analyzed (Fig. 5). In contrast, granulomatous lesions were observed in lungs infected with Mtb H37Rv or Mtb H37RvΔRv3484::Rv3484 from day 21 on until day 180 p.i.

Histopathology of lung sections of mice infected with the Mtb H37Rv Rv3484 deletion mutant. HE stained sections of lungs prepared at day 21 (a), 42 (b), 90 (c) and 180 (d) post-infection scanned with 1× (original scan area 1 cm × 1 cm) using a slide scanner and 100 × (scale ≙ 100 µm) magnification of wild type (H37Rv; confirmed infection dose 88 CFU/lung), the Rv3484 deletion mutant (H37Rv∆Rv3484; confirmed infection dose 210 CFU/lung) and the complemented mutant strain (H37Rv∆Rv3484::Rv3484; confirmed infection dose 171 CFU/lung).
Discussion
Our results demonstrate that Rv3484 is essential for growth and survival of Mtb H37Rv in vivo, in mice infected by aerosol. In contrast to Mtb H37Rv and Mtb H37RvΔRv3484::Rv3484, the marker free Mtb H37RvΔRv3484 was not able to establish a progressive infection in the aerosol mouse infection model.
Recently, Rv3484 has been proposed to encode for a LCP protein responsible for the transfer of AG to PGN. However, no major differences of the single mutant’s glycosyl compositions were detected, which was likely due to mutual functional compensation by these LCP proteins23. A detailed biochemical characterization using a conditional Rv3267/Rv3484 Mtbc double knockout strain in order to evaluate the proposed function is currently lacking. In addition, in vivo data are not available to date, and the relevance of this particular LCP protein homologue, out of the four LCP proteins found in Mtb, was yet studied. Indeed, functional redundancy between Rv3484 and Rv3267 was proposed. However, by establishing an unmarked Rv3484 deletion mutant, we were able to demonstrate an exclusive function of this LCP protein for in vivo virulence in mice.
Our own previous experiments showed that the expression of Rv3484 was upregulated universally in 17 representative strains of the Mtbc in response to conditions they encounter in mBMDMs4, indicating its importance for virulence of strains across distinct Mtbc lineages. Indeed, the Mtbc was proven to be more diverse than previously anticipated, which is presumably reflected in the successful spread of certain lineages in different areas of the world34,35,36. This resulted in particular host-pathogen-coevolution leading to differences in strain pathobiology as well as functional heterogeneity in in vitro and in vivo infection models; e.g. lineages of the Mtbc show specific virulence patterns in human primary macrophages and aerogenically infected C57BL/6 mice37. The conservation of putative cell wall synthesis associated domains in LCPs and the role of Rv3484 homologues in other bacteria6,7,8,9,10,11,12,13,14,15 together with its universal upregulation in response to macrophage infection in a diverse set of clinical Mtbc isolates4, prompted us to investigate the significance of this gene for Mtbc virulence in the reference strain, H37Rv. Indeed, our experiments showed, that the deletion mutant failed to establish infection in mice, thus, identifying Rv3484 as an essential gene for growth in vivo.
The mycobacterial cell wall is an important virulence determinant providing resistance to stresses Mtbc strains encounter during the course of infection, either simply by its rigidity, by masking pathogen associated molecular patterns e.g. by PDIM, or by immune modulatory compounds38,39. Defects in cell wall synthesis, especially disturbed homeostasis of the PGN backbone, can also lead to differential sensitivities to enzymes and antibiotics, which target this important structural component of the bacterial cell. Various antibiotics target PGN of bacterial cell walls. One example recently attracting much interest due to its mycobactericidal activity is meropenem, which shows increased efficacy in the presence of the β-lactamase inhibitor clavulanate33. We found increased resistance to meropenem/clavulanate of Mtb H37RvΔRv3484, while results for rifampicin were slightly ambiguous; however, Mtb H37RvΔRv3484 seemed to have increased resistance to this antibiotic, too. As rifampicin is not targeting the cell wall, increased resistance of the mutant strain could be due to reduced access of the drug to the mycobacterial cytoplasm to target the DNA-dependent RNA polymerase. Moreover, and in contrast to all the other antibiotics tested, to which both, mutant and wild-type strain, were equally susceptible, meropenem and rifampicin are both only sparingly soluble in water. However, analysis of Mtb H37RvΔRv3484 and Mtb H37Rv surface hydrophobicity using a biphasic hexadecane system did not reveal differences between both strains. Accordingly, differences in cell wall hydrophobicity that can determine permeability to compounds depending on their hydrophobic/hydrophilic properties cannot explain this observation. Grzegorzewicz et al. reported no differences between the Rv3484 mutant and wild type regarding the sensitivity to rifampicin. Slight differences in the experimental set-up, e.g. the usage of an auxotrophic H37Rv and the CDC1551 background, Tween 80 instead of Tyloxapol as a medium supplement, which was interchangeably used by Grzegorzewicz et al. for mutants derived from the two wild-type strains used in this work23, or other factors, might also influence the differential outcome. Taken together, we believe that rifampicin resistance, in contrast to the one against lysozyme and meropenem/clavulanate, does not represent a clear biological property which can characterize the Rv3484 deletion phenotype but rather a secondary effect.
The fact that Grzegorzewicz et al. did not observe a phenotype for the Rv3484 single KO, including no effects on the biochemical composition of the cell wall in vitro, let us refrain from thorough analytical chemistry approaches23. However, as we found a strong phenotype, which manifested predominantly in vivo we assume that the phenotype causing the attenuation might only be detectable in the in vivo situation e.g. directly from materials isolated from the lungs of infected mice at early time points or in response to stressors not yet identified. The experimental procedure and methodology to address this issue still need to be established. Furthermore, we cannot yet exclude the possibility that the in vivo attenuation we observed might be due to factors other than modified cell wall composition. Our results clearly show that Rv3484 function is essential for infection of mice. The phenotype, based on Rv3484 deficiency, becomes obvious already at early time points p.i. and results in almost complete elimination of the bacteria; thus, Rv3484 awaits further evaluation for its suitability as target for anti-TB treatment.
Only recently, another study supported the proposed function of the LCP homologues in C. glutamicum where biochemical analyses revealed a decreased amount of arabinogalactan attached to the cell wall in a mutant strain. Moreover, the importance of the C-terminal LytR_C domain was shown. Either concerted action of both domains on one substrate or subsequent cascade of two catalytic reactions has been surmised40. To answer whether the LytR_C domain plays a distinct role for Mtb survival in vivo requires additional genetic approaches.
In contrast to the impaired growth of the M. marinum mutant defective in the Rv3484 orthologue observed in vitro22, but in agreement with Grzegorzewicz et al.23, we found similar growth rates for Mtb H37Rv, Mtb H37Rv∆Rv3484, Mtb H37Rv∆Rv3484::Rv3484 and Mtb H37Rv∆Rv3484::pMV306. In contrast Rv3484 is essential for Mtbc survival in the TB mouse model, as only in some mice and organs very few bacteria of Mtb H37RvΔRv3484 could be detected in the lungs, spleen or liver of aerosol infected C57BL/6 mice. This severe attenuation of Mtb H37RvΔRv3484 could be complemented by the introduction of a single copy of Rv3484 under the control of its native promoter in the chromosome of Mtb H37RvΔRv3484 using an integrative vector. This finding sheds a different light on the role of this particular LCP protein Rv3484 and is in clear contrast to the functional redundancy proposed by Grzegorzewicz et al.23. The exact mechanism underlying our observation is currently addressed by structural and molecular biological approaches.
Methods
Bacterial strains and culture conditions
Mycobacterium tuberculosis H37Rv ATCC 27294 was used as a parental strain for genetic manipulations. Escherichia coli HB101 served as a host for molecular cloning procedures. Middlebrook 7H10 agar (Becton Dickinson, Franklin Lakes, USA) containing 0.5% glycerol (Applichem, Darmstadt, Germany), 10% OADC (Becton Dickinson, Franklin Lakes, USA), and 0.05% Tyloxapol (Sigma-Aldrich, St. Louis, USA) for 7H9 broth if required supplemented with 50 µg/ml hygromycin (Carl Roth, Karlsruhe, Germany) was used to cultivate Mtb strains, whereas Luria Bertani (LB) medium (Carl Roth, Karlsruhe, Germany) was used to cultivate E. coli. LB supplemented with Ampicillin (Carl Roth, Karlsruhe, Germany) was used as a selective medium for E. coli. For animal infection experiments Mtb stocks were prepared as follows. Bacteria were grown in 7H9 broth to mid-exponential growth phase and aliquots frozen at −80 °C. Titers of viable bacteria were determined after thawing by plating serial dilutions of the aliquots on 7H10 agar plates. Single cell suspensions were prepared by pushing the bacteria 10 times through a Microlance 3 26-gauge needle (Becton Dickinson, Franklin Lakes, USA). These suspensions were used for aerosol infection.
Generation of the Rv3484 deletion mutant of Mtb H37Rv
Flanking regions of Rv3484 were amplified using the primer pair Rv3484_KO_fus1 5′ - cggtgtcgggtcggtggtggt – cagcccgatgagcaagatg −3′, Rv3484_KO_PacI3 5′ - gagtct - ttaattaa - cgtgctccattcaacagtc −3′ and the primer pair Rv3484_KO_fus2 5′ - acatcttgctcatcgggctg – accaccaccgacccgacac −3′, Rv3484_KO_PacI_4 5′ - agtatg - ttaattaa - cgtcgaacgtgaactgag −3′. The resulting fragments were fused in a third self-primed PCR reaction via their complementary 5 prime end extensions and the primers Rv3484_KO_PacI_3 and Rv3484_KO_PacI_4. This final PCR fragment was cloned via PacI into the mycobacterial suicide plasmid pYUB65741 (kindly provided by WR Jacobs) to obtain pSvM2 and used to transform Mtb H37Rv ATCC 27294. The mutant strain was generated using the two step allelic exchange methodology and successful deletion of Rv3484 from the genome was confirmed by PCR, Southern blotting and sequencing. The Rv3484 mutant was complemented using the integrative mycobacterial shuttle plasmid pMV306 providing a single wild-type copy of Rv348442. Briefly, Rv3484 including 400 bp of its upstream region and 208 bp of its downstream region was amplified using HindIII-restriction site containing primers Rv3484_HindIII1 5′ - gagtctaagctt-cacacaggccaggaccac −3′ and Rv3484_HindIII2 5′ - gtatgaaagctt-gggtctgcacgccattac −3′ and cloned in pMV306 cut with HindIII. The resulting plasmid pSM14 was used to transform the Rv3484 deletion mutant of Mtb ATCC 27294. Southern blotting was performed according to standard procedures. The probe was amplified from genomic DNA using the primer pair #103 5′ - gtcaatctcgtcagacacctaac −3′ and #104 5′ – gcccgatgagcaagatg −3′ yielding a probe of 518 bp. PCR for validation of the strains used in this study was performed using primer pair #172 5′ - cctgccctggtcggtct −3′ and #185 5′ - gggtctgcacgccattac −3′ specific for the wild-type copy of Rv3484 as one primer binds in the deletion and the other one in the genome/the genome fragment cloned into the integrative vector resulting in the amplification of 1481 bp in case a wild-type copy is present. A second PCR was set up in order to validate the genomic situation at the Rv3484 locus using the primers #184 5′ - cacacaggccaggaccac −3′ and #190 5′ – gaacgtcgaacgtgaactgag −3′. These primers do not amplify from the integrative complementation vector as one primer does not bind in the construct and give a fragment of 2913 bp for the wild type and 1680 bp in case Rv3484 is deleted.
Ziehl-Neelsen staining
Acid-fast staining was performed according to the Ziehl-Neelsen standard procedure. Briefly, bacteria were fixed on glass slides by heat fixation followed by staining with carbol-fuchsin, destaining with HCl–ethanol, washing with dH2O and counterstaining using methylene blue followed by a final wash step with dH2O.
In vitro susceptibility testing of Mtb H37RvΔRv3484 to antibiotics and lysozyme
The resazurin assay was used to determine the susceptibility to antibiotics and certain stress conditions43,44. The strains were cultured in fully supplemented 7H9 medium until they reached the exponential growth phase. After adjusting bacterial suspensions to an OD600 of 0.3, inoculi were further diluted 1:20 before submitting 100 µl to the assay. Lysozyme or antibiotic dilutions (lysozyme 0, 0.094, 0.125, 0.188, 0.250, 0.375, 0.500, 0.750, 1 [mg/ml]; meropenem (+2.5 µg/ml clavulanate) 0, 0.04, 0.08, 0.16, 0.32, 0.63, 1.25, 2.5, 5 [µg/ml]; or as indicated in the supplementary information) were prepared in 100 µl volume in a 96-well microtiter plate (Corning, Corning, USA) and mixed with the bacterial suspensions. After 7 days of incubation at 37 °C 30 µl 0.01% (w/v) resazurin (Sigma-Aldrich, St. Louis, USA) was added to the wells. After further incubation at 37 °C o/n plates were read in a Biotek Synergy 2 plate reader using the Gen5 software (Biotek, Winooski, USA) and emission detected at 590 nm using an excitation wavelength of 540 nm or MICs were evaluated by eye and pictures were taken for documentation of negative results. Fluorescence reads were corrected for background fluorescence by subtracting values obtained from wells only containing dH2O (lysozyme) or medium (antibiotics) and resazurin for each plate. Data were plotted as background corrected (normalized) fluorescence reads. Experiments were performed in 5–11 independent replicates (lysozyme 5–9, meropenem/clavulanate 6, rifampicin 8–11) each designed in technical duplicates, in case no effect was observed at least 2 independent experiments in technical duplicates were performed and pictures of representative 96-well plates taken for documentation. Data were evaluated statistically using two-way ANOVA followed by the Bonferroni post-hoc test as indicated in the figure legends and p-values ≤ 0.05 (*), ≤0.01 (**), ≤0.001 (***) and ≤0.0001 (****) were considered as significant. GraphPad Prism software (GraphPad, La Jolla, USA) was used for plotting and statistical analysis of the data. Adobe Photoshop CS5 (Adobe Systems, San Jose, USA) was used to crop images which, if they were adjusted, only as whole pictures.
In vivo infection experiments
10–12 week old female C57BL/6 mice (Charles River, Sulzfeld, Germany) were infected via aerosol (Glas-Col, Terre-Haute, Indiana, USA) with approximately 100 CFU of each Mtb strain. The actual infection dose was confirmed one day p.i. by plating lung homogenates on 7H10 agar plates supplemented with glycerol and 10% bovine serum (Biowest, Nuaillé, France). At further time points (21, 42, 90 and 180 days p.i.) lung, liver and spleen were removed and dilutions of homogenates plated on 7H10 agar plates to determine the bacterial load (2 lobes of the lungs, ca. ½ of liver and spleen) and to assess the pathology based on hematoxylin and eosin (HE) stained sections of fixed and paraffin embedded organs. The detection limit was defined for samples in which at least for one sample no CFU could be detected by plating 100 µl organ homogenate. In this case, raw CFU counts were set to 0.99, the theoretical CFU/organ calculated and defined as detection limit. The experiments were approved by the Ministry of Energy, Agriculture, the Environmental and Rural Areas of the state Schleswig-Holstein, Germany, and comply with the legal requirements of the German Animal Protection law.
References
Global tuberculosis report 2015.
Marais, B. J. et al. Epidemic Spread of Multidrug-Resistant Tuberculosis in Johannesburg, South Africa. J. Clin. Microbiol. 51, 1818–1825 (2013).
Jackson, M., McNeil, M. R. & Brennan, P. J. Progress in targeting cell envelope biogenesis in Mycobacterium tuberculosis. Future Microbiol. 8, 855–875 (2013).
Homolka, S., Niemann, S., Russell, D. G. & Rohde, K. H. Functional Genetic Diversity among Mycobacterium tuberculosis Complex Clinical Isolates: Delineation of Conserved Core and Lineage-Specific Transcriptomes during Intracellular Survival. PLoS Pathog. 6 (2010).
Marchler-Bauer, A. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 (2017).
Lazarevic, V., Margot, P., Soldo, B. & Karamata, D. Sequencing and analysis of the Bacillus subtilis lytRABC divergon: A regulatory unit encompassing the structural genes of the N-acetylmuramoyl-L-alanine amidase and its modifier. J. Gen. Microbiol. 138, 1949–1961 (1992).
Johnsborg, O. & Håvarstein, L. S. Pneumococcal LytR, a Protein from the LytR-CpsA-Psr Family, Is Essential for Normal Septum Formation in Streptococcus pneumoniae. J. Bacteriol. 191, 5859–5864 (2009).
Chatfield, C. H., Koo, H. & Quivey The putative autolysin regulator LytR in Streptococcus mutans plays a role in cell division and is growth-phase regulated. Microbiology 151, 625–631 (2005).
Guidolin, A., Morona, J. K., Morona, R., Hansman, D. & Paton, J. C. Nucleotide sequence analysis of genes essential for capsular polysaccharide biosynthesis in Streptococcus pneumoniae type 19F. Infect. Immun. 62, 5384–5396 (1994).
Cieslewicz, M. J., Kasper, D. L., Wang, Y. & Wessels, M. R. Functional Analysis in Type Ia Group B Streptococcus of a Cluster of Genes Involved in Extracellular Polysaccharide Production by Diverse Species of Streptococci. J. Biol. Chem. 276, 139–146 (2001).
Hanson, B. R., Lowe, B. A. & Neely, M. N. Membrane Topology and DNA-Binding Ability of the Streptococcal CpsA Protein. J. Bacteriol. 193, 411–420 (2011).
Rossi, J., Bischoff, M., Wada, A. & Berger-Bächi, B. MsrR, a Putative Cell Envelope-Associated Element Involved in Staphylococcus aureus sarA Attenuation. Antimicrob. Agents Chemother. 47, 2558–2564 (2003).
Chan, Y. G. Y., Frankel, M. B., Dengler, V., Schneewind, O. & Missiakas, D. Staphylococcus aureus Mutants Lacking the LytR-CpsA-Psr Family of Enzymes Release Cell Wall Teichoic Acids into the Extracellular Medium. J. Bacteriol. 195, 4650–4659 (2013).
Kawai, Y. et al. A widespread family of bacterial cell wall assembly proteins. EMBO J. 30, 4931–4941 (2011).
Chan, Y. G.-Y., Kim, H. K., Schneewind, O. & Missiakas, D. The Capsular Polysaccharide of Staphylococcus aureus Is Attached to Peptidoglycan by the LytR-CpsA-Psr (LCP) Family of Enzymes. J. Biol. Chem. 289, 15680–15690 (2014).
Schaefer, K., Matano, L. M., Qiao, Y., Kahne, D. & Walker, S. In vitro reconstitution demonstrates the cell wall ligase activity of LCP proteins. Nat. Chem. Biol. 13, 396–401 (2017).
Liszewski Zilla, M., Chan, Y. G. Y., Lunderberg, J. M., Schneewind, O. & Missiakas, D. LytR-CpsA-Psr Enzymes as Determinants of Bacillus anthracis Secondary Cell Wall Polysaccharide Assembly. J. Bacteriol. 197, 343–353 (2015).
Wu, C. et al. Lethality of sortase depletion in Actinomyces oris caused by excessive membrane accumulation of a surface glycoprotein: Bacterial cell killing by glyco-stress. Mol. Microbiol. 94, 1227–1241 (2014).
Burkovski, A. Cell Envelope of Corynebacteria: Structure and Influence on Pathogenicity. ISRN Microbiol. 2013, 1–11 (2013).
Angala, S. K., Belardinelli, J. M., Huc-Claustre, E., Wheat, W. H. & Jackson, M. The cell envelope glycoconjugates of Mycobacterium tuberculosis. Crit. Rev. Biochem. Mol. Biol. 49, 361–399 (2014).
Jankute, M., Cox, J. A. G., Harrison, J. & Besra, G. S. Assembly of the Mycobacterial Cell Wall. Annu. Rev. Microbiol. 69, 405–423 (2015).
Wang, Q. et al. CpsA, a LytR-CpsA-Psr Family Protein in Mycobacterium marinum, Is Required for Cell Wall Integrity and Virulence. Infect. Immun. 83, 2844–2854 (2015).
Grzegorzewicz, A. E. et al. Assembling of the Mycobacterium tuberculosis Cell Wall Core. J. Biol. Chem. jbc.M116.739227, https://doi.org/10.1074/jbc.M116.739227 (2016).
Harrison, J. et al. Lcp1 Is a Phosphotransferase Responsible for Ligating Arabinogalactan to Peptidoglycan in Mycobacterium tuberculosis. mBio 7, e00972–16 (2016).
Nakayama, G. R., Caton, M. C., Nova, M. P. & Parandoosh, Z. Assessment of the Alamar Blue assay for cellular growth and viability in vitro. J. Immunol. Methods 204, 205–208 (1997).
Franzblau, S. G. et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis 92, 453–488 (2012).
Vandevelde, N. M., Tulkens, P. M. & Van Bambeke, F. Antibiotic Activity against Naive and Induced Streptococcus pneumoniae Biofilms in an In Vitro Pharmacodynamic Model. Antimicrob. Agents Chemother. 58, 1348–1358 (2014).
Collins, L. & Franzblau, S. G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 41, 1004–1009 (1997).
Banfi, E. Development of a microdilution method to evaluate Mycobacterium tuberculosis drug susceptibility. J. Antimicrob. Chemother. 52, 796–800 (2003).
Coban, A. Y., Deveci, A., Sunter, A. T., Palomino, J. C. & Martin, A. Resazurin microtiter assay for isoniazid, rifampicin, ethambutol and streptomycin resistance detection in Mycobacterium tuberculosis: Updated meta-analysis. Int. J. Mycobacteriology 3, 230–241 (2014).
Noncommercial Culture and Drug-Susceptibility Testing Methods for Screening Patients at Risk for Multidrug-Resistant Tuberculosis: Policy Statement. (World Health Organization, 2011).
Flores, A. R. Genetic analysis of the β-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to β-lactam antibiotics. Microbiology 151, 521–532 (2005).
Hugonnet, J.-E., Tremblay, L. W., Boshoff, H. I., Barry, C. E. & Blanchard, J. S. Meropenem-Clavulanate Is Effective Against Extensively Drug-Resistant Mycobacterium tuberculosis. Science 323, 1215–1218 (2009).
Hershberg, R. et al. High Functional Diversity in Mycobacterium tuberculosis Driven by Genetic Drift and Human Demography. PLoS Biol 6, e311 (2008).
Comas, I. et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 42, 498–503 (2010).
Wirth, T. et al. Origin, Spread and Demography of the Mycobacterium tuberculosis Complex. PLoS Pathog 4, e1000160 (2008).
Reiling, N. et al. Clade-specific virulence patterns of Mycobacterium tuberculosis complex strains in human primary macrophages and aerogenically infected mice. mBio 4 (2013).
Cambier, C. J. et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 505, 218–222 (2013).
Källenius, G., Correia-Neves, M., Buteme, H., Hamasur, B. & Svenson, S. B. Lipoarabinomannan, and its related glycolipids, induce divergent and opposing immune responses to Mycobacterium tuberculosis depending on structural diversity and experimental variations. Tuberculosis 96, 120–130 (2016).
Baumgart, M., Schubert, K., Bramkamp, M. & Frunzke, J. Impact of LytR-CpsA-Psr Proteins on Cell Wall Biosynthesis in Corynebacterium glutamicum. J. Bacteriol. 198, 3045–3059 (2016).
Pavelka, M. S. & Jacobs, W. R. Comparison of the Construction of Unmarked Deletion Mutations in Mycobacterium smegmatis, Mycobacterium bovis Bacillus Calmette-Guérin, and Mycobacterium tuberculosis H37Rv by Allelic Exchange. J. Bacteriol. 181, 4780–4789 (1999).
Stover, C. K. et al. New use of BCG for recombinant vaccines. Nature 351, 456–460 (1991).
Palomino, J.-C. et al. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 46, 2720–2722 (2002).
Yajko, D. M. et al. Colorimetric method for determining MICs of antimicrobial agents for Mycobacterium tuberculosis. J. Clin. Microbiol. 33, 2324–2327 (1995).
Acknowledgements
We thank Dr. Bianca Schneider, Research Center Borstel – Leibniz Center for Medicine and Biosciences, for her support. We thank Dr. Nicolas Gisch, Research Center Borstel – Leibniz Center for Medicine and Biosciences for fruitful discussions about bioanalytical chemistry. In addition, we acknowledge excellent assistance of the technical staff of the Molecular and Experimental Mycobacteriology of the Research Center Borstel – Leibniz Center for Medicine and Biosciences. This work was funded by the DFG grant number NI 1131/4-1.
Author information
Authors and Affiliations
Contributions
Sven Malm conceived the study and experiments, performed experiments, analyzed and interpreted the data, wrote the paper. S.M. performed experiments, S.E., U.E.S. and S.N. conceived the study and wrote the paper. All authors reviewed the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Malm, S., Maaß, S., Schaible, U.E. et al. In vivo virulence of Mycobacterium tuberculosis depends on a single homologue of the LytR-CpsA-Psr proteins. Sci Rep 8, 3936 (2018). https://doi.org/10.1038/s41598-018-22012-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22012-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
