
Abstract
Although many genome-wide association studies (GWASs) of hyperuricemia or gout have been reported, the related genetic factors and the mechanisms from hyperuricemia to gouty attack remain unclear. This study aimed to identify genetic factors and pathogenesis of gout from hyperuricemia by genome-wide association study (GWAS). 747 gout patients, 747 hyperuricemia and 2071 age-matched controls were recruited and analyzed with Affymetrix 650 K chip to find the related genetic variants. The functions of the related genes were investigated in an endothelial cell (EC) with urate crystal stimulation. The GWAS results showed 36 SNPs to be strongly associated with gout compared to controls (all p-values < 10−7). Whereas the rs2231142 in ABCG2 gene had significant associations between gout and controls, between gout and hyperuricemia, and between hyperuricemia and controls (all p-values < 10−7), and the ORs were 4.34, 3.37 and 2.15 (all p-values < 0.001) after adjustment of potential confounders, respectively. The cell model showed significantly higher IL-8 release from EC combined with ABCG2 knockdown. We concluded that ABCG2 gene contributed to hyperuricemia but also gout, and that it was involved in the inflammation dysregulation via augmented IL-8 release in EC.
Similar content being viewed by others


Introduction
Gout is a common form of inflammatory arthritis characterized by recurrent attacks of acute inflammatory arthritis caused by monosodium urate (MSU) deposition and inflammation dysregulation. Since serum urate levels (SUA) and gout are heritable1, and most cases with hyperuricemia are asymptomatic, the genetic component of pathogenic mechanism remains unclear. Many genes are involved in urate transporter; SLC22A122, SLC2A93,4 and ABCG25,6,7 have been reported to play important roles in the regulation of SUA, and their dysfunctions cause aberrant urate transport disorders leading to hyperuricemia. Hyperuricemia and gouty arthritis have different pathogenic mechanisms. A number of genes have been reported only to be associated with gouty inflammation or urate phagocytosis, but not to be related to hyperuricemia, such as tumor necrosis factor alpha (TNF-α), toll-like receptor II (TLR-2), NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome and type 2 cyclic GMP-dependent protein kinase (cGKII)8,9,10,11. However, the related genes of pathogenic processes from hyperuricemia to gouty inflammation are also unclear.
Genome-wide association studies (GWASs) have explored many genes associated with serum uric acid levels or gout, for instance, SLC2A9, SLC2A12, SLC22A1212,13 for urate transporter, and ABCG2, SLC2A9, BCAS3, RFX3, KCNQ1, SLC22A12 and SLC17A1 for gout disease with individuals of Asian or European descent14,15,16,17,18. However, common variants identified by GWASs associated both with serum urate levels and gout were reported only in individuals of European ancestry19,20,21 and in a Japanese population14. Because most gout-related genes were also associated with hyperuricemia, in the present study we intended to identify the risk loci related to gout from the status of hyperuricemia, and then investigated their function in a cell model.
Results
In our study, 747 gout patients, 747 male individuals with hyperuricemia and 2071 male normal controls were included from the Taiwan biobank database which provided gout history, uric acid, creatinine, biochemical markers, demographic and Affymetrix TWB 650 K SNP chip data. The mean ages of gout patients, hyperuricemia patients and controls were 50.29 years (±10.49), 49.19 years (±10.73) and 49.72 years (±11.11), respectively, which did not show any significant difference (p = 0.149; Table 1). Uric acid, creatinine, triglycerides, fasting sugar, total cholesterol, high-density lipoprotein cholesterol (HDL-c), low-density lipoprotein cholesterol (LDL-c), body mass index (BMI), body fat rate, GOT and GPT showed higher significant differences among these three groups (all p-values < 0.010; Table 1). The aforementioned variables were then taken as potential confounders for further analysis. For the GWAS analysis, the results showed that 36 SNPs located on chromosome 4 were significantly associated with gout disease compared to normal control (Supplementary Table 1), and one SNP (polymorphism rs2231142) showed significant associations between gout and hyperuricemia, and between hyperuricemia and normal controls (all p-values < 1 × 10−7). As known that data overfitting is always a challenge in biomarker discovery22, therefore we did a re-sampling to perform a non-matched design to analysis the related gene markers for gout. The result showed a total of 37 SNPs were related to gout while compared to normal controls (Supplementary Figure 1), ie, there were 36 SNPs between these two designs showed the same significant association (p < 10−7). All these significant SNPs passed 1% of false discovery rate (FDR) for association with gout or hyperuricemia.
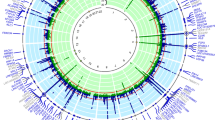
The Manhattan plot displayed all p-values of SNPs across 23 pairs and mitochondria chromosomes and showed the genes on chromosome 4 were the hot spot for gout-susceptible genes (Fig. 1A). Figure 1B shows the p-values after negative logarithm transformation for the SNPs of gout compared to normal control in chromosome 4. It shows two spots positioned near in the region of 1 × 107 base-pair and in the region between 8 × 107 and 9 × 107 bases. The latter region revealed that ABCG2 gene was the major gene contributing to gout disease, and MEPE, SPP1, and PKD2 were also involved. However, the first region consisted of four non-redundant genes encoded around rs9999470 within one mega-bases (1 Mb) and the expression quantitative trait loci (eQTL) analysis revealed higher association between the expressions of SLC2A9 and polymorphism rs9999470, but not WDR1, ZNF518B or CLNK genes (Supplementary Figure 2).

The Manhattan plot displays p-values of SNPs from 23 pairs of chromosomes and mitochondria and shows the genes on chromosome 4 were the hot spot for gout-susceptible genes (A). The log (1/p) of SNPs associated with gout compared to normal control in chromosome 4 (B). It showed two spots positioned near in 1 × 107 (SLC2A9 gene) and in region of 8 × 107 and 9 × 107 near or in MEPE, SPP1 PKD2 and ABCG2 genes involved in gout occurrence.
A linkage disequilibrium test was further performed to eliminate redundant SNPs with genetic associations during genetic recombination (Fig. 2). Since the polymorphism rs2231142 in ABCG2 gene showed most significant association with gout or hyperuricemia, we displayed the LD according to different rs2231142 genotypes. Figure 2A shows the LD association between gout and normal control ignoring rs2231142 genotypes, while Fig. 2-B1, B2 and B3 display the same LD maps with different rs2231142 genotypes in GG, GT and TT, respectively.

The linkage disequilibrium (LD) maps. Figure A is the result of comparing gout to normal control ignoring rs2231142 genotype, while Figure B1, B2 and B3 displays the same LD maps with different rs2231142 genotypes, namely GG, GT, and TT, respectively. Parenthesis indicates the gene name and position. Green line blocks the SNPs of the same genes.
Concerning the effect of polymorphism rs2231142 with different genotypes on the occurrence of gout disease from hyperuricemia, the OR was 1.84 (95% C I = 1.45–2.34) in genotype GT of rs2231142, and increased to 3.37 in genotype TT as compared to genotype GG after adjustment of potential confounders (Table 2). The same phenomenon of increased OR also was observed in comparisons of between gout and controls as well as between hyperuricemia and controls (Table 2). Moreover, the polymorphism rs9999470 showed weak association effect for the developments of gout and hyperuricemia after adjustment of potential confounders (Table 2).
Furthermore, since polymorphism rs2231142 provided the highest association with development of gout disease, we performed another analysis for polymorphism rs2231142 with representative SNPs of the other four genes (Table 3). The polymorphism rs9999470 showed a significant association with polymorphism rs2231142 on gout occurrence; it showed that those with rs9999470 genotypes CT and CC interacted with rs2231142 genotype GT to increase gout risk in a dose-dependent manner. ORs increased from 2.07 to 2.67 and 4.94 in genotypes TT, CT and CC of rs9999470 among those with rs2231142 genotype GT after adjusting potential confounders, respectively (all p- values < 0.001; Table 3). Moreover, in those with rs2231142 genotype TT, the associated effect after adjusting potential confounders was clearer; the ORs increased from 4.24 to 4.64 and 21.32 (all p- values < 0.001; Table 3). An additive interaction effect was observed in those combined with genotype CC of rs9999470 and genotypes GT and TT of rs2231142 (p < 0.05; Table 3). This implied that polymorphism rs9999470 acted as an additive associated factor for developing gout, which was dependent on the genotypes of polymorphism rs2231142.
Our study found that ABCG2 contributed not only to hyperuricemia but also to gouty inflammation. Previously, some studies demonstrated that ABCG2 contributed to hyperuricemia, which was caused by renal urate overload and intestinal urate under-excretion23,24,25,26. In addition, ABCG2 knockout mice showed increased inflammatory responses, including increased IL-8 release in the brain via regulating NF-κB activation27. Therefore, we performed cell study to validate ABCG2-associated inflammatory responses instead of urate excretion, IL-1β and IL-8 cytokines release with MSU crystals stimulation. PMA-primed THP-1 (under a macrophage M1-like inflammatory stage) and EC line EA. HY296 were used with and without ABCG2 gene knockdown (ABCG2 RNAi). The results showed that IL-8 release was significantly higher from ABCG2 knockdown EA. HY296 cell after urate crystal stimulation (p < 0.05), but this was not observed in PMA-primed THP-1 cells (Fig. 3A). Meanwhile, the IL-1β release did not show significantly increases in either THP-1 or EA. HY296 cells even under the condition of ABCG2 knockdown or urate crystal stimulation (both p-values > 0.05, Fig. 3B). These data suggested that ABCG2 dysfunction (knockdown) promoted MSU-induced IL-8 release from EC, but not in M1-like macrophages. Furthermore, with a combination of THP-1 and EA. HY296, the results showed the releases of both IL-8 and IL-1β were significantly increased after urate crystal stimulation and ABCG2 knockdown (both p-values < 0.05, Fig. 3C,D). These results implied that, under conditions of ABCG2 knockdown and MSU crystals-stimulation, the release of IL-8 from EC (EA. HY296) was able to enhance inflammatory cytokine IL-1β release with the involvement of macrophages (THP-1). Overall, the cell model showed that ABCG2 knockdown increased the MSU crystals-induced inflammation responses through IL-8 release in EC.

PMA-primed THP-1 and endothelial cell line EA. HY296 were used under conditions of with or without ABCG2 gene knockdown (ABCG2 RNAi). It showed IL-8 released was significantly higher from ABCG2 knockdown EA. HY296 cell after urate stimulation (p < 0.05), but not in PMA-primed THP-1 cells (p > 0.05) (A). The IL-1β release did not show significant increases either in THP-1 or EA. HY296 cells even under the condition of ABCG2 knockdown or urate stimulation (both p-values > 0.05) (B). A co-culture of THP-1 and EA. HY296 showed the releases of both IL-8 and IL-1β were significantly increased after urate stimulation and ABCG2 knockdown (both p-values < 0.05) (C and D). *p < 0.05.
Discussion
This study demonstrates that ABCG2 gene contributes to the development of not only hyperuricemia but also gout with solid evidence from the GWAS and endothelial cell model. Moreover, this might be the first research to clarify the mechanism of ABCG2 leading to gouty inflammation involving the release of IL-8 upon MSU crystals-stimulation in EC. Previously, many studies showed that ABCG2 played an important role in renal urate overload and extra-renal urate under-excretion, especially intestinal excretion23,24,28,29,30. However, the development of gout needs both hyperuricemia and aberrant inflammatory mechanism, possibly involving IL-1β, TNF-α and IL-8, etc. In our knowledge, few previous studies determined whether ABCG2 gene dysfunction could be associated with aberrant production of inflammatory cytokines in gout. Shen et al. showed that ABCG2 gene could relieve oxidative stress and inflammatory response via inhibiting NF-κB signaling pathway in cell models and brain27, and recently oxidative stress has proved to be a key for gouty arthropathy31,32. Our current study showed that ABCG2 knockdown induced increase of IL-8 release after urate crystal stimulation. This finding suggested that ABCG2 dysfunction contributed to gouty inflammation.
The mechanism causing acute inflammation in gout patients initiates from MSU crystal deposition, and then MSU interacts with macrophages to activate NLRP3 inflammasome and IL-1β release. Once IL-1β is released, it orchestrates a series of events leading to EC activation and neutrophils recruitment. Upon EC activation, several cytokines are released, particularly IL-8, which is the key for neutrophils recruitment. Nishimura et al. provided evidence that IL-8 release was related to MSU crystal-induced gouty arthritis33. Gagne et al. demonstrated that reduced myeloid inhibitory C-type lectin (MICL) expression was associated with augmented inflammatory responses from neutrophils, and that a higher level of neutrophil MICL expression was associated with reduced IL-8 production34,35. Therefore, the molecular basis for IL-8 regulation in EC may be the key to clarifying the mechanisms of gout flare. Here, we propose that ABCG2 gene may play an important role in release of IL-8 for further neutrophilic recruitment. Therefore, in case of ABCG2 dysfunction, the subjects have hyperuricemia first by renal urate overload and intestinal urate under-excretion; and then increased release of IL-1β (by macrophages) and IL-8 (by ECs) invoke further neutrophilic recruitment and gouty flare.
The polymorphism rs2231142 in ABCG2 gene, which causes a Glu141Lys amino acid substitution, accounted for 0.57% of the variation in serum urate36, and a functional study of the rs2231142 has shown that it causes a 53% reduction in the rate of ABCG2-mediated urate transport compared with wild-type5,37. Even though polymorphism rs2231142 acted as a risk factor for gout development, our study also found that polymorphism in SLC2A9 gene revealed associated effect dependent on rs2231142 genotypes. For example, polymorphisms rs9999470 which regulates SLC2A9 function showed an additive significant association with gout in those with polymorphism rs2231142 genotype TT. SLC2A9 coding for the glucose-facilitated transporter GLUT936, some studies showed renal hypouricemia was caused by dysfunction in the SLC2A9 gene via its decreased urate reabsorption on the renal proximal tubules3,4. Here, our study demonstrates that those with mutants in both ABCG2 and SLC2A9 genes cause an additive interaction effect for gout occurrence.
This study also explores whether PKD2 genes may interact with rs2231142 genotypes in the development of gout. Polymorphism rs10516801 in PKD2 gene showed risk effects in those with rs2231142 genotypes GT or TT. PKD2 gene encodes a member of the polycystin protein family; the protein is an integral membrane protein involved in cell-cell/matrix interactions, which may function in renal tubular development, morphology and function, and modulate intracellular calcium homoeostasis and other signal transduction pathways38. Lee et al. applied pathway analysis to GWAS data on uric acid levels and found PKD2 contributed to uric acid levels by affecting ion transmembrane transporter activity39. According to the Genecards, PKD2 is not only expressed in kidney but also in white blood cells, especially in B cells. Surprisingly, the role of B lymphocytes related to gout development has only recently been recognized40,41, it may be involved in the gouty inflammation mediated by B cells.
In conclusion, based upon GWAS of gout and hyperuricemia simultaneously, we revealed that ABCG2 gene contributed to the development of not only hyperuricemia but also gout, affecting the IL-8 release from EC to cause dysregulation of inflammation and the development of gouty attack.
Methods
Study population
Our study aimed to provide risk genome components of developing gout and hyperuricemia based on GWAS. We recruited 747 male gout patients, 747 male individuals with hyperuricemia (uric acid levels > = 7.0 mg/dl; without gout disease history) and 2071 male normal controls (with neither gout nor hyperuricemia) from the Taiwan biobank database which provided gout history, uric acid, creatinine, biochemical, demographic and Affymetrix TWB 650 K SNP chip data.
The cohort was selected based on community population initiated by Academia Sinica in Taiwan from 2012, which collect the DNA specimen of a large group to understand the relationships between genetics, environmental exposure and the etiology/progression of disease, including gout, diabetes, and hypertension. Enrollees of the Taiwan biobank have undergone physical examination, and provided blood and urine samples for biochemical test and detailed health information about their lifestyles and gout disease history for further analysis. Gout status was obtained by self-report, which has been evidenced to be the best test performance characteristics of existing definitions with sensitivity 80% and specificity 72%42. This research project was approved by the ethics committee of Antai-Tian-Sheng memorial Hospital Institutional Review Board (TSMHIRB 16-006-C0). The study was conducted in accordance with the principles of the Declaration of Helsinki and the Good Clinical Practice Guidelines, and all the participants were informed consent.
SNP genotyping and quality control
Blood DNA samples from Taiwan biobank participants were genotyped using TWB 650 K SNPs array designed by Affymetrix (Affymetrix, Santa Clara, California, USA) for selected SNPs43. For TWB SNPs array, we used the Affymetrix Power Tools (APT) and performed a standard quality control procedure to exclude SNPs with low call rate (<99%), p value for the Hardy-Weinberg equilibrium test of <1.0 × 10−4 for controls and minor allele frequency of <0.01. In total, we obtained 537,478 SNPs for analysis in the screening stage.
Cell culture
EA. HY296, an immortalized human endothelial cell (EC), was cultured in DMEM medium with 10% FBS at 37 °C in a humidified incubator with 5% CO2 atmosphere, with or without 7.0 mg/dl of monosodium urate crystals (MSU; Sigma, St. Louis, MO) treatment for 48 hours. THP-1, a human acute monocytic leukemia cell line, was cultured in RPMI 1640 medium with 10% FBS at 37 °C in a humidified incubator with 5% CO2 atmosphere. THP-1 monocyte has the potential to polarize into macrophage-like cells and functionally regulates purified T cells44. We treated THP-1 with 0.2 μg/ml of phorbol myristate acetate (PMA; Sigma, St. Louis, MO) for 3 days to activate it to a macrophage-like (M1-like) phenotype. The M1-like THP-1 cells were treated with 7.0 mg/dl of MSU crystals (Sigma, St. Louis, MO) for 48 hours.
ABCG2 siRNA synthesis and transfection
Using the siGENOME SMARTpool system, human ABCG2 siRNA was chemically synthesized and one tube containing a mixture of SMART selection-designed siRNA targeting gene for in-vitro transfection was prepared. The siGENOME non-targeting siRNA pool was also performed in the same way (Thermo, Dharmacon, Inc. DBA). EA. HY296 or THP-1 cells were seeded into 24-well culture plates and transfected with 100 nM of siRNA per well using XfectTM siRNA transfection reagent (Clontech, CA, USA), according to the manufacturer’s instruction. Cells were analyzed at 48 hours after transfection.
ELISA tests for interleukin-8 (IL-8) and IL-1β
We selected IL-8 and IL-1β as detection targets based on the identified gouty inflammatory factors. After treatment, the cell-free supernatant from cultured EA. HY296 or THP-1 cells was obtained for IL-8 and IL-1β release measurement using commercially available ELISA kits (Quantikine, R&D System, Minneapolis, MN) according to the manufacturer’s instructions. IL-8 and IL-1β cytokine concentrations were calculated based on linear regression standard curves where r2 was higher than 0.99. The detection ranges were 1.6–1000 pg/ml for ELISA kits.
Statistical and expression quantitative trait loci (eQTL) analysis
The associations of SNPs for gout or hyperuricemia occurrence in the GWAS were tested by chi-square tests, odds ratios (ORs) and 95% confident intervals (95% CI), and a Manhattan Plot was made to show the p-values across all the 22 pairs, x, y and mitochondria chromosomes. A false discovery rate (FDR) was applied for analysis of further association with gout, and a linkage-disequilibrium (LD) test was drawn using the R program (LDheatmap). A one-way analysis of variance (ANOVA) was applied to estimate the mean differences of demographic and biochemical data among those gout patients, hyperuricemia and controls. A logistic regression model was applied to estimate the associations between gout phenotype and genetic variants after adjusting potential variables, such as age, uric acid, creatinine, body mass index (BMI), body fat rate, total cholesterol, triglycerides, high-density lipoprotein cholesterol (HDL-c), low-density lipoprotein cholesterol (LDL-c), serum glutamic oxaloacetic transaminase (GOT), serum glutamic pyruvic transaminase (GPT) and fasting glucose. The genotypes associated with genes expression in whole-blood samples were retrieved from genotype-tissue expression (GTEx) Portal V6p (www.gtexportal.org; eQTL), and statistical significance was determined using the website’s algorithm25. Significance after Bonferroni correction for multiple testing was considered for p values less than 1 × 10−7 in the part of GWAS, and it was 0.05 in the analysis of SNPs with demographic data.
References
Yang, Q. et al. Genome-wide search for genes affecting serum uric acid levels: the Framingham Heart Study. Metabolism 54, 1435–1441, https://doi.org/10.1016/j.metabol.2005.05.007 (2005).
Enomoto, A. et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 417, 447–452, https://doi.org/10.1038/nature742 (2002).
Matsuo, H. et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet 83, 744–751, https://doi.org/10.1016/j.ajhg.2008.11.001 (2008).
Dinour, D. et al. Homozygous SLC2A9 mutations cause severe renal hypouricemia. J Am Soc Nephrol 21, 64–72, https://doi.org/10.1681/ASN.2009040406 (2010).
Woodward, O. M. et al. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci USA 106, 10338–10342, https://doi.org/10.1073/pnas.0901249106 (2009).
Matsuo, H. et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci Transl Med 1, 5ra11, https://doi.org/10.1126/scitranslmed.3000237 (2009).
Matsuo, H. et al. Common dysfunctional variants in ABCG2 are a major cause of early-onset gout. Sci Rep 3, 2014, https://doi.org/10.1038/srep02014 (2013).
Cai, Y. et al. Association of Toll-like receptor 2 polymorphisms with gout. Biomed Rep 2, 292–296, https://doi.org/10.3892/br.2014.224 (2014).
Kingsbury, S. R. et al. The role of the NLRP3 inflammasome in gout. J Inflamm Res 4, 39–49, https://doi.org/10.2147/jir.s11330 (2011).
Chang, S. J. et al. The polymorphism -863C/A in tumour necrosis factor-alpha gene contributes an independent association to gout. Rheumatology (Oxford) 46, 1662–1666, https://doi.org/10.1093/rheumatology/kem235 (2007).
Liao, W. T. et al. Cyclic GMP-dependent protein kinase II is necessary for macrophage M1 polarization and phagocytosis via toll-like receptor 2. J Mol Med (Berl) 93, 523–533, https://doi.org/10.1007/s00109-014-1236-0 (2015).
Voruganti, V. S. et al. Genome-wide association analysis confirms and extends the association of SLC2A9 with serum uric acid levels to Mexican Americans. Front Genet 4, 279, https://doi.org/10.3389/fgene.2013.00279 (2013).
Tin, A. et al. Genome-wide association study for serum urate concentrations and gout among African Americans identifies genomic risk loci and a novel URAT1 loss-of-function allele. Hum Mol Genet 20, 4056–4068, https://doi.org/10.1093/hmg/ddr307 (2011).
Kamatani, Y. et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet 42, 210–215, https://doi.org/10.1038/ng.531 (2010).
Li, C. et al. Genome-wide association analysis identifies three new risk loci for gout arthritis in Han Chinese. Nat Commun 6, 7041, https://doi.org/10.1038/ncomms8041 (2015).
Nakayama, A. et al. GWAS of clinically defined gout and subtypes identifies multiple susceptibility loci that include urate transporter genes. Ann Rheum Dis 76, 869–877, https://doi.org/10.1136/annrheumdis-2016-209632 (2016).
Sulem, P. et al. Identification of low-frequency variants associated with gout and serum uric acid levels. Nat Genet 43, 1127–1130, https://doi.org/10.1038/ng.972 (2011).
Kottgen, A. et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet 45, 145–154, https://doi.org/10.1038/ng.2500 (2013).
Yang, Q. et al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet 3, 523–530, https://doi.org/10.1161/CIRCGENETICS.109.934455 (2010).
Vitart, V. et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet 40, 437–442, https://doi.org/10.1038/ng.106 (2008).
Dehghan, A. et al. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet 372, 1953–1961, https://doi.org/10.1016/S0140-6736(08)61343-4 (2008).
Li, J. et al. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat Commun 1, 34, https://doi.org/10.1038/ncomms1033 (2010).
Matsuo, H. et al. Hyperuricemia in acute gastroenteritis is caused by decreased urate excretion via ABCG2. Sci Rep 6, 31003, https://doi.org/10.1038/srep31003 (2016).
Matsuo, H. et al. ABCG2 dysfunction increases the risk of renal overload hyperuricemia. Nucleosides Nucleotides Nucleic Acids 33, 266–274, https://doi.org/10.1080/15257770.2013.866679 (2014).
Consortium, G. T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660, https://doi.org/10.1126/science.1262110 (2015).
Dong, Z. et al. Effects of multiple genetic loci on the pathogenesis from serum urate to gout. Sci Rep 7, 43614, https://doi.org/10.1038/srep43614 (2017).
Shen, S. et al. ABCG2 reduces ROS-mediated toxicity and inflammation: a potential role in Alzheimer’s disease. J Neurochem 114, 1590–1604, https://doi.org/10.1111/j.1471-4159.2010.06887.x (2010).
Nagura, M. et al. Uric acid metabolism of kidney and intestine in a rat model of chronic kidney disease. Nucleosides Nucleotides Nucleic Acids 35, 550–558, https://doi.org/10.1080/15257770.2016.1163379 (2016).
Kannangara, D. R. et al. Hyperuricaemia: contributions of urate transporter ABCG2 and the fractional renal clearance of urate. Ann Rheum Dis 75, 1363–1366, https://doi.org/10.1136/annrheumdis-2015-208111 (2016).
Takada, T. et al. ABCG2 dysfunction increases serum uric acid by decreased intestinal urate excretion. Nucleosides Nucleotides Nucleic Acids 33, 275–281, https://doi.org/10.1080/15257770.2013.854902 (2014).
Zamudio-Cuevas, Y. et al. Molecular basis of oxidative stress in gouty arthropathy. Clin Rheumatol 34, 1667–1672, https://doi.org/10.1007/s10067-015-2933-y (2015).
Kim, S. K. et al. Rebamipide Suppresses Monosodium Urate Crystal-Induced Interleukin-1beta Production Through Regulation of Oxidative Stress and Caspase-1 in THP-1 Cells. Inflammation 39, 473–482, https://doi.org/10.1007/s10753-015-0271-5 (2016).
Nishimura, A. et al. Attenuation of monosodium urate crystal-induced arthritis in rabbits by a neutralizing antibody against interleukin-8. J Leukoc Biol 62, 444–449 (1997).
Gagne, V. et al. Modulation of monosodium urate crystal-induced responses in neutrophils by the myeloid inhibitory C-type lectin-like receptor: potential therapeutic implications. Arthritis Res Ther 15, R73, https://doi.org/10.1186/ar4250 (2013).
So, A. How to regulate neutrophils in gout. Arthritis Res Ther 15, 118, https://doi.org/10.1186/ar4316 (2013).
Kolz, M. et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet; 5, e1000504, https://doi.org/10.1371/journal.pgen.1000504 (2009).
Reginato, A. M. et al. The genetics of hyperuricaemia and gout. Nat Rev Rheumatol 8, 610–621, https://doi.org/10.1038/nrrheum.2012.144 (2012).
Koulen, P. et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 4, 191–197, https://doi.org/10.1038/ncb754 (2002).
Lee, Y. H. & Song, G. G. Pathway analysis of genome-wide association studies on uric acid concentrations. Hum Immunol 73, 805–810, https://doi.org/10.1016/j.humimm.2012.05.004 (2012).
Lai, S. & Zhou, X. Inflammatory cells in tissues of gout patients and their correlations with comorbidities. Open Rheumatol J 7, 26–31, https://doi.org/10.2174/1874312901307010026 (2013).
Kanevets, U. et al. A role of IgM antibodies in monosodium urate crystal formation and associated adjuvanticity. J Immunol 182, 1912–1918, https://doi.org/10.4049/jimmunol.0803777 (2009).
Dalbeth, N. et al. Survey Definitions of Gout for Epidemiologic Studies: Comparison With Crystal Identification as the Gold Standard. Arthritis Care Res (Hoboken) 68, 1894–1898, https://doi.org/10.1002/acr.22896 (2016).
Fan, C. T. et al. Taiwan Biobank: a project aiming to aid Taiwan’s transition into a biomedical island. Pharmacogenomics 9, 235–246, https://doi.org/10.2217/14622416.9.2.235 (2008).
Tsuchiya, S. et al. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer 26, 171–176 (1980).
Acknowledgements
This work was supported by the Ministry of Science and Technology in Taiwan (105-2634-F-390 -001).
Author information
Authors and Affiliations
Contributions
Chang S.J. and Chen C.J. conceived, designed and led the study. Chen C.J., Tseng C.C. and Yen J.H. interpreted the main findings. Chang S.J., Chen C.J. and Liao W.T. drafted the manuscript. Chang S.J. and Chang J.G. processed the bioinformatics/statistical analysis. Liao W.T. undertook the cell experiments. Chou W.C. and Chu H.W. processed eQTL analysis and interpreted the results.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, CJ., Tseng, CC., Yen, JH. et al. ABCG2 contributes to the development of gout and hyperuricemia in a genome-wide association study. Sci Rep 8, 3137 (2018). https://doi.org/10.1038/s41598-018-21425-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21425-7
This article is cited by
-
Polygenic risk score trend and new variants on chromosome 1 are associated with male gout in genome-wide association study
Arthritis Research & Therapy (2022)
-
Susceptibility genes of hyperuricemia and gout
Hereditas (2022)
-
A meta-analysis of genome-wide association studies using Japanese and Taiwanese has revealed novel loci associated with gout susceptibility
Human Cell (2022)
-
An atlas connecting shared genetic architecture of human diseases and molecular phenotypes provides insight into COVID-19 susceptibility
Genome Medicine (2021)
-
Pleiotropic effect of the ABCG2 gene in gout: involvement in serum urate levels and progression from hyperuricemia to gout
Arthritis Research & Therapy (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
