
Abstract
Somatostatin receptor targeting is considered the standard nuclear medicine technique for visualization of neuroendocrine tumors (NET). Since not all NETs over-express somatostatin receptors, the search for novel targets, visualizing these NETs, is ongoing. Many NETs, expressing low somatostatin receptor levels, express glucose-dependent insulinotropic polypeptide (GIP) receptors (GIPR). Here, we evaluated the performance of [Lys37(DTPA)]N-acetyl-GIP1-42, a newly synthesized GIP analogue to investigate whether NET imaging via GIPR targeting is feasible. Therefore, [Lys37(DTPA)]N-acetyl-GIP1-42 was radiolabeled with 111In with specific activity up to 1.2 TBq/µmol and both in vitro and in vivo receptor targeting properties were examined. In vitro, [Lys37(111In-DTPA)]N-acetyl-GIP1-42 showed receptor-mediated binding to BHK-GIPR positive cells, NES2Y cells and isolated islets. In vivo, both NES2Y and GIPR-transfected BHK tumors were visualized on SPECT/CT. Furthermore, co-administration of an excess unlabeled GIP1-42 lowered tracer uptake from 0.7 ± 0.2%ID/g to 0.6 ± 0.01%ID/g (p = 0.78) in NES2Y tumors and significantly lowered tracer uptake from 3.3 ± 0.8 to 0.8 ± 0.2%ID/g (p = 0.0001) in GIPR-transfected BHK tumors. In conclusion, [Lys37(111In-DTPA)]N-acetyl-GIP1-42 shows receptor-mediated binding in various models. Furthermore, both GIPR-transfected BHK tumors and NES2Y tumors were visible on SPECT/CT using this tracer. Therefore, [Lys37(111In-DTPA)]N-acetyl-GIP1-42 SPECT seems promising for visualization of somatostatin receptor negative NETs.
Similar content being viewed by others
Introduction
Targeting of peptide hormone receptors, expressed on tumors, is a valuable tool for in vivo molecular imaging and radionuclide therapy of a variety of neuroendocrine tumors (NET). Successful tumor targeting relies on over-expression of target receptors on cancer cells when compared to their expression levels in healthy tissues1,2. Nowadays, somatostatin receptor targeting is a standard procedure for NET detection3,4,5,6 and radionuclide therapy with 90Y- and 177Lu-labeled somatostatin analogues was proven beneficial to patients with NET7. However, not all NETs show elevated somatostatin receptor expression levels. Therefore, false negative results may occur in all NET types and somatostatin receptor imaging displays limited sensitivity for some types of NET, such as insulinomas (80–90% on 68Ga-DOTATATE PET)3,5,8,9,10 and medullary thyroid carcinoma (MTC) for which the detection rate is below 40–60%11.
A variety of alternative peptide receptors, such as cholecystokinin (CCK) receptors12, vasoactive intestinal peptide (VIP) receptors13 and glucagon-like peptide 1 (GLP-1) receptors (GLP-1R)14 are expressed on NET. Recent findings showed glucose-dependent insulinotropic polypeptide (GIP) receptor (GIPR) expression in gastric, duodenal and bronchial NET15,16. Furthermore, approximately 90% of somatostatin receptor negative NET are GIPR positive16. In 2013, Sherman et al. pointed out that GIPR is a promising target for NET imaging and therapy due to its favorable expression pattern17 and the feasibility of GIPR targeting was shown in a GIPR over-expressing tumor model using a truncated GIP1-30 analogue18.
GIP is secreted from the intestinal K cells after nutrient ingestion19 and together with GLP-1, it enhances glucose-induced insulin secretion upon receptor binding on pancreatic beta cells, the so-called incretin effect20,21. Since insulinomas are NETs derived from pancreatic beta cells, incretin receptors are expected to be ideal candidate receptors for insulinoma imaging. Indeed, GLP-1R targeting tracers, such as [68Ga]- or [111In]-labeled exendin, can be applied for non-invasive in vivo insulinoma detection22,23,24. However, since malignant insulinomas display differential GLP-1R and somatostatin receptor expression patterns25, detection rates of these tumors remain limited to 50% by scintigraphy. Interestingly, GLP-1R negative malignant insulinomas and a majority of somatostatin negative NETs express enhanced GIPR levels16, rendering this receptor an interesting target for NET and insulinoma imaging.
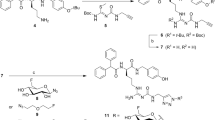
Since it was described that GIP1-30 exhibits reduced receptor binding affinity when compared to the full length peptide GIP1-4226,27, we have investigated the potential of a newly synthesized GIP1-42 analogue, [Lys37(111In-DTPA)]N-acetyl-GIP1-42 (Fig. 1) as a radiotracer for NET imaging, starting from the initial hypothesis that a full-length peptide-based tracer might show improved characteristics for in vivo NET imaging when compared to GIP1-30. Therefore, we optimized the radiolabeling procedure and investigated its binding and internalization kinetics using GIPR-positive tumor cells (BHK-GIPR). Furthermore, we have also explored the tracers binding characteristics to NES2Y cells (a human beta cell-derived cell line, representing a more realistic model in terms of receptor expression)28 and isolated islets of Langerhans. Finally, subcutaneous BHK-GIPR and NES2Y tumors were visualized by SPECT after injection of [Lys37(111In-DTPA)]N-acetyl-GIP1-42.

Amino acid sequence and molecular modifications of [Lys37(111In-DTPA)]N-acetyl-GIP1-42.
Results
Radiolabeling and Serum Stability
[Lys37(DTPA)]N-acetyl-GIP1-42 could be labeled with 111In with a specific activity (now referred to as molar activity29) of up to 1.2 TBq/µmol. Radiochemical purity exceeded 95% as determined by RP-HPLC and ITLC, resulting in a final molar activity exceeding 142.5 MBq/µg or 712.5MBq/nmol when starting with 150 MBq [111In]Cl3. Figure 2a shows the HPLC analysis of the labeling mixture. 111In-EDTA eluted with a retention time of 3 minutes, whereas 111In labeled [Lys37(DTPA)]N-acetyl-GIP1-42 had a retention time of 14 minutes. After 12 minutes, a very small impurity (<2%) eluted from the column. Since GIP is known to be prone to inactivation by dipeptidyl peptidase IV (DPP IV), the stability of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 was analyzed in human serum. The results of this stability analysis are shown in Fig. 2b and c. Up to 4 hours of incubation in human serum, the [Lys37(111In-DTPA)]N-acetyl-GIP1-42 remained intact. After 24 hours of incubation in human serum, 73% of the activity was still found as intact radiolabeled peptide, as determined by HPLC.

(a) Radiochemical purity analysis of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 as determined by HPLC, immediately after labeling. (b) HPLC profile of 111In-GIP after 24 hours of incubation in human serum. (c) Overview of stability behavior after various incubation times in human serum.
In vitro binding and internalization kinetics
The results of the apparent IC50 determination are shown in Fig. 3a. The apparent IC50 of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 is 4.8 µM (95% confidence interval: 0.7–32.8 µM). However, the low internalization rate described below, render the observed value more likely to be a true IC50 rather than an apparent IC50. Figure 3b summarizes the binding and internalization kinetics of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 by BHK-GIPR transfected cells and NES2Y cells as determined in vitro. Both cell lines displayed similar binding and internalization characteristics. After 1 hour, 0.6 ± 0.1% and 0.6 ± 0.02% of the added [Lys37(111In-DTPA)]N-acetyl-GIP1-42 was bound to BHK-GIPR transfected cells and NES2Y cells, respectively. At this time point, the internalized fractions were 1.1 ± 0.1% and 1.5 ± 0.3%, respectively. After 24 hours, the binding values had increased to 13.8 ± 1.7% and 8.6 ± 1.5%, respectively and the internalization values to 4.2 ± 0.6% and 4.7 ± 3.6% for BHK-GIPR and NES2Y cells, respectively. All values were corrected for non-specific binding, as determined by the addition of 25 µg unlabeled GIP1-42 which significantly reduced the binding and internalization at all time points indicating GIPR-mediated binding in both cell lines (p < 0.05). Figure 4 shows the in vitro binding characteristics of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 to 200 isolated islets of C3H mice. After 24 hours, 0.22 ± 0.01 fmol [Lys37(DTPA)]GIP1-42 bound to the islets. Addition of 25 µg unlabeled GIP1-42 reduced the binding to 0.03 ± 0.01 fmol, indicating specific binding of the tracer to the GIPR on the islets of Langerhans.

In vitro characterization (a) Competition binding assay (apparent IC50) of [Lys37(DTPA)]GIP1-42 on NES2Y cells. [Lys37(111In-DTPA)]N-acetyl-GIP1-42 was used as a radiotracer (b) binding and internalization kinetics of 111In-GIP in BHK-GIPR positive cells and NES2Y cells. Cell bound and internalized fractions are corrected for non-specific binding and accumulation, as determined by co-incubation with an excess unlabeled GIP1-42.

Binding of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 to isolated islets. The solid bar represents binding of [Lys37(111In-DTPA)]N-acetyl-GIP1-42. The white bar represents binding of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 in the presence of an excess GIP1-42.
Peptide Dose Escalation Study
The effect of the peptide dose on tumor targeting was studied in BALB/c nude mice with subcutaneous BHK-GIPR tumors and the results are summarized in Fig. 5a. The [Lys37(DTPA)]N-acetyl-GIP1-42 dose had a pronounced effect on tumor uptake, which decreased significantly at peptide doses exceeding 0.2 µg (Fig. 5a and b). Highest tumor uptake of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 was observed at peptide doses ≤ 0.2 µg (0.04 nmol) per mouse: 5.2 ± 1.4%ID/g at 4 hours post-injection. Administration of 0.5 µg (0.1 nmol) as a peptide dose resulted in a significantly lower tumor uptake: 2.4 ± 0.7%ID/g (p = 0.008) (Fig. 5b). Four hours post-injection, [Lys37(111In-DTPA)]N-acetyl)GIP1-42 also showed uptake in various organs such as the lung, spleen, pancreas, liver, stomach and duodenum. The high uptake in the kidneys has been described as being the result of tubular reabsorption through the scavenger receptors cubilin and megalin, which is the case for many peptides30. As the kidney uptake of radiolabeled Lys37(111In-DTPA)]N-acetyl-GIP1-42 cannot be inhibited by an excess of unlabeled peptide, tubular reabsorption indeed appears to be the cause of the high kidney uptake. Raw data of this biodistribution study are shown in supplementary Table 1.

(a) Peptide dose escalation study in BALB/c nude mice with subcutaneous BHK-GIPR tumors. Values are expressed as percentage injected dose per gram tissue (%ID/g) (n = 5). (b) Tumor uptake of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 using different peptide doses. Tumor uptake was significantly higher (**p = 0.008) at peptide doses smaller than 0.5 µg (0.1 nmol).
Biodistribution Studies
Figure 6a summarizes the biodistribution of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 in BALB/c nude mice bearing a subcutaneous BHK-GIPR transfected tumor. One hour after injection, highest tumor uptake was observed: 4.7 ± 0.8%ID/g. After 4 and 24 hours, tumor uptake decreased to 3.3 ± 0.8%ID/g and 2.0 ± 0.2%ID/g, respectively (Fig. 6a). Co-administration of an excess unlabeled GIP1-42 significantly lowered tumor uptake to 0.8 ± 0.2%ID/g (p = 0.0001) 4 hours after injection, indicating GIPR-mediated tumor uptake. One hour after injection, tumor uptake in NES2Y tumors was 0.7 ± 0.2%ID/g (Fig. 6b). Co-administration of an excess unlabeled GIP1-42 lowered tumor uptake to 0.6 ± 0.01%ID/g (p = 0.78) Raw data of these biodistribution studies are shown in supplementary Tables 2 and 3.

(a) Biodistribution study of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 in BALB/c nude mice bearing subcutaneous BHK-GIPR tumors at 1, 4 and 24 hours after tracer injection. Values are expressed as percentage injected dose per gram tissue (%ID/g) (n = 5). Blocking was performed by co-injected of 25 µg unlabeled GIP1-42 (n = 5). (b) Biodistribution study of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 in BALB/c nude mice bearing subcutaneous NES2Y tumors, 1 hour after tracer injection. Values are expressed as percentage injected dose per gram tissue (%ID/g) (n = 5). Blocking was performed by co-injection of 25 µg unlabeled GIP1-42 (n = 2).
SPECT/CT imaging
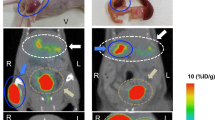
One and four hours after injection of [Lys37(111In-DTPA)]N-acetyl-GIP1-42, BHK-GIPR transfected tumors were clearly visible on SPECT/CT (Fig. 7a and b). Co-injection of an excess unlabeled GIP1-42 completely blocked tracer uptake in the BHK-GIPR transfected tumor (Fig. 7c). Also in mice bearing NES2Y tumors, clear tumor uptake was observed 1 hour after tracer injection (Fig. 8a and b). Furthermore, co-injection of an excess unlabeled GIP1-42 prevented tracer uptake in NES2Y tumors, suggesting GIPR-mediated tracer uptake in these tumors.

Maximum intensity projection (MIP) SPECT/CT images of a BALB/c nude mouse bearing a subcutaneous BHK-GIPR tumor 1 hour after injection (a), 4 hours after injection (b) and 4 hours after injection and co-injection with 25 µg unlabeled GIP1-42 (c).

SPECT/CT (a) and SPECT (b) of a BALB/c nude mouse bearing a subcutaneous NES2Y tumor, 1 hour after injection and SPECT/CT (c) and SPECT (d) after co-injection with 25 µg unlabeled GIP1-42.
Discussion
In this study, we investigated the potential of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 for NET detection. The tracer showed GIPR-mediated binding to BHK-GIPR positive cells and NES2Y cells, and to isolated islets in vitro. Optimal GIPR-mediated tumor targeting of BHK-GIPR positive tumors in vivo, was observed 1 hour post injection using a peptide dose of 0.2 µg (0.04 nmol). Furthermore, both BHK-GIPR transfected tumors and NES2Y tumors were visualized by SPECT.
It has been described that C-terminally truncated GIP analogues, such as GIP1-30, exhibit reduced receptor binding affinity when compared to intact GIP1-42, while retaining their full insulinotropic effect26,27. Since the goal of this study was to apply the analogue for in vivo imaging, high receptor affinity should be preserved rather than biological activity. Therefore, we selected GIP1-42 as a basis for our radiotracer. Another point of consideration while designing a GIPR targeting radiotracer, is the susceptibility of the native peptide to DPP IV degradation31. Since it was shown that several N-terminal modifications showed increased half-life in vitro32,33, we acetylated the N-terminal tyrosine residue to obtain a better DPP IV degradation-resistant tracer. Indeed, we observed that the tracer remained stable in serum for at least 4 hours after incubation and even after 24 hours of incubation 73% of the tracer remained intact.
In order to obtain high quality images, high target-to-background ratios are required. Tracer metabolites labeled with a radiometal via a chelator, can be trapped in the lysosomes upon tracer internalization and degradation, resulting in enhanced tracer accumulation in the target cells23,34. Therefore, we investigated tracer internalization rates in vitro. Internalization kinetics of the tracer were comparable and relatively slow: after 24 hours, both BHK-GIPR positive and NES2Y cell showed similar internalization rates of 4.2 ± 0.6%ID/g and 4.7 ± 3.6%ID/g, respectively. Interestingly, the majority of the tracer was bound to the cell surface in both cell lines. These observations could be explained by very recent findings of Ismail et al. indicating that N-terminal acetylation of the peptide hampers receptor internalization35.
In BHK-GIPR positive tumors, GIP1-42 doses lower than 0.5 µg (0.1 nmol) resulted in maximal tumor accumulation. This indicates that administration of higher peptide doses results in partial saturation of the GIPR in vivo. However, since the tracer could be labeled with a specific activity up to 1.2 TBq/µmol, administration of an activity dose for imaging dose is still feasible. For in vivo SPECT imaging, high tumor-to-blood ratios are required. However, since tracer uptake in the blood was still very low at a peptide dose of 0.2 µg, we selected this dose as the optimal dose for in vivo imaging since it resulted in the highest absolute tumor uptake.
Although [Lys37(111In-DTPA)]N-acetyl-GIP1-42 showed high and specific tumor uptake in GIPR-transfected GIPR tumors, tumor uptake in NES2Y tumors was lower and co-injection of an excess unlabeled GIP1-42 did not result in significantly lower tracer uptake. This clear difference in tracer uptake might be explained by the completely different origin and purpose of both cell lines. While the BHK-GIPR cell line was created to express artificially high receptor levels, the NES2Y cells are derived from patients with congenital hyperinsulinism of infancy (28). Since these cells do not overexpress the target receptor and it is known that the natural expression levels of the GIPR are much lower compared to its brother incretin peptide GLP-136, the lower uptake is to be expected. As for the blocking study, the small size of the blocking group (n = 2) impairs generation of representative results. Therefore, the obtained significance level (p = 0.78) might be distorted. On the SPECT images however, tracer accumulation was observed in NES2Y tumors but was no longer observed after co-injection of an excess unlabeled GIP1-42 (Fig. 8b), suggesting specific tumor targeting. Nevertheless, several tracer characteristics might be optimized to further improve tumor targeting. Firstly, other N-terminal modifications than acetylation, such as glucitol or pyroglutamil insertion, have been shown to improve the half-life of the peptide in the presence of DPP IV32,33 and might also result in higher internalization rates. Secondly, receptor binding and internalization might increase with longer circulation times. As shown in our biodistribution studies, [Lys37(111In-DTPA)]N-acetyl-GIP1-42 is cleared from the blood very rapidly, hampering longer tracer circulation and thus higher tumor uptake. Several methods such as PEGylation, multimerization or introduction of free fatty acid tails might increase circulation time37 and thereby tracer accumulation in the tumor. However, a good balance between tracer retention in blood and tracer targeting dynamics has to be found in order to obtain favorable target-to-background ratios for in vivo imaging.
[Lys37(111In-DTPA)]N-acetyl-GIP1-42 was produced at a high molar activity and showed receptor mediated binding to various GIPR expressing cells in vitro. Subcutaneous BHK-GIPR positive tumors showed GIPR-mediated tracer accumulation with optimal in vivo targeting 1 hour post-injection, at a peptide dose of 0.2 µg (0.04 nmol). Furthermore, although very low, uptake of a GIP-based tracer was demonstrated for the first time in a human beta-cell derived tumor. Unfortunately, specificity of tracer binding could not be shown due to the small size of the blocking group. In conclusion, [Lys37(111In-DTPA)]N-acetyl-GIP1-42 shows promising results as a radiotracer for NET imaging. However, the initial working hypothesis that a full-length peptide-based tracer might show improved characteristics for in vivo NET imaging when compared to GIP1-30 could not be confirmed.
Methods
Radiolabeling
[Lys37(DTPA)]N-acetyl-GIP1-42 (MW = 5422 g/mol) and GIP1-42 (MW = 5046.7 g/mol) were purchased from Peptide Specialty Laboratories (PSL GmbH, Heidelberg, Germany). DTPA was conjugated to the ε-amino group of the Lysine (K37) and the N-terminal tyrosine was acetylated to reduce its susceptibility to endopeptidases (Fig. 1). [Lys37(DTPA)]GIP1-42 was dissolved in 0.5 M MES (2-(N-morpholino)ethanesulfonic acid) buffer, pH 5.5 and stored at 4 °C until use. 150 MBq [111In]Cl3 (Mallinckrodt Pharmaceuticals, ‘s Hertogenbosch, The Netherlands) was added to 1 µg [Lys37(DTPA)]N-acetyl-GIP1-42 together with 5 volumes of 0.5 M MES buffer (1:5 relation 111In:buffer), pH 5.5 and incubated for 20 min at room temperature (RT). After incubation, EDTA (Sigma Aldrich, St. Louis, MO, USA) and Tween80 (Sigma Aldrich) were added to a final concentration of 5 mM and 0.1%, respectively. The labeling mixture was purified by solid-phase extraction using a hydrophilic-lipophilic balance (HLB) cartridge (30 mg, Waters Oasis, Milford, MA, USA), to eliminate unincorporated 111In. The cartridge was activated with 1 ml ethanol, washed with 2 ml water and conditioned with 1 ml of 0.5 M MES, pH 5.5. Subsequently, the cartridge was loaded with the labeling mixture, washed with 1 ml 0.5 M MES and 2 ml water and [Lys37(111In-DTPA)]N-acetyl-GIP1-42 was eluted from the cartridge with 200 µL 100% ethanol. For in vivo use, purified labeled peptide was diluted in phosphate buffered saline (PBS), 0.5% bovine serum albumin (BSA) (w/v) to obtain a final ethanol concentration of less than 10%. Radiochemical purity of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 was determined by instant thin-layer chromatography (ITLC) (ITLC-SG, Agilent Technologies, Lake Forest, CA, USA), using 0.1 M EDTA in 0.1 M NH4Ac (Sigma Aldrich), pH 5.5 as a mobile agent and by reversed-phase high performance liquid chromatography (RP-HPLC) using an Eclipse XDB C18 column (Agilent Technologies). The column was eluted with 0.1% trifluoroacetic acid (TFA) in H2O (0-5 minutes) followed by a linear gradient from 3% to 100% acetonitrile with 0.1% TFA over 10 minutes (flow rate: 1 ml/minute).
Serum Stability Analysis
For the serum stability analysis, [Lys37(DTPA)]N-acetyl-GIP1-42 was labeled with [111In]Cl3 at a specific activity of 27 GBq/µmol. Radiochemical purity of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 directly after labeling was determined by ITLC-SG (Agilent Technologies) and RP-HPLC as described above. A sample of the labeling mixture was incubated in human serum (1:10) at 37 °C. After 1, 2, 4 and 24 hours, a sample was taken and serum proteins were precipitated by adding acetonitrile. This mixture was centrifuged and after dilution, the radiochemical purity of the compound in the supernatant was analyzed using ITLC and RP-HPLC as described above.
Cell Culture
Baby hamster kidney cells, transfected with the human GIP (BHK-GIPR cells) were maintained in DMEM Glutamax (cat. Nr. 6195, GIBCO, BRL Life Sciences Technologies, Bleiswijk, The Netherlands) supplemented with 10% fetal bovine serum (FCS), (HyClone, Celbio, Logan, UT, USA) and 1 mg/ml G418, in a humidified atmosphere containing 5% CO2 at 37 °C. The human beta cell line NES2Y was cultured as described previously (28). Briefly, cells were maintained in RPMI 1640 (R0883, GIBCO) supplemented with 10% FCS, (HyClone, Celbio), 2 mM L-glutamine (Sigma Aldrich), 100 U/ml penicillin (Sigma Aldrich) and 100 µg/ml streptomycin (Sigma Aldrich) (complete RPMI), in a humidified atmosphere containing 5% CO2 at 37 °C.
IC50 Determination
The 50% inhibitory concentration (IC50) of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 was determined using NES2Y cells. NES2Y cells were seeded at a density of 200,000 cells/well in 24-well plates and incubated overnight at 37 °C. The cells were washed with binding buffer (RPMI + 0.5% BSA (w/v)) and [Lys37(DTPA)]N-acetyl-GIP1-42 was added at a final concentration ranging from 1.0 – 100 µM along with radiolabeled [Lys37(111In-DTPA)]N-acetyl-GIP1-42 (4.6 kBq = 2.7 fmol). After o/n (15 h) incubation at 37 °C, cells were washed with binding buffer and cell-associated activity was measured in a well type gamma counter (Wallac 2480 wizard, Perkin Elmer). Under these conditions, internalization may occur. We therefore designate the results of this competitive binding assay as “apparent IC50” values rather than IC50.
Internalization kinetics
The internalization kinetics of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 was determined as previously described38 using BHK-GIPR transfected cells and NES2Y cells. BHK-GIPR transfected cells and NES2Y cells were seeded in 24-well plates with a density of 50,000 and 200,000 cells/well, respectively and incubated overnight at 37 °C. Cells were washed with binding buffer and incubated with approximately 1.6 kBq [Lys37(111In-DTPA)]N-acetyl-GIP1-42 (2.7 fmol) for 1, 2, 4 and 24 h at 37 °C. After incubation, cells were washed twice with binding buffer. To determine the surface-bound fraction, ice-cold acid buffer (0.1 M acetic acid, 154 mM NaCl, pH 2.5) was added and cells were incubated for 10 minutes at 4 °C. After washing the cells twice with binding buffer to makes sure that all surface-bound tracer was removed from the cells, the cell-associated activity was measured in a well type gamma counter. After removal of the surface-bound activity, the internalized fraction was represented by the remaining cell-associated activity. The internalized fraction (remaining cell-associated activity after acid wash) and the receptor bound fraction (activity removed by acid wash) were determined in a well-type gamma counter.
Animals
All animal experiments were approved by the Animal Welfare Body of the Radboud University, Nijmegen, The Netherlands and carried out in accordance with their guidelines. Six to eight weeks old, female BALB/c nude mice and C3H mice were purchased from Janvier labs (Le Genest Saint Isle, France). For all in vivo experiments, 6-8 weeks old female BALB/c nude mice were injected subcutaneously in the right shoulder with 0.2 ml of a 2.5 × 107 cells/ml suspension of BHK-GIPR transfected cells or with 0.2 ml of a 2.5 × 107 cells/ml suspension of NES2Y cells mixed with matrigel (2:1) in the right shoulder. When the tumor reached a diameter of 2-5 mm, mice were randomly divided in groups.
Islet Isolation
Islets of Langerhans were isolated from donor C3H mice by collagenase digestion as follows. Mice were euthanized by CO2/O2 suffocation and 2 ml of ice cold RPMI 1640 containing 1 mg/ml collagenase type V (Sigma Aldrich) were infused via the pancreatic duct in situ. After dissection, the perfused pancreata were collected in serum free medium containing collagenase and kept on ice until digestion. Pancreata were digested for 12 min at 37 °C. Digestion was stopped by adding RPMI medium containing 10% FCS, 2 mM L-glutamine, 100 U/ml penicillin and 100 U/ml streptomycin (complete RPMI). After washing, digested pancreata were passed through a mesh. Afterwards, islets were purified on a discontinuous Ficoll gradient (densities: 1.108; 1.096; 1.037; Cellgro by Mediatech Inc, Manassas, VA, USA) by centrifugation at 625 × g for 16 min without brake. Islets were collected from the intersection of the second and third layer and remaining Ficoll was removed by washing with complete RPMI. Isolated islets were cultured overnight in complete RPMI medium in a humidified atmosphere containing 5% CO2 at 37 °C.
In Vitro Binding to Isolated Islets
After overnight recovery from the isolation procedure, islets from the donor C3H mice were collected, counted and resuspended in complete RPMI. After transwell saturation using binding buffer, islets were transferred to 24 well transwell plates (200 islets per transwell) (Corning Inc. Tewksbury, MA, USA) and washed with binding buffer. Subsequently, approximately 8 kBq [Lys37(111In-DTPA)]N-acetyl-GIP1-42 (13.5 fmol) was added followed by incubation for 24 hours at 37 °C. To investigate the GIPR-mediated binding, 25 µg of unlabeled GIP1-42 was added together with [Lys37(111In-DTPA)]N-acetyl-GIP1-42 in a separate set of wells. After incubation, islets were washed extensively with binding buffer and islet-associated radioactivity was measured in a well type gamma counter.
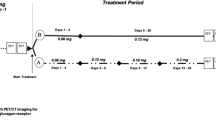
Peptide Dose Escalation Study
To determine the optimal peptide dose of [Lys37(DTPA)]N-acetyl-GIP1-42, BHK-GIPR transfected tumor bearing BALB/c nude mice (n = 5/group) were injected intravenously with approximately 3 MBq [Lys37(111In-DTPA)]N-acetyl-GIP1-42 (0.05 µg) in the tail vein and were co-injected with escalating amounts of unlabeled [Lys37(DTPA)]N-acetyl-GIP1-42 (0.05 µg – 1.95 µg), resulting in final peptide doses ranging from 0.1 to 2 µg/mouse (0.02 – 0.4 nmol/mouse) (5 groups, n = 5/group). Four hours after injection, mice were euthanized by CO2/O2 suffocation and tumors and other relevant tissues (heart, muscle, lung, spleen, pancreas, kidney, liver, stomach and duodenum) were dissected, weighed and measured in a well type gamma counter. The percentage injected dose per gram of tissue (%ID/g) was determined for each tissue.
Biodistribution Studies
BALB/c nude mice bearing BHK-GIPR transfected tumors were injected intravenously with approximately 3 MBq (peptide dose 0.2 µg = 0.04 nmol) of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 via the tail vein. At various time points after injection (1, 4 and 24 hours), mice were euthanized by CO2/O2 suffocation and blood, tumor and other relevant tissues were dissected, weighed and measured in a well type gamma counter. The percentage injected dose per gram (%ID/g) was calculated for each tissue. To determine whether the GIP1-42 uptake was GIPR mediated, 25 µg of unlabeled GIP1-42 was co-injected in a separate group (n = 5) and mice were euthanized four hours post-injection. BALB/c nude mice bearing NES2Y tumors (n = 5) were injected intravenously with approximately 3 MBq (peptide dose 0.2 µg = 0.04 nmol) of [Lys37(111In-DTPA)]N-acetyl-GIP1-42 via the tail vein. One hour after injection, mice were euthanized by CO2/O2 suffocation and blood, tumor and other relevant tissues were dissected, weighed and measured in a well type gamma counter. The percentage injected dose per gram (%ID/g) was determined for each tissue. To determine whether the GIP1-42 uptake was GIPR mediated, 25 µg of unlabeled GIP1-42 was co-injected in a separate group (n = 2) and mice were euthanized one hours post-injection.
SPECT/CT
One (BHK-GIPR tumors and NES2Y tumors) or four (BHK-GIPR tumors)hours (n = 2/group) prior to SPECT imaging, BALB/c nude mice were injected intravenously in the tail vein with 20.6 ± 0.7 MBq [Lys37(111In-DTPA)]N-acetyl-GIP1-42 (peptide dose 0.2 µg ( = 0.04 nmol) in 200 µl injection fluid). SPECT/CT scans were acquired on a dedicated small animal SPECT/CT scanner (U-SPECT-II, Milabs, Utrecht, The Netherlands) with a 1.0 mm mouse collimator, using 36 bed positions and an acquisition time of 30 minutes (BHK-GIPR tumors) or 45 minutes (NES2Y tumors). After SPECT acquisition, CT (65 kV, 615 µA, 1 bed position) was acquired as an anatomical reference. SPECT scans were reconstructed using U-SPECT-II reconstruction software (U-SPECT-Rec, Milabs, Utrecht, The Netherlands) with the following settings: selection of the lower 111In photopeak (152–183 keV), corrected for two backgrounds (135–151 keV and 184–211 keV), pixel based OSEM, voxel size 0.4 mm3 and 1 iteration over 16 subsets.
Statistics
All values are expressed as mean ± standard deviation (SD). Statistical analysis was performed using unpaired two-tailed t-test using GraphPad Prism v5.03 (GraphPad Softwarem Inc., San Diego, CA, USA). The level of significance was set at p < 0.05.
Ethical approval
All animal experiments were approved by the Animal Welfare Body of the Radboud University, Nijmegen, The Netherlands and carried out in accordance with their guidelines.
Data availability statement
all data are available at the department of Nuclear medicine of the Radboud University medical center, Nijmegen.
References
Reubi, J. C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocrine reviews 24, 389–427, https://doi.org/10.1210/er.2002-0007 (2003).
Schottelius, M. & Wester, H. J. Molecular imaging targeting peptide receptors. Methods 48, 161–177, https://doi.org/10.1016/j.ymeth.2009.03.012 (2009).
Krenning, E. P. et al. Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123I-Tyr3]-octreotide: the Rotterdam experience with more than 1000 patients. European journal of nuclear medicine 20, 716–731 (1993).
Lamberts, S. W., Bakker, W. H., Reubi, J. C. & Krenning, E. P. Somatostatin-receptor imaging in the localization of endocrine tumors. The New England journal of medicine 323, 1246–1249, https://doi.org/10.1056/NEJM199011013231805 (1990).
Modlin, I. M. & Tang, L. H. Approaches to the diagnosis of gut neuroendocrine tumors: the last word (today). Gastroenterology 112, 583–590 (1997).
Yang, J. et al. Diagnostic role of Gallium-68 DOTATOC and Gallium-68 DOTATATE PET in patients with neuroendocrine tumors: a meta-analysis. Acta Radiol 55, 389–398, https://doi.org/10.1177/0284185113496679 (2014).
Bodei, L. et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. European journal of nuclear medicine and molecular imaging 40, 800–816, https://doi.org/10.1007/s00259-012-2330-6 (2013).
Joseph, K. et al. [Receptor scintigraphy using 111In-pentetreotide in endocrine gastroenteropancreatic tumors]. Nuklearmedizin. Nuclear medicine 32, 299–305 (1993).
Prasad, V. et al. Role of (68)Ga somatostatin receptor PET/CT in the detection of endogenous hyperinsulinaemic focus: an explorative study. Eur J Nucl Med Mol Imaging 43, 1593–1600, https://doi.org/10.1007/s00259-016-3331-7 (2016).
Nockel, P. et al. Localization of Insulinoma Using 68Ga-DOTATATE PET/CT Scan. J Clin Endocrinol Metab 102, 195–199, https://doi.org/10.1210/jc.2016-3445 (2017).
Gotthardt, M. et al. Improved tumour detection by gastrin receptor scintigraphy in patients with metastasised medullary thyroid carcinoma. European journal of nuclear medicine and molecular imaging 33, 1273–1279, https://doi.org/10.1007/s00259-006-0157-8 (2006).
Reubi, J. C., Schaer, J. C. & Waser, B. Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer research 57, 1377–1386 (1997).
Reubi, J. C. et al. Vasoactive intestinal peptide/pituitary adenylate cyclase-activating peptide receptor subtypes in human tumors and their tissues of origin. Cancer research 60, 3105–3112 (2000).
Reubi, J. C. & Waser, B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. European journal of nuclear medicine and molecular imaging 30, 781–793, https://doi.org/10.1007/s00259-003-1184-3 (2003).
Sherman, S. K. et al. GIPR expression in gastric and duodenal neuroendocrine tumors. The Journal of surgical research 190, 587–593, https://doi.org/10.1016/j.jss.2014.01.044 (2014).
Waser, B., Rehmann, R., Sanchez, C., Fourmy, D. & Reubi, J. C. Glucose-dependent insulinotropic polypeptide receptors in most gastroenteropancreatic and bronchial neuroendocrine tumors. The Journal of clinical endocrinology and metabolism 97, 482–488, https://doi.org/10.1210/jc.2011-2454 (2012).
Sherman, S. K. et al. Gastric inhibitory polypeptide receptor (GIPR) is a promising target for imaging and therapy in neuroendocrine tumors. Surgery 154, 1206–1213; discussion 1214, https://doi.org/10.1016/j.surg.2013.04.052 (2013).
Gourni, E. et al. The glucose-dependent insulinotropic polypeptide receptor: a novel target for neuroendocrine tumor imaging-first preclinical studies. Journal of nuclear medicine: official publication, Society of Nuclear Medicine 55, 976–982, https://doi.org/10.2967/jnumed.113.133744 (2014).
Mortensen, K., Christensen, L. L., Holst, J. J. & Orskov, C. GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regulatory peptides 114, 189–196 (2003).
Elrick, H., Stimmler, L., Hlad, C. J. Jr. & Arai, Y. Plasma Insulin Response to Oral and Intravenous Glucose Administration. The Journal of clinical endocrinology and metabolism 24, 1076–1082, https://doi.org/10.1210/jcem-24-10-1076 (1964).
McIntyre, N., Holdsworth, C. D. & Turner, D. S. New Interpretation of Oral Glucose Tolerance. Lancet 2, 20–21 (1964).
Brom, M., Oyen, W. J., Joosten, L., Gotthardt, M. & Boerman, O. C. 68Ga-labelled exendin-3, a new agent for the detection of insulinomas with PET. European journal of nuclear medicine and molecular imaging 37, 1345–1355, https://doi.org/10.1007/s00259-009-1363-y (2010).
Gotthardt, M. et al. Use of the incretin hormone glucagon-like peptide-1 (GLP-1) for the detection of insulinomas: initial experimental results. European journal of nuclear medicine and molecular imaging 29, 597–606, https://doi.org/10.1007/s00259-002-0761-1 (2002).
Wild, D., Macke, H., Christ, E., Gloor, B. & Reubi, J. C. Glucagon-like peptide 1-receptor scans to localize occult insulinomas. The New England journal of medicine 359, 766–768, https://doi.org/10.1056/NEJMc0802045 (2008).
Wild, D. et al. Glucagon-like peptide-1 versus somatostatin receptor targeting reveals 2 distinct forms of malignant insulinomas. Journal of nuclear medicine: official publication, Society of Nuclear Medicine 52, 1073–1078, https://doi.org/10.2967/jnumed.110.085142 (2011).
Maletti, M., Altman, J. J., Hoa, D. H., Carlquist, M. & Rosselin, G. Evidence of functional gastric inhibitory polypeptide (GIP) receptors in human insulinoma. Binding of synthetic human GIP 1-31 and activation of adenylate cyclase. Diabetes 36, 1336–1340 (1987).
Wheeler, M. B. et al. Functional expression of the rat pancreatic islet glucose-dependent insulinotropic polypeptide receptor: ligand binding and intracellular signaling properties. Endocrinology 136, 4629–4639, https://doi.org/10.1210/endo.136.10.7664683 (1995).
Adams, G. G., Uddin, A., Vives-Pi, M., Pujol-Borrell, R. & James, R. F. Characterisation of the NES2Ycell line and its use in the production of human glucose-responsive insulin producing (hGRIP) cell lines by cell-cell fusion. Islets 1, 117–123, https://doi.org/10.4161/isl.1.2.9432 (2009).
Coenen, H. H. et al. Consensus nomenclature rules for radiopharmaceutical chemistry - Setting the record straight. Nucl Med Biol 55, v–xi, https://doi.org/10.1016/j.nucmedbio.2017.09.004 (2017).
Gotthardt, M. et al. Indication for different mechanisms of kidney uptake of radiolabeled peptides. J Nucl Med 48, 596–601 (2007).
Kieffer, T. J., McIntosh, C. H. & Pederson, R. A. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 136, 3585–3596, https://doi.org/10.1210/endo.136.8.7628397 (1995).
O’Harte, F. P., Mooney, M. H. & Flatt, P. R. NH2-terminally modified gastric inhibitory polypeptide exhibits amino-peptidase resistance and enhanced antihyperglycemic activity. Diabetes 48, 758–765 (1999).
Gault, V. A., Flatt, P. R. & O’Harte, F. P. Glucose-dependent insulinotropic polypeptide analogues and their therapeutic potential for the treatment of obesity-diabetes. Biochemical and biophysical research communications 308, 207–213 (2003).
Gotthardt, M., Boermann, O. C., Behr, T. M., Behe, M. P. & Oyen, W. J. Development and clinical application of peptide-based radiopharmaceuticals. Current pharmaceutical design 10, 2951–2963 (2004).
Ismail, S. et al. Internalization and desensitization of the human glucose-dependent-insulinotropic receptor is affected by N-terminal acetylation of the agonist. Molecular and cellular endocrinology. https://doi.org/10.1016/j.mce.2015.07.001 (2015).
De Marinis, Y. Z. et al. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab 11, 543–553, https://doi.org/10.1016/j.cmet.2010.04.007 (2010).
Pathak, V., Vasu, S., Gault, V. A., Flatt, P. R. & Irwin, N. Sequential induction of beta cell rest and stimulation using stable GIP inhibitor and GLP-1 mimetic peptides improves metabolic control in C57BL/KsJ db/db mice. Diabetologia 58, 2144–2153, https://doi.org/10.1007/s00125-015-3653-1 (2015).
Laverman, P. et al. Targeting of a CCK(2) receptor splice variant with (111)In-labelled cholecystokinin-8 (CCK8) and (111)In-labelled minigastrin. European journal of nuclear medicine and molecular imaging 35, 386–392, https://doi.org/10.1007/s00259-007-0604-1 (2008).
Acknowledgements
The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/under REA grant agreement n° 289932. The authors declare that there is no conflict of interest. SW is a postdoctoral fellow of the research foundation Flanders (FWO). We thank the biotechnicians of PRIME for their assistance in the animal experiments, Jacob Hecksher-Sørensen of Novo Nordisk for kindly providing us with BHK-GIPR cells and Roger James of the University of Leicester for kindly providing us with NES2Y cells.
Author information
Authors and Affiliations
Contributions
S.M.A.W. and L.J. designed and performed the experiments; acquired, analyzed and interpreted data; and drafted and critically revised the manuscript. O.C.B., M.B. and M.G. provided a substantial contribution to conception and design, and data interpretation. O.C.B., M.B. and M.G. critically revised the manuscript for intellectual content. All authors approved the final version of the manuscript. M.G. is responsible for the integrity of the manuscript as a whole.
Corresponding author
Ethics declarations
Competing Interests
M.G. is patent holder in the field.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Willekens, S.M.A., Joosten, L., Boerman, O.C. et al. Characterization of 111In-labeled Glucose-Dependent Insulinotropic Polypeptide as a Radiotracer for Neuroendocrine Tumors. Sci Rep 8, 2948 (2018). https://doi.org/10.1038/s41598-018-21259-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21259-3
This article is cited by
-
Molecular imaging phenotyping for selecting and monitoring radioligand therapy of neuroendocrine neoplasms
Cancer Imaging (2022)
-
The pathogenic role of the GIP/GIPR axis in human endocrine tumors: emerging clinical mechanisms beyond diabetes
Reviews in Endocrine and Metabolic Disorders (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
