
Abstract
RNA alternative splicing (AS) is an important post-transcriptional mechanism enabling single genes to produce multiple proteins. It has been well demonstrated that viruses deploy host AS machinery for viral protein productions. However, knowledge on viral AS is limited to a few disease-causing viruses in model species. Here we report a novel approach to characterizing viral AS using whole transcriptome dataset from host species. Two insect transcriptomes (Acheta domesticus and Planococcus citri) generated in the 1,000 Insect Transcriptome Evolution (1KITE) project were used as a proof of concept using the new pipeline. Two closely related densoviruses (Acheta domesticus densovirus, AdDNV, and Planococcus citri densovirus, PcDNV, Ambidensovirus, Densovirinae, Parvoviridae) were detected and analyzed for AS patterns. The results suggested that although the two viruses shared major AS features, dramatic AS divergences were observed. Detailed analysis of the splicing junctions showed clusters of AS events occurred in two regions of the virus genome, demonstrating that transcriptome analysis could gain valuable insights into viral splicing. When applied to large-scale transcriptomics projects with diverse taxonomic sampling, our new method is expected to rapidly expand our knowledge on RNA splicing mechanisms for a wide range of viruses.
Similar content being viewed by others

Introduction
As increasing number of next-generation sequencing (NGS) datasets are being produced from various-omics initiatives, transcriptome sequencing of flora and fauna for a specific developmental stage/condition gains its popularity in biological research. Transcriptomics is implemented in discoveries of novel transcripts, SNPs, gene splicing and fusion, in determination of gene structure, function and regulation, and in quantification of expression levels1. It has already contributed a great deal of understanding to the mechanisms of functional elements, genes and transcripts2,3.
RNA splicing plays a vital role in genetics by increasing mRNA and protein diversities and by regulating gene expressions, providing an important link between genetic variation and disease4,5,6,7. Alternative splicing (AS) is one of the major mechanisms in increasing the diversity of proteins translated from a limited number of genes in metazoans8,9. The spliceosome complex, composed of at least 170 proteins and several small nuclear RNAs (snRNAs), is the key structure responsible for splicing in eukaryotes10. The complex defines exons/introns in transcribed RNAs by three major sequence elements: the 5′ splice site (donor site), the 3′ splice site (acceptor site), and the branch point4,11. When compared with annotated genome sequences, transcriptome sequencing could identify gene splicing isoforms and expression patterns associated with biological functions12,13.
Deep sequencing experiments often detect gene expressions not only for the focal taxon but also for pathogens infecting the host14,15. This feature has been known as dual-sequencing16. Viruses and endogenous viral elements occur in most organisms, including fungi, plants and animals17,18,19. Viruses also play pivotal roles in ecological systems20. In recent years, many novel viral infections have been discovered using NGS21,22,23,24. Several methods and tools have been developed for virus detection25,26, viral gene expression and host adaptation15 using NGS datasets. Among these, viral sequences are often assembled de novo using all reads or those not matched to the host genome27,28,29,30.
Transcriptome datasets are one of the most preponderant resources, in which host and viral components are both recorded12,31. Combined with genomic data, transcriptome sequencing has been used to detect known and novel disease-causing viruses27,32,33, to observe viral mutagenesis and recombination26,34,35,36, and to understand virus-host interactions26,37. RNA splicing plays important roles in viral replication and virus-host interactions38. Viral gene expression and RNA splicing are exclusively dependent of the host genomics machinery39,40, therefore the whole transcriptome datasets generated from host species (containing mRNA from both host and infecting virus) are good resources for revealing viral RNA splicing characteristics. There are more than 4,000 viral genomes publically accessible, even though more viruses are yet to be described24,41.
Two transcriptomes obtained from the 1000 Insect Transcriptome Evolution project (1KITE, www.1kite.org)42 were analyzed in this study: house cricket (Acheta domesticus (Linnaeus), Orthoptera, Gryllidae) and citrus mealybug (Planococcus citri (Risso), Hemiptera, Pseudococcidae). Previous studies suggest these insects often carry closely related viruses - Acheta domesticus densovirus (AdDNV) and Planococcus citri densovirus (PcDNV), respectively43,44. Densoviruses (family Parvoviridae) are widely distributed among arthropods19,45 with linear single-stranded DNA genomes of approximately 5,000-nucleotides, including two major gene cassettes encoding viral nonstructural (NS or Rep) and structural proteins (VP or CP)43,45. Densoviruses employ RNA AS to produce the nonstructural protein 1 (NS1) endonuclease using a rolling-hairpin mechanism to regulate replication46,47. Two NS transcripts and one VP transcript were detected in AdDNV and four splicing junctions were reported48.
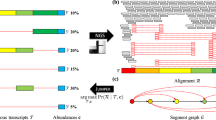
In the present study, we completed a NGS-based informatics pipeline to: 1) detect virus from the whole assembled transcriptome; 2) obtain viral genome sequence by calling consensus sequence from virus reads; 3) characterize virus AS codes and reveal gene expression patterns of the virus (Fig. 1). Using the 1KITE transcriptome, we characterized the splicing patterns in AdDNV and PcDNV, demonstrating both shared and unique splicing patterns in closely related viruses.

Analysis framework. (A) Analysis framework; (B) Detailed analytical pipeline. Virus detection and viral expression analyses: this pipeline was designed to detect and obtain viral sequences from transcriptome datasets; all in house Perl scripts used in the pipeline are available on web (https://github.com/linzhi2013/Virusfishing).
Results
Data description
The transcriptomes of A. domesticus and P. citri were generated by the 1KITE project42. In brief, total RNA was isolated from one A. domesticus juvenile female, collected in Hamburg, Germany, February 2013, and ca. 150 P. citri individuals, collected in Brandenburg, Germany, November 2011, respectively, using TRIzol (Invitrogen, Grand Island, NY, USA). The mRNA was isolated using the Dynabeads mRNA Purification Kit following the manufacturer’s protocol (Invitrogen, Grand Island, NY, USA). The mRNA extracts were treated with RNA fragmentation reagent (Ambion, Austin, Texas, US). Two cDNA libraries were constructed using SuperScript™II Reverse Transcriptase (Invitrogen, Grand Island, NY, USA), random N6 primer (IDT), RNase H (Invitrogen, Grand Island, NY, USA) and DNA polymerase I (New England BioLabs, Ipswich, MA, USA). The cDNA libraries were sequenced with the 150 bp paired-end strategy and 250 bp insert-size using Illumina’s HiSeq. 2000 platform (Illumina, San Diego, CA) at BGI-Shenzhen. The resulting sequences were subject to Illumina’s read quality control pipeline. 2.36 Gb (16,898,600 reads with 150 bp read size) and 2.89 Gb (20,670,410 reads with 150 bp read size) of high quality sequence data were obtained for A. domesticus (NCBI Accession No: PRJNA286330) and P. citri (NCBI Accession No: PRJNA219593, published by Misof et al.42), respectively.
Viral sequence detection and calling
Both viral and Nt databases applied in our study were downloaded from the GenBank (accessed in Nov. 2014). The virus database contains 1,561,606 viral sequences (2.2 Gb) including nearly 5,000 complete viral genomes and the Nt database contains 29,059,038 sequences reaching a data size of 84.0 Gb.
After virus detection and false positive (sequence matched to non-viral subjects) removal, the assembled transcriptome sequences had best matches with AdDNV in A. domesticus and with PcDNV in P. citri (Fig. 1B, Supplementary Table S1 and Supplementary Text S1). Near full-length consensus genomes of AdDNV (NCBI Accession No: KX145610) and PcDNV (NCBI Accession No: KX145609) were called based on the templates of viral reference genome, with 5,259 bases (96.94% of the AdDNV reference KF015278.1 with 6,084 mapped reads) and 5,220 bases (97.03% of the PcDNV reference NC004289 with 33,604 mapped reads), respectively. The missing regions were located at 5′ and 3′ ends for both viruses and were replaced by Ns for the following analysis. Eleven single nucleotide variants (SNVs) were detected for AdDNV and eight SNVs for PcDNV (Fig. 2A and B, Supplementary Table S2 and Supplementary Data file S1). None of these SNVs were located in splicing sites while some SNVs resulted in nonsynonymous mutations in ORF translations, i.e., six out of eleven SNVs in AdDNV and five out of eight in PcDNV (Supplementary Data file S1 Column N). Phylograms based on genome sequences and deduced proteins confirmed that AdDNV and PcDNV were closely related species within Ambidensovirus (Supplementary Fig. S1 and Supplementary Text S1)43,49,50,51,52,53,54,55,56,57.

Genome coverage and annotations of AdDNV and PcDNV. Genome coverage of (A) AdDNV and (B) PcDNV. Log2 scale of read density was based on genomic sequences of AdDNV and PcDNV. Vertical bars highlight mutation sites against the reference sequences. Annotations of (C) AdDNV and (D) PcDNV. Coverage (Y-axis) of each nucleotide position (X-axis) was plotted for AdDNV_1KITE and PcDNV_1KITE. Six reading-frames and previously described genes were represented using information provided by NCBI, including: start/stop codons (short blue/red vertical bars), transcription directions (black arrows, from top to bottom: forward reading frames +1, +2, +3 and reverse reading frames −1, −2, −3), ORFs (solid gray boxes). Virus genes (blue boxes), proteins (red boxes) and conserved motifs (black boxes) were represented according to the NCBI annotations. BWA mapping profiles (green lines), TopHat2 mapping profiles (purple lines), TopHat2 gap mapping profiles (yellow lines, the number of both splicing and non-splicing reads, correspond to splicing junctions) were represented according to the mapping results. AdDNV introns reported in existing studies include: In (nt 223 to 855), Ia (nt 4403 to 4758), Ib (nt 4403 to 4544) and II (nt 4260 to 4434).
Expression of viral genes in transcriptome
Compared to the unspliced aligner BWA58 (Supplementary Table S2), TopHat259,60 obtained much greater depth coverages (Supplementary Table S3 and Supplementary Data file S2) due to successful alignments of fragmented (gapped) transcripts onto reference genomes. AdDNV_1KITE obtained 6,090 mapped reads (998,487 bases) with an average depth coverage of 189×, and a highest coverage of 659× on a single site in the NS region (Fig. 2C). The PcDNV_1KITE consensus sequence had 40,101 reads mapped against the genome at an average coverage of 1,325× and a highest coverage of 2,785× on a single site in the VP region (Fig. 2D). To examine whether the difference in sequencing depth may affect the viral gene expression patterns, we carried out downsampling analyses using proportions (1/10 and 1/20) of the PcDNV dataset (details provided in the Validation Section).
Several conserved protein domains play important roles in DNA replication, gene expression, infection and transfection in DNVs46,61,62. Conserved domains, including Parvo_NS1 located in NS proteins, Pavo_coat_N, Denso_VP4 and phospholipase A2 (PLA2) motif located in VP proteins appeared to be highly expressed in both transcriptomes63,64 (Fig. 2 and Supplementary Text S1). In the meantime, the unevenness of coverage also suggested possibilities of novel transcripts or other special characters in particular regions that were prone or hard to be enriched during the library construction.
Splicing profiles and introns of two viruses
Seven AS patterns are commonly reported in many species: exon skipping (SE), mutually exclusive exons (MXE), intron retention (IR), alternative 3′ sites (A3SS), alternative 5′ sites (A5SS), alternative first exon (AFE) and alternative last exon (ALE)8,65,66. In our findings, five introns were detected in AdDNV_1KITE that involved three AS patterns, i.e., A5SS, A3SS and IR (Table 1, Supplementary Table S4). All five AdDNV introns belonged to the canonical intron GT-AG type67.
Detected splicing junction regions matched to previously reported AdDNV introns In (nt 223 to 855), Ib (nt 4403 to 4544) and II (nt 4260 to 4434), which were determined by Sanger sequencing of RT-PCR products48. Two A5SS introns, AdDNV_I1 (Table 1, nt 223 to 855, identical to the previously described In) and AdDNV_I2 (Table 1, nt 431 to 855) occurred in NS transcriptions. Two A3SS introns (Table 1, AdDNV_I3, nt 4245 to 4533; and AdDNV_I5, nt 4403 to 4533, the same as previously described Ib) and an IR intron (Table 1, AdDNV_I4, nt 4260 to 4434, previously described as II) occurred in the VP transcriptions48. In short, two novel introns (AdDNV_I2, _I3) and three known introns (AdDNV_I1, _I4, _I5) were detected while a previously reported intron (Ia, nt 4403 to 4758) was not detected in this research. Reads supported splicing junction models were showed in Fig. 3A by Integrative Genomics Viewer (IGV)68. Nine long open reading frames (ORFs) ranging from 207 to 2,451 nt were detected in the AdDNV_1KITE genome (Table 2, Fig. 4), six of which had been previously validated by experiments48. Five of the ORFs did not require splicing whereas the other four were splicing products.

Splicing profiles of AdDNV and PcDNV. (A) Detected splicing junctions of AdDNV_1KITE. (B) Detected splicing junction models of PcDNV_1KITE: Solid gray areas represented the TopHat2 mapping profiles and each color-coded block represented a splicing junction. Red and purple blocks were forward and reverse junctions, respectively. The edge of each block represented the coverage of supporting reads and the length of a block represented the location of a splicing event. The number near each block was the coverage of supporting reads. The middle bridge showed the intron region from the splicing event. The block thickness represented frequency (the number of supporting reads) of the intron. Splice site compositions for donor sites, branch sites and acceptor sites of all GT-AG type introns in AdDNV_1KITE (Panel C) and PcDNV_1KITE (Panel D) were displayed using WebLogo. The overall height of each stack indicated the sequence conservation at that position, measured in bits. Proteins mediating the GT-AG splicing were labelled as snRNP (small nuclear ribonucleoproteins) and SR (splicing regulatory proteins). (E) Log2 scale of reads density of introns in the genome alignment: The Y-axis showed the expression levels (the number of reads) of intron related splicing events. Introns with forward junctions (red labels, at NS region) and reverse junctions (blue labels, at VP region) of PcDNV_1KITE (top half) and AdDNV_1KITE (bottom half) were shown in the genome alignment. Multiple splicing events (orange labels) were also displayed.
Introns of AdDNV_I1, _I4 and _I5 were supported by 88, 135 and 18 reads, respectively (Table 1, Supplementary Table S4). These detected introns were congruent with experimental results from a previous study48, demonstrating that the NGS approach and bioinformatics applied in our study were reliable. The missing of a previously reported AdDNV (Intron Ia, nt 4403 to 4758) might be caused by inter-individual differences. In addition, two novel introns (AdDNV_I2 and AdDNV_I3) were detected at low read numbers (3 and 7, respectively, Table 1). Three out of four introns were validated using RT-PCR (details provided in the Validation Section). AdDNV_I2 was confirmed demonstrating that NGS was sensitive in detecting introns at very low expression levels.
The splicing junctions and AS pattern of PcDNV are reported in the present study for the first time (Fig. 3B). The A5SS and A3SS introns occurred in PcDNV with high frequencies. Seven out of eight NS introns belonged to A5SS and all shared the same 3′ site at nt 879 (Fig. 3E). In the VP encoding region, four splicing islands were detected and all of them contained either A5SS or A3SS or both modes (Fig. 3E and Table 1). Five in 23 introns had only a single read support suggesting limited function if there were any. All introns belonged to canonical GT-AG introns69. Twenty-eight ORFs ranging from 531 to 2,517 nt were detected in the PcDNV genome (Table 2, Fig. 4). Five of the ORFs, as in AdDNV, did not require RNA splicing whereas the others (23 out of 28) were splicing products.

Inferred viral gene products. Viral gene products were annotated according to viral genome positions. The NS genes were represented in forward direction (Panels A and C) and the VP genes were represented in reverse direction (Panels B and D). For the splicing products, numbers of the detected splicing reads/non-splicing reads (over the intron) were listed next to the gene ID (covering the donor and receptor junctions). Numbers of the non-splicing reads of the donor (d) and receptor (r) sites were also labeled. Positions of start codons, stop codons and amino acids at splicing junctions were shown in the reading frames of forward (NS) and reverse (VP) polarities.
Different from AdDNV, PcDNV lacked the major AdDNV_I1 intron in the NS region. Instead, PcDNV displayed a set of seven A5SS introns with a shared receptor junction at nt 879. With different donor positions, the PcDNV NS introns produced a set of novel proteins composing NS ORF1 and NS ORF2 (Fig. 4C). The dominant NS splicing was PcDNV_NS_ORF7_I2. Other seven splicing events in NS region produced more additional isoforms of the ORF1-ORF2 protein and ORF1-ORF3 protein (Fig. 4C). 17 out of the 28 ORFs were located in VP region. The most common VP splicing in PcDNV was PcDNV_VP_ORF8_I16 (Fig. 4D). Like the previously described AdDNV_VP_ORF6_I4, PcDNV_VP_ORF8_I16 eliminated the stop codon of PcDNV_VP_ORF5 and joined PcDNV_VP_ORF4 (homologue of AdDNV-VP2) reading frame. Downsampling tests of PcDNV showed similar results (details in Validation Section), demonstrating that the differences observed between the two viruses were not solely caused by sequencing depth. More detailed descriptions of gene products of the two viruses could be found in Supplementary Text S1.
Gene expression features of AdDNV and PcDNV
Gene expressions of AdDNV_1KITE
By calculating the ratio of the number of splicing and non-splicing reads spanning the exon-intron regions and the FPKM/TPM values of the viral ORFs, expression patterns were compared between AdDNV and PcDNV. Both RSEM70 (Table 2) and Kallisto71 (Supplementary Table S6) produced similar profiles. Junctions with high splicing/non-splicing ratios also produced transcripts with high FPKM/TPM values. In AdDNV, the ratio of AdDNV_I4 was the highest in AdDNV (Supplementary Table S4) and AdDNV_NS_ORF2 (Fig. 4A) had the highest FPKM indicating that the encoded AdDNV_NS1 was the most abundantly expressed protein (Table 2) while AdDNV_VP_ORF6_I4 was the mostly expressed VP isoform (VP1) (Fig. 4B).
Gene expressions of PcDNV_1KITE
PcDNV_I16 had the highest splicing/non-splicing ratio in PcDNV, followed by PcDNV_I23 (Supplementary Table S4). Based on the FPKM values, PcDNV_NS_ORF7_I2, PcDNV_NS_ORF6_I1 and PcDNV_NS_ORF1 were the most abundantly expressed PcDNV_NS proteins (Fig. 4C, Table 2). Although PcDNV_VP_ORF8_I16 encoded a VP protein similar to AdDNV_VP_ORF6_I4 (AdDNV_VP1), its FPKM value was second to that of PcDNV_VP_ORF10_I23 which encoded a novel protein without any conserved Ambiensovirus VP motifs48,57,72 (Fig. 4B and D, Table 2). The PcDNV_NS proteins had smaller FPKM values than the PcDNV_VP proteins, suggesting that PcDNV_NS proteins were expressed less abundantly than the PcDNV_VP proteins (Table 2). This was different to the situation found in AdDNV.
Expression patterns of viral genes
Expression patterns of the viral transcripts were significantly different between AdDNV and PcDNV on two levels, i.e., transcript isoforms and expression abundance (Fig. 4 and Table 2). Most of the spliced transcripts had low FPKM values or even effectively zero count (for those with small effective lengths), suggesting that these rare splicing products were unlikely to be responsible for any fundamental viral function48,57,72. On the other hand, as splicing products dominated in both NS and VP of AdDNV and PcDNV, RNA splicing played essential roles in these two densoviruses. Differential splicing resulted in remarkable divergence of the viral transcriptome (Figs 3 and 4).
The two viruses were phylogenetically closely related (Supplementary Fig. S1) therefore were expected to adopt similar gene expression strategies including AS48,52. Indeed, the splicing junctions were located in both NS and VP regions in AdDNV and PcDNV (Fig. 3A and B) and IR, A5SS and A3SS modes of alternative splicing were also detected for PcDNV in this study. The majority of the PcDNV GT-AG splicing occurred at a region similar to the AdDNV splicing hotspot (Fig. 3E), i.e., PcDNV_I16 (covered by 1,994 reads) and AdDNV_I4 (covered by 135 reads) were both positioned at the genome alignment region from nt 4,596 to 4,418. This structural consistency indicated that this splicing event was conserved across species and likely played an important role in densoviruses (Fig. 3E, Table 1, Supplementary Table S4).
On the other hand, novel splicing events, including the canonical GT-AG introns with SE modes (Table 1), were discovered for PcDNV. There were more splicing events in PcDNV than in AdDNV, even if the same level of sequencing depth was tested in each sample. The question whether or not such a difference may be caused by the number of host individuals sampled remains open. In general, the PcDNV consensus was less restricted than that of AdDNV, particularly at both donor (+4G and +5T) and acceptor positions (−3C and −5T) (Fig. 3C and D, Supplementary Fig. S3, Supplementary Text S1). All five AdDNV introns belonged to the canonical intron GT-AG type67. It is worth noting that the consensus sequences of these GT-AG introns (nG|GTAnGTnG for donor and TnTTGCAG|An for acceptor, Fig. 3C) were different to the corresponding consensus sequences of GT-AG introns in PcDNV (AG|GTAAnnnn for donor and AnTTACAG|AT for acceptor with all junctions, AG|GTAAnnnn for donor and AATTACAG|AT for acceptor when junctions of very low frequencies (<3 reads) were excluded, Fig. 3D, Supplementary Fig. S3). Both viral consensus sequences had differences with the conserved consensus of U2 type GT-AG introns (AG|GTRAGT for donor and YYTTYYYYYYNCAG|G for acceptor) or U12 type introns (|RTATCCTTT for donor and TTCCTTRAY for branch sites) (Fig. 3C and D, Supplementary Fig. S3, Supplementary Text S1)69,73.
Validating splicing products using bioinformatics and experimental methods
Downsampling of the PcDNV dataset
To examine potential effects of sequencing depth on resultant splicing patterns, we randomly subsampled 1/10 and 1/20 of the PcDNV dataset and performed identical analyses on these subsamples. The 1/10 subsample had 2,067,041 reads in total, with 8,428 reads mapped to PcDNV after filtering (Supplementary Table S3). 12 out of 23 splicing junctions recovered from the full dataset were detected, including all major junctions (read depth > 50) and some (4 out of 14) minor junctions with low read depths (read depth < 10, Supplementary Tables S3 and S4). Similarly, the 1/20 subsample had 1,034,316 reads in total with 4,806 reads mapped to PcDNV after filtering. Although the mapped viral reads in the 1/20 subsample of PcDNV was fewer than that from AdDNV (6,090), more splicing junctions were detected (9 as oppose to 5 in AdDNV) (Supplementary Tables S3 and S4). These results showed that increases of sequencing depth enhanced the number of rare splicing junctions but had no impact on the detection of major splicing junctions.
Splicing results from RNA-seq aligner STAR
We also analyzed splicing patterns using STAR74. In AdDNV, the five introns detected by TopHat2 were also supported by STAR (Supplementary Tables S3 and S4). In PcDNV, STAR detected 28 introns (23 by TopHat2), including 23 GT-AG introns that were also detected by TopHat2 and five additional rare splicing junctions supported by single read (Supplementary Table S4).
Junctions validation using RT-PCR method
RNA extracts remaining from the production of the two transcriptomes were used for RT-PCR validations (Supplementary Table S7). Primers were designed based on ORF sequences revealed by viral genome assemblies and annotation from this study to amplify regions of the splicing junctions. Several ORFs could share the same primers (Table 3, Supplementary Table S8, Supplementary Fig. 4). As expected, RT-PCR amplified ORFs with the most abundant junctions or highest expression levels sharing the same primers. AdDNV_I2, _I4 and _I5 (read depths of 3, 135 and 18, respectively) and PcDNV_I1, _I2, _I16 and _I23 (read depths of 43, 275, 1994 and 1699, respectively) were validated by Sanger sequencing of the RT-PCR products75. However, some rare junctions with low coverage depths were not confirmed by this method. The results showed that deep sequencing was more sensitive in detecting rare junctions than the RT-PCR based approach. On the other hand, it is also possible that splicing junctions supported by a single read might be caused by sequencing and/or mapping errors.
Discussion
Virus infections are common in most eukaryotic organisms30. Deep sequencing of transcriptomes coupled with bioinformatics pipeline developed in the present study can readily detect transcripts of the target organism (host) as well as those of the pathogens. In our pipeline, we firstly screened the assembled sequences (scaffolds) for potential viral sequences against a customized viral database instead of the full Nt database. The resulting viral candidate sequences were then screened against the Nt database to remove false positives (sequences matched to non-viral subjects). The outcome of this two-steps virus screening is the same as directly using the assembled sequences to screen against the Nt database. However, virus screening using two large datasets (assembled sequences and Nt database) consumes much greater computer resource. Therefore, our pipeline for virus detection improved computational efficiency without compromising on accuracy. Given that a wide range of transcriptomes for non-model organisms have been de novo assembled by a series of large-scale transcriptome projects, including the 1KITE42,76,77, this practice provides an effective pathway to characterize virus diversity across various lineages of life.
In addition, our study demonstrated that transcriptome sequencing was an effective and accurate approach to improve the understandings of gene catalogues, expression levels, and RNA splicing patterns for pathogens. Viral gene expression profiles can be identified from the transcriptomes (Figs 3 and 4). Transcriptomic analysis in a phylogenetic context can help to elucidate functional conservativeness and novelty of expressed genes. For instance, phylogenetically closely related viruses, AdDNV and PcDNV, exhibited both conserved transcriptions and lineage-specific profiles. Common VP1 isoforms were abundantly transcribed using highly conserved splicing events (AdDNV_VP_ORF6_I4 and PcDNV_VP_ORF6_I16, Fig. 4), suggesting that these proteins were essential for the virus survival. On the other hand, differences in structural and non-structural proteins inferred from viral transcripts were found between the two viruses, including novel sequence divergences and distinctive variations in expression ratios and levels.
The diverse splicing mechanisms in PcDNV seemed to have directly led to more varieties of gene splicings than that in AdDNV (Fig. 3E, Table 1, Supplementary Tables S4 and S5) and these novel introns inturn produced new protein isoforms. The number of pooled individual specimens in the 1KITE project was justified according to the body weight of insects to provide sufficient RNA for transcriptome sequencing, and it could cause limitations in sample variation and inadequacies in detecting of inter-individual differences in the current study. Variations introduced by sequencing depth (1,325× versus 189×), number of pooled individual specimens (150 versus 1), temporal variation in gene expressions and inter-individual difference may have also contributed to some of the unique patterns observed in PcDNV, especially some of the rare introns. In future research, analyses on more virus-host pairs with controlled individual numbers could help to clarify the effects of different factors. RT-PCR based validations verified the majority of major novel splicing junctions, but most junctions with low read support could not be confirmed. Therefore, the possibility of artificial errors cannot be completely ruled out for expressed junctions at very low read coverage. Empirical validation of rare splicing junctions is a challenging task. Although the Sanger sequencing of RT-PCR products has successfully verified a number of major and minor junctions, the conventional method is far not as sensitive as NGS. Therefore, interpretation of rare slicing junctions should be cautious until additional technologies are developed for validation and functional testing.
Nevertheless, transcriptome sequencing of host and co-expressed pathogen creates a unique opportunity to examine host-virus association. To our knowledge, our report on AS consensus sequences were the first description for PcDNV and AdDNV. Viruses rely on the host machinery for RNA biology and can co-evolve with the host splicing78. It is likely that the observed divergence of viral RNA splicing patterns (Fig. 3) were influenced by both host and viral factors. Additional pipelines for analyzing the host splicing patterns may help us to understand the virus-host interaction and co-evolution in the future.
Methods
Virus detection
Raw reads were assembled using SOAPdenovo-Trans79 with the following settings: “-K 31 –i 20 -e 3 –M 3 –L 100”42. We then searched for matches against a customized virus database (described below) using the Basic Local Alignment Search Tool (BLASTn, version 2.2.26)80, including 6 steps (Fig. 1B).
Customized virus database
All known viruses in non-redundant nucleotide (Nt) databases and their corresponding taxonomic identity were downloaded from the GenBank (November 2014) and served as the virus reference database, which was much smaller than the complete Nt database but more dedicated to viruses. This modification improved screening efficiency and alleviated computational demand.
Virus search
Assembled transcriptome scaffolds were searched for sequence homology against the customized virus reference database using a local BLASTN algorithm (e value < 1e-5). Only query sequences with a match-length ≥200 nt and identity ≥90% were retained as candidate viral sequences for further analyses. Overlapping candidate viral sequences were merged by combining the BLASTN results using an in house Perl script (available at https://github.com/linzhi2013/Virusfishing) with improved procedures in query selection (merged candidate viral sequences versus whole scaffolds or sequencing reads)81.
Removal of false positives
Candidate viral sequences were compared to the complete Nt database using BLASTN aiming to identify false positives. Sequences with higher scores to non-virus subjects were deemed as false positives and subsequently removed from downstream analyses.
Viral consensus genome calling
Virus genomes were achieved by calling consensus sequences. Candidate reference genomes (based on sequence homology from previous steps) were chosen from viral database to aid the following calling of relevant virus genome. Burrows-Wheeler Aligner (BWA, version 0.7.10)58 was applied to align all raw reads of the transcriptome onto virus reference genomes with default parameters. After removal of PCR redundancy using SAMtools (If multiple read pairs have identical external coordinates, only the pair with highest mapping quality was retained) and reads with more than two mismatches, consensus viral sequences were obtained using SAMtools (version 0.1.19)82, with ambiguous sites substituted by the base of highest allele frequency and with missing sites replaced by Ns.
Genome coverage and annotation
ReSeqTools (version 0.23)83 and self-developed Perl scripts were used to calculate the genome coverage and read depth. Viral gene information was downloaded from the GenBank. All translation start site and translation termination site at both directions were annotated based on prediction of amino acid sequences.
Phylogram construction
The virus phylogram was inferred using both genome sequences and protein sequences of the assembled viruses, reference viruses and other nine related viruses from the same family downloaded from GenBank. MUSCLE84 was applied to conduct multiple sequence alignments (MSA) and MEGA5 was used for tree construction using the maximum likelihood method85 and Neighbour-Joining method86 with 1000 bootstraps (MEGA5 software)87.
Gene expression analyses
Gene expressions were analysed in five steps:
Detection of alternative splicing
TopHat2 (version tophat-2.0.7)59,88 was applied to indentify RNA splicing patterns based on junction signals. Alternative splicing events were identified accroding to gene splicing patterns. All transcriptome reads were mapped to reference viral genome assemblies and host database using paramters “-r 10 -i 50 -I 2000–library-type fr-unstranded –G” according to the TopHat2 manual, respectively. TopHat2 aligns reads that are spanning across gaps onto a reference more efficiently than the unspliced aligners, such as BWA and Bowtie60,89. RNA mapping characteristics were obtained from high-quality BAM files with unique mapping reads after removing of PCR redundancy and reads with more than two mismatches.
Determination of AS junctions, introns and gene expression patterns
Splicing junction locations and counts were obtained from spliced sequences that might contain one or two junctions per read. Non-canonical splicing sites with low number of supporting (n < 3) reads were filtered out from further analysis. And the putative branch site regions were identified in introns by searching regions relative to the 3′ splicing site67, using an in house Perl script. Sequence logos of all detected junctions and junctions with coverage depth ≥ 3 were generated by WebLogo90 and consensus sequences were generated by the tool cons from EMBOSS91, respectively. Introns and splicing patterns were identified according to the letters on donor and acceptor sites of the splice junctions69,73,92,93. The splicing level of an intron was recorded as read counts mapped to a junction, similar to the method by MATS94 in calculating the exon inclusion level. Integrative Genomics Viewer (IGV)68,95 was used to show the putative viral AS and intron models.
Annotation of viral open reading frames
Firstly, the putative coding regions of the viral genome were identified using an in house Perl script. Putative expression products with amino acid length > 30 aa were defined according to the open reading frames (ORFs) from the unspliced/spliced sequences. The resulting amino acid sequences were annotated using the online searching tool BLASTp at NCBI80. ORFs and spliced isoforms were named following the names described in the NCBI annotation with splicing sites noted as: I for intron and MI for multiple introns of a splicing event.
Comparison between related viral species
Multiple aligments were conducted for genome sequences using ClustalW96 to compare splicing junction locations between viral species. The graphical mapping details of final alignments were drawn using R.
Calculation of expression levels
A local estimation of each junctions were used to show their expression levels. The number of splicing and non-splicing reads located in junction regions were caculated based on the alignment Bam files. The number of non-splicing reads with donor or acceptor sites (reads spanning the exon-intron) were also caculated. We considered one ORF as one gene to calculate the expression levels. The expression level related values: i.e., the counts of positions of valid fragment (effective length), the sum of the posterior probability of each read coming from this part over all reads (expected count), the transcripts per million (TPM), and fragments per kilobase of transcript per million mapped reads (FPKM), were calculated using RSEM70 and Kallisto71. A relative expression level of each viral gene was measured using the FPKM value of one gene divided by the FPKM value of the highest expressed gene of the same virus.
Validation of splicing products
Downsampling
1/10 and 1/20 subsamples of PcDNV related whole transcriptome reads were randomly selected from total reads using the toolkit Seqtk97 and analyzed using the same analytical pipeline conducted for the full transcriptomes to examine potential impact of sequencing depth.
Junction detection using STAR
STAR (version 2.5.3a) was applied to check the detected junctions using the parameters “–runMode genomeGenerate–sjdbOverhang 149–genomeChrBinNbits 12–genomeSAindexNbases 7 –sjdbGTFfile …” for STAR index construction and “–alignSJoverhangMin 5–alignIntronMin 50–alignIntronMax 2000–outSJfilterOverhangMin 5 5 5 5” for alignment. Then the alignment BAM file was used to define junctions using the script “Splicing_Search.pl” in pipeline after filtering with the same parameters as described in the TopHat2 step.
DNaseI treatment and reverse transcription
Total mRNAs were obtained from the 1KITE project directly and the quality was evaluated using a Qubit Fluorometer. To eliminate DNA contamination, 1 μg of total RNA was treated with 1 U DNaseI (Promega, US) and incubated at 37 °C for 30 min. Then samples (1 μg) of total RNA were reversely transcribed in a 25 μL reaction mixtures with oligo(dT)15 primers and M-MLV reverse transcriptase (Promega, US).
PCR
Splicing was validated by PCR of the cDNAs using primers designed from viral genome sequences from this study (Supplementary Table S8). The PCR mixture (50 μL) consisted of 4 μL cDNA template, 0.2 μM primers, 200 μM dNTP mix and 2.5 U TaKaRa LA Taq polymerase. The amplification conditions were denaturing at 94 °C for 5 min, then 40 cycles of denaturing at 94 °C for 30 s, annealing at 50 °C for 30 s, and extension at 72 °C (the extension time depends on the length of PCR amplicon, 1 kb/min), and final extension at 72 °C for 10 min.
Cloning and Sanger sequencing
PCR products were cloned into pEASY-T1 simple cloning vector (Transgen, China) and transformed into Trans1-T1 Phage resistant chemically competent cells (Transgen, China) for Sanger sequencing at Ruibiotech Company (Beijing, China).
Data Availability
Raw data are available from NCBI bioprojects PRJNA286330 (A. domesticus) and PRJNA219593 (P. citri). The virus screening pipeline is available from https://github.com/linzhi2013/Virusfishing.
References
Qi, Y.-X., Liu, Y.-B. & Rong, W.-H. RNA-Seq and its applications: a new technology for transcriptomics. Hereditas (Beijing) 33, 1191–1202 (2011).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63, https://doi.org/10.1038/nrg2484 (2009).
Mutz, K.-O., Heilkenbrinker, A., Lönne, M., Walter, J.-G. & Stahl, F. Transcriptome analysis using next-generation sequencing. Current Opinion in Biotechnology 24, 22–30 (2013).
Kelemen, O. et al. Function of alternative splicing. Gene 514, 1–30 (2013).
Graveley, B. R. Alternative splicing: increasing diversity in the proteomic world. TRENDS in Genetics 17, 100–107 (2001).
Li, Y. I. et al. RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604 (2016).
Lareau, L. F., Brooks, A. N., Soergel, D. A., Meng, Q. & Brenner, S. E. The coupling of alternative splicing and nonsense-mediated mRNA decay. Alternative splicing in the Postgenomic Era 623 (2007).
Feng, H., Qin, Z. & Zhang, X. Opportunities and methods for studying alternative splicing in cancer with RNA-Seq. Cancer Lett 340, 179–191, https://doi.org/10.1016/j.canlet.2012.11.010 (2013).
Black, D. L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336 (2003).
Matera, A. G. & Wang, Z. A day in the life of the spliceosome. Nature reviews. Molecular cell biology 15, 108–121, https://doi.org/10.1038/nrm3742 (2014).
Burset, M., Seledtsov, I. A. & Solovyev, V. V. SpliceDB: database of canonical and non-canonical mammalian splice sites. Nucleic Acids Res 29, 255–259 (2001).
Griffith, M. et al. Alternative expression analysis by RNA sequencing. Nat Methods 7, 843–847 (2010).
Marquez, Y., Brown, J. W., Simpson, C., Barta, A. & Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome research 22, 1184–1195 (2012).
Hardy, J. & Singleton, A. Genomewide association studies and human disease. N Engl J Med 360, 1759–1768, https://doi.org/10.1056/NEJMra0808700 (2009).
Tang, K. W., Alaei-Mahabadi, B., Samuelsson, T., Lindh, M. & Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun 4, 2513, https://doi.org/10.1038/ncomms3513 (2013).
Guo, L. et al. A De Novo-Assembly Based Data Analysis Pipeline for Plant Obligate Parasite MetatranscriptomicStudies. Frontiers in Plant Science 7 (2016).
Katzourakis, A. & Gifford, R. J. Endogenous viral elements in animal genomes. PLoS Genet 6, e1001191–e1001191 (2010).
Geering, A. D. et al. Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat Commun 5, 5269, https://doi.org/10.1038/ncomms6269 (2014).
Liu, H. et al. Widespread endogenization of densoviruses and parvoviruses in animal and human genomes. Journal of virology 85, 9863–9876, https://doi.org/10.1128/JVI.00828-11 (2011).
Rohwer, F., Prangishvili, D. & Lindell, D. Roles of viruses in the environment. Environmental microbiology 11, 2771–2774 (2009).
Mokili, J. L., Rohwer, F. & Dutilh, B. E. Metagenomics and future perspectives in virus discovery. Curr Opin Virol 2, 63–77, https://doi.org/10.1016/j.coviro.2011.12.004 (2012).
Liu, S., Vijayendran, D. & Bonning, B. C. Next generation sequencing technologies for insect virus discovery. Viruses 3, 1849–1869, https://doi.org/10.3390/v3101849 (2011).
Roossinck, M. J. Plant virus metagenomics: biodiversity and ecology. Annu Rev Genet 46, 359–369, https://doi.org/10.1146/annurev-genet-110711-155600 (2012).
Pennisi, E. Going Viral: Exploring the Role Of Viruses in Our Bodies. Science 331, 1513–1513, https://doi.org/10.1126/science.331.6024.1513 (2011).
Chen, Y. et al. VirusSeq: software to identify viruses and their integration sites using next-generation sequencing of human cancer tissue. Bioinformatics 29, 266–267, https://doi.org/10.1093/bioinformatics/bts665 (2013).
Wang, H. et al. Sequence recombination and conservation of Varroa destructor virus-1 and deformed wing virus in field collected honey bees (Apis mellifera). PloS one 8, e74508 (2013).
Radford, A. D. et al. Application of next-generation sequencing technologies in virology. The Journal of general virology 93, 1853–1868, https://doi.org/10.1099/vir.0.043182-0 (2012).
Routh, A. & Johnson, J. E. Discovery of functional genomic motifs in viruses with ViReMa-a Virus Recombination Mapper-for analysis of next-generation sequencing data. Nucleic Acids Res 42, e11, https://doi.org/10.1093/nar/gkt916 (2014).
Wang, Q., Jia, P. & Zhao, Z. VirusFinder: Software for Efficient and Accurate Detection of Viruses and Their Integration Sites in Host Genomes through Next Generation Sequencing Data. PLOS ONE 8, e64465, https://doi.org/10.1371/journal.pone.0064465 (2013).
Wang, Y. et al. A survey of overlooked viral infections in biological experiment systems. PLoS One 9, e105348, https://doi.org/10.1371/journal.pone.0105348 (2014).
Skalsky, R. L. & Cullen, B. R. Viruses, microRNAs, and host interactions. Annu Rev Microbiol 64, 123–141, https://doi.org/10.1146/annurev.micro.112408.134243 (2010).
Gilbert, J. A. & Dupont, C. L. Microbial metagenomics: beyond the genome. Ann Rev Mar Sci 3, 347–371, https://doi.org/10.1146/annurev-marine-120709-142811 (2011).
Edwards, R. A. & Rohwer, F. Viral metagenomics. Nature Reviews Microbiology 3, 504–510 (2005).
Cullen, B. R. The virology-RNA biology connection. RNA 21, 592–594, https://doi.org/10.1261/rna.049882.115 (2015).
Cook, S. et al. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS One 8, e80720, https://doi.org/10.1371/journal.pone.0080720 (2013).
Ma, J., Pallett, D., Jiang, H., Hou, Y. & Wang, H. Mutational bias of Turnip Yellow Mosaic Virus in the context of host anti-viral gene silencing. Virology 486, 2–6 (2015).
Donaldson, E. F. et al. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. Journal of virology 84, 13004–13018 (2010).
Johansson, C. & Schwartz, S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nature reviews Microbiology 11, 239–251 (2013).
Gatherer, D. et al. High-resolution human cytomegalovirus transcriptome. Proceedings of the National Academy of Sciences 108, 19755–19760 (2011).
Concha, M. et al. Identification of new viral genes and transcript isoforms during Epstein-Barr virus reactivation using RNA-Seq. Journal of virology 86, 1458–1467 (2012).
Breitbart, M. & Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends in microbiology 13, 278–284 (2005).
Misof, B. et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 346, 763–767, https://doi.org/10.1126/science.1257570 (2014).
Pham, H. T. et al. Comparative genomic analysis of Acheta domesticus densovirus isolates from different outbreaks in Europe, North America, and Japan. Genome announcements 1, e00629–00613 (2013).
Thao, M. L., Wineriter, S., Buckingham, G. & Baumann, P. Genetic characterization of a putative Densovirus from the mealybug Planococcus citri. Current microbiology 43, 457–458, https://doi.org/10.1007/s002840010339 (2001).
Tijssen, P. et al. Evolution of densoviruses. Parvoviruses 55, (2005).
Cotmore, S. F. & Tattersall, P. Parvovirus DNA Replication. Cold Spring Harbor Monograph Archive 31, 799–813 (1996).
Berns, K. I. Parvovirus replication. Microbiological reviews 54, 316–329 (1990).
Liu, K. et al. The Acheta domesticus densovirus, isolated from the European house cricket, has evolved an expression strategy unique among parvoviruses. Journal of virology 85, 10069–10078, https://doi.org/10.1128/JVI.00625-11 (2011).
Mukha, D. V., Chumachenko, A. G., Dykstra, M. J., Kurtti, T. J. & Schal, C. Characterization of a new densovirus infecting the German cockroach, Blattella germanica. The Journal of general virology 87, 1567–1575, https://doi.org/10.1099/vir.0.81638-0 (2006).
Sukhumsirichart, W., Attasart, P., Boonsaeng, V. & Panyim, S. Complete nucleotide sequence and genomic organization of hepatopancreatic parvovirus (HPV) of Penaeus monodon. Virology 346, 266–277, https://doi.org/10.1016/j.virol.2005.06.052 (2006).
Kapelinskaya, T. V., Martynova, E. U., Schal, C. & Mukha, D. V. Expression strategy of densonucleosis virus from the German cockroach, Blattella germanica. Journal of virology 85, 11855–11870, https://doi.org/10.1128/JVI.05523-11 (2011).
Yu, Q. & Tijssen, P. Gene expression of five different iteradensoviruses: Bombyx mori densovirus, Casphalia extranea densovirus, Papilio polyxenes densovirus, Sibine fusca densovirus, and Danaus plexippus densovirus. Journal of virology 88, 12152–12157, https://doi.org/10.1128/JVI.01719-14 (2014).
Yamagishi, J., Hu, Y., Zheng, J. & Bando, H. Genome organization and mRNA structure of Periplaneta fuliginosa densovirus imply alternative splicing involvement in viral gene expression. Archives of virology 144, 2111–2124 (1999).
Fediere, G., Li, Y., Zadori, Z., Szelei, J. & Tijssen, P. Genome organization of Casphalia extranea densovirus, a new iteravirus. Virology 292, 299–308, https://doi.org/10.1006/viro.2001.1257 (2002).
Zhai, Y. G. et al. Isolation and characterization of the full coding sequence of a novel densovirus from the mosquito Culex pipiens pallens. The Journal of general virology 89, 195–199, https://doi.org/10.1099/vir.0.83221-0 (2008).
Baquerizo-Audiot, E. et al. Structure and expression strategy of the genome of Culex pipiens densovirus, a mosquito densovirus with an ambisense organization. Journal of virology 83, 6863–6873, https://doi.org/10.1128/JVI.00524-09 (2009).
Tijssen, P. et al. Organization and Expression Strategy of the Ambisense Genome of Densonucleosis Virus of Galleria mellonella. Journal of virology 77, 10357–10365, https://doi.org/10.1128/jvi.77.19.10357-10365.2003 (2003).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36, https://doi.org/10.1186/gb-2013-14-4-r36 (2013).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111, https://doi.org/10.1093/bioinformatics/btp120 (2009).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res 42, D222–230, https://doi.org/10.1093/nar/gkt1223 (2014).
Marchler-Bauer, A. et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res 43, D222–226, https://doi.org/10.1093/nar/gku1221 (2015).
Nüesch, J. P. & Tattersall, P. Nuclear targeting of the parvoviral replicator molecule NS1: evidence for self-association prior to nuclear transport. Virology 196, 637–651 (1993).
Tullis, G. E., Burger, L. R. & Pintel, D. J. The minor capsid protein VP1 of the autonomous parvovirus minute virus of mice is dispensable for encapsidation of progeny single-stranded DNA but is required for infectivity. Journal of virology 67, 131–141 (1993).
Gerstein, M. B. et al. Comparative analysis of the transcriptome across distant species. Nature 512, 445–448, https://doi.org/10.1038/nature13424 (2014).
Sammeth, M., Foissac, S. & Guigó, R. A general definition and nomenclature for alternative splicing events. PLoS Comput Biol 4, e1000147 (2008).
Burge, C. B., Padgett, R. A. & Sharp, P. A. Evolutionary fates and origins of U12-type introns. Molecular cell 2, 773–785 (1998).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192, https://doi.org/10.1093/bib/bbs017 (2013).
Sharp, P. A. & Burge, C. B. Classification of introns: U2-type or U12-type. Cell 91, 875–879 (1997).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 12, 323, https://doi.org/10.1186/1471-2105-12-323 (2011).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nature biotechnology 34, 525–527 (2016).
Ward, A. J. & Cooper, T. A. The pathobiology of splicing. The Journal of pathology 220, 152–163, https://doi.org/10.1002/path.2649 (2010).
Burset, M., Seledtsov, I. & Solovyev, V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic acids research 28, 4364–4375 (2000).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Clementi, M. et al. Quantitative PCR and RT-PCR in Virology. Genome Research 2, 191–196 (1993).
Matasci, N. et al. Data access for the 1,000 Plants (1KP) project. GigaScience 3, 1 (2014).
Sun, Y. et al. Fish-T1K (Transcriptomes of 1,000 Fishes) Project: large-scale transcriptome data for fish evolution studies. GigaScience 5, 1 (2016).
Dubois, J., Terrier, O. & Rosa-Calatrava, M. Influenza viruses and mRNA splicing: doing more with less. MBio 5, e00070–00014 (2014).
Xie, Y. W. et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 6 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool.pdf. journal of molecular biology 215, 403–410 (1990).
Moustafa, A. et al. The blood DNA virome in 8,000 humans. PLoS pathogens 13, e1006292 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
He, W. et al. ReSeqTools: an integrated toolkit for large-scale next-generation sequencing based resequencing analysis. Genetics and Molecular Research 12, 6275–6283 (2013).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 5, https://doi.org/10.1093/nar/gkh340 (2004).
Kishino, H. & Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. Journal of Molecular Evolution 29, 170–179 (1989).
Nei, N. S. A. M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4 (1987).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution 28, 2731–2739, https://doi.org/10.1093/molbev/msr121 (2011).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 7, 562–578 (2012).
Garber, M., Grabherr, M. G., Guttman, M. & Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nature methods 8, 469–477 (2011).
Crooks, G. E., Hon, G., Chandonia, J.-M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome research 14, 1188–1190 (2004).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European molecular biology open software suite. Trends in genetics 16, 276–277 (2000).
Sparks, M. E. & Brendel, V. Incorporation of splice site probability models for non-canonical introns improves gene structure prediction in plants. Bioinformatics 21(Suppl 3), iii20–30, https://doi.org/10.1093/bioinformatics/bti1205 (2005).
Parada, G. E., Munita, R., Cerda, C. A. & Gysling, K. A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res 42, 10564–10578, https://doi.org/10.1093/nar/gku744 (2014).
Shen, S. et al. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic acids research, gkr1291 (2012).
Robinson, J. T. et al. Integrative genomics viewer. Nature biotechnology 29, 24–26 (2011).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948, https://doi.org/10.1093/bioinformatics/btm404 (2007).
Seqtk, January 2017. Heng Li, Broad Institute, USA. https://github.com/lh3/seqtk (2017).
Acknowledgements
Authors thank the 1KITE initiative (Bernhard Misof, Karen Meusemann, Alexander Donath, Kai Schuette and the 1KITE Polyneoptera group) for granting access to transcriptome sequences and RNA materials. The authors thank colleagues from the China National Genebank and BGI-Shenzhen for their effort in sampling, infrastructure support, data operation and thoughtful comments. Great thanks to the two reviewers for constructive suggestions on improving the manuscript. This work was supported by the China National GeneBank, BGI-shenzhen, China Agricultural University and Sichuan University, and by the Natural Environment Research Council (UK) (NE/I000593/1 and NE/L012863/1 to HW) and the National Natural Science Foundation of China (No. J1103518). X.Z. is also supported by the Chinese Universities Scientific Fund, 2017QC114 and by grant through the Beijing Advanced Innovation Center for Food Nutrition and Human Health.
Author information
Authors and Affiliations
Contributions
X.Z., H.W. and Y.Z. conceived and designed the study. C.Z. led data analyses and pipeline construction. S.L., W.S., J.W. and H.M.Y. coordinated scientific questions. S.Q.L. led RT-PCR validations. G.M., C.Y. and J.M. participated in pipeline construction. W.S., C.Y. and H.Y. assisted writing. L.W. and S.G. helped with RNA splicing analysis. C.Z. wrote first drafts and X.Z., W.H., S.L. improved the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, C., Liu, S., Song, W. et al. Characterization of viral RNA splicing using whole-transcriptome datasets from host species. Sci Rep 8, 3273 (2018). https://doi.org/10.1038/s41598-018-21190-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21190-7
This article is cited by
-
Reference gene and small RNA data from multiple tissues of Davidia involucrata Baill
Scientific Data (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
