
Abstract
To examine for a causal relationship between vitamin D and glioma risk we performed an analysis of genetic variants associated with serum 25-hydroxyvitamin D (25(OH)D) levels using Mendelian randomisation (MR), an approach unaffected by biases from confounding. Two-sample MR was undertaken using genome-wide association study data. Single nucleotide polymorphisms (SNPs) associated with 25(OH)D levels were used as instrumental variables (IVs). We calculated MR estimates for the odds ratio (OR) for 25(OH)D levels with glioma using SNP-glioma estimates from 12,488 cases and 18,169 controls, using inverse-variance weighted (IVW) and maximum likelihood estimation (MLE) methods. A non-significant association between 25(OH)D levels and glioma risk was shown using both the IVW (OR = 1.21, 95% confidence interval [CI] = 0.90–1.62, P = 0.201) and MLE (OR = 1.20, 95% CI = 0.98–1.48, P = 0.083) methods. In an exploratory analysis of tumour subtype, an inverse relationship between 25(OH)D levels and glioblastoma (GBM) risk was identified using the MLE method (OR = 0.62, 95% CI = 0.43–0.89, P = 0.010), but not the IVW method (OR = 0.62, 95% CI = 0.37–1.04, P = 0.070). No statistically significant association was shown between 25(OH)D levels and non-GBM glioma. Our results do not provide evidence for a causal relationship between 25(OH)D levels and all forms of glioma risk. More evidence is required to explore the relationship between 25(OH)D levels and risk of GBM.
Similar content being viewed by others
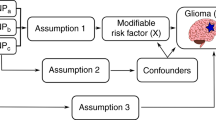


Introduction
While glioma accounts for around 80% of malignant primary brain tumours1, thus far exposure to ionising radiation is the only well-established exogenous risk factor2. Vitamin D provides many health benefits, including increased bone strength and protection against autoimmune diseases and type 2 diabetes3. In-vitro studies have also suggested an anti-neoplastic role for vitamin D4. Several epidemiological studies have shown that vitamin D may indeed afford protection against the development of some cancers, including colon, prostate and breast cancer5. Associations in such observational studies do not however constitute evidence for a causal relationship and in some studies bias from confounding and reverse causation cannot be excluded.
Mendelian randomisation (MR) uses genetic markers as proxies for environmental exposures to determine the effect of the exposure on disease risk6. It therefore provides a strategy for establishing causal relationships where randomised control trials (RCTs) would involve either high cost or impractical study design. In the case of a possible relationship between vitamin D and glioma, the rarity of the cancer would limit any RCT to small sample sizes and would require lengthy follow up times.
We implemented two-sample MR analysis to examine the relationship between vitamin D and glioma risk in order to avoid the limitations of follow up time, reverse causation and confounding. Genotypes are randomly assigned at conception, thereby limiting confounding. Furthermore an individual’s genotype will always be established before the onset of disease, excluding the possibility of reverse causation. The genotype is in part equivalent to a lifetime vitamin D deficiency, and hence a lifetime follow-up time in a RCT. We determine the relationship between vitamin D and glioma risk using genetic variants associated with 25(OH)D levels, rather than measuring 25(OH)D levels directly.
Genetic variants identified by the Study of Underlying Genetic Determinants of Vitamin D and Highly Related Traits (SUNLIGHT) Consortium7 and the Canadian Multicentre Osteoporosis Study (CaMOS)8 were used as an instrumental variable (IV). We performed an MR analysis to test for a causal relationship between 25(OH)D levels and glioma, using summary data from a recent genome-wide association study (GWAS) meta-analysis performed by the Glioma International Case-Control Consortium (GICC)9.
Methods
Two-sample MR was undertaken using GWAS data. Ethical approval was not sought for this specific project because all data came from the summary statistics of previously published GWAS, and no individual-level data were used.
Genetic variant instruments for 25(OH)D level
Genetic variants used as IVs were selected from the previously published SUNLIGHT study7. The SUNLIGHT Consortium GWAS identified four genetic variants associated with lowered 25(OH)D levels in 33,996 individuals of European descent from 15 cohorts. These variants were rs2282679 in GC (vitamin D binding carrier protein), rs10741657 near CYP2R1 (converter of vitamin D to the active ligand for the vitamin D receptor), rs12785878 near DHCR7 (7-dehydrocholesterol synthesis from cholesterol, a precursor to vitamin D) and rs6013897 in CYP24A1 (degrader of active 1,25-dihydroxyvitamin D3 to inactive vitamin D)10. The roles of GC, CYP2R1, DHCR7 and CYP24A1 in the vitamin D pathway are shown in Fig. 1. Association estimates (per-allele log-ORs) for SNPs were taken from previously published studies, which used data from the CaMOS study, a population based cohort study of 2,347 Canadians, genotyped and assayed for 25(OH)D levels8,10,11. None of the SNPs were in linkage disequilibrium (i.e. r2 ≥ 0.001). For each SNP, we recovered the chromosome position, risk allele, genetic locus, F-statistic and association estimates (Table 1). Standard errors (SE) were calculated from F-statistics calculated by previous studies, which derive from the CaMOS cohort11. The risk allele was taken to be the 25(OH)D decreasing allele. Allele frequencies for these SNPs were compared between the 25(OH)D and glioma data sets to ensure that the effect estimates were recorded with respect to the same allele. This study calculated the variants to account for about 2% of the variation in circulating 25(OH)D levels, and have a combined F-statistic of 12.5712.

Effect of SNPs chosen as IVs on the vitamin D pathway. Genes that contain, or are in proximity to, variants chosen as IVs are highlighted green. P values for the association of these variants with 25(OH)D levels were 1.9 × 10−109 for GC, 2.1 × 10−27 for DHCR7, 3.3 × 10−20 for CYP2R1, and 6.0 × 10−10 for CYP24A1.
Glioma genotyping data
Association data between the four genetic variants and glioma were taken from the most-recent meta-analysis of GWAS in glioma9, which related >10 million genetic variants (after imputation) to glioma (Supplementary Table 1). This meta-analysis comprised eight GWAS datasets of individuals of European descent: FRE, GER, GICC, MDA, GliomaScan (NIH), UCSF-Mayo, UCSF and UK (Supplementary Table 2). All diagnoses were confirmed in accordance with WHO guidelines. Full quality control details are provided in previously published work9. Gliomas are heterogeneous and different tumour subtypes, defined in part by malignancy grade (for example, pilocytic astrocytoma World Health Organization (WHO) grade I, diffuse ‘low-grade’ glioma WHO grade II, anaplastic glioma WHO grade III and glioblastoma (GBM) WHO grade IV) can be distinguished13. To avoid diagnostic ambiguity and for simplicity we considered glioma subtypes as being either GBM or non-GBM.
Statistical analyses
We examined the association between circulating 25(OH)D levels and glioma (including subtypes) using MR on summary statistics using the inverse variance weighted (IVW) and maximum likelihood estimation (MLE) methods, as described by Burgess et al.14. The combined ratio estimate (\(\hat{\beta }\)) of all SNPs associated with 25(OH)D levels on glioma risk was calculated under a fixed-effects model:
\({X}_{k}\) is the association between SNP k with 25(OH)D levels, \({Y}_{k}\) is the association between SNP k and glioma risk with standard error \({\sigma }_{Y}\). The standard error of this association is given by:
We also conducted a likelihood based analysis using the same genetic summary data15. For this maximum likelihood estimate, a bivariate normal distribution for the genetic associations was assumed, and the R function optim was used to estimate \(\beta \). SE \((\beta )\) was calculated using observed information.
With the estimates from the two analyses calculated for each of the eight cohorts in the glioma data, we performed a meta-analysis under a fixed-effect model to derive final odds ratios (ORs) and confidence intervals (CIs)16.
To test whether the variants chosen as instruments were valid under MR assumptions, we examined the instruments for pleiotropy (multiple traits influenced by one gene) between the exposure and disease risk. This would be revealed as deviation from a linear relationship between SNPs and their effect size for 25(OH)D levels and glioma risk. We performed MR-Egger regression to test the average pleiotropic effect caused by the variants combined, as well as to provide a third association estimate between 25(OH)D level and glioma17. As per Dimitrakopoulou et al.18, we further evaluated the presence of horizontal pleiotropy by conducting stratified MR analyses using only the genetic variants influencing vitamin D synthesis (rs12785878, rs10741657) and vitamin D metabolism (rs2282679, rs6013897). rs12785878 has been associated with non-European status10 and we therefore also undertook a sensitivity analysis excluding rs12785878.
For each statistical test, we considered a global significance level of P < 0.05 as being satisfactory to derive conclusions. To assess the robustness of our conclusions, we imposed a conservative Bonferroni-corrected significance threshold of 0.017 (i.e. 0.05/3 tumour classifications).
The power of a MR investigation depends greatly on the proportion of variance in the risk factor that is explained by the IV. We therefore estimated study power to assess the strength of the results19. The detectable ORs at 80% power were 1.26 or 0.79 in the all glioma analysis, 1.34 or 0.75 in the GMB analysis and 1.35 or 0.74 in the non-GBM analysis. All power calculations were completed at a significance level of 0.05 and assumed the variants explained 2% of the total variance of 25(OH)D levels.
Results
The results of the IVW and MLE methods are summarised in Table 2. Results of the MR-Egger analysis are summarised in Table 3. Forest plots of all results from the IVW and MLE methods are shown in Figs 2 and 3. There was no evidence to support an association (i.e. P > 0.05) between circulating 25(OH)D levels and risk of all glioma using either the IVW (OR = 1.21, 95% CI = 0.90–1.62, P = 0.201) or MLE (OR = 1.20, 95% CI = 0.98–1.48, P = 0.083) methods. MR-Egger regression produced an intercept of −0.001 (95% CI = −0.019–0.017, P = 0.893) and therefore provided no evidence for pleiotropy amongst the genetic variants chosen as IVs (Supplementary Fig. 1). Hence there was no evidence of violation of MR assumptions.

Individual cohort and meta-analysis ORs calculated using the IVW method. (a) All glioma, (b) GBM and (c) non-GBM glioma. Boxes are OR point estimates with area proportional to the weight of the study. Diamonds are overall summary estimates, with 95% CIs given by the width. Vertical line is null value (OR = 1.0).

Individual cohort and meta-analysis ORs calculated using the MLE method. (a) All glioma, (b) GBM and (c) non-GBM glioma. Boxes are OR point estimates with area proportional to the weight of the study. Diamonds are overall summary estimates, with 95% CIs given by the width. Vertical line is null value (OR = 1.0).
We explored the possibility that a relationship between vitamin D and glioma may be subtype specific, considering GBM and non-GBM separately. We imposed a stronger significance threshold of P = 0.017 (i.e. 0.05/3), to correct for multiple testing. The MLE method identified an inverse relationship between 25(OH)D levels and risk of the GBM subtype, with an OR of 0.62 (95% CI = 0.43–0.89, P = 0.010). The IVW method provided a similar, but non-significant effect size (OR = 0.62, 95% CI = 0.37–1.04, P = 0.070). No evidence for an association between 25(OH)D levels and the non-GBM subtype was identified using either the IVW or MLE methods. MR-Egger regression provided intercepts of −0.013 (95% CI = −0.039–0.012, P = 0.307) for GBM and −0.005 (95% CI = −0.035–0.026, P = 0.768) for non-GBM, again providing no evidence of pleiotropy.
Stratified MR analyses using separate allelic scores for vitamin D synthesis and metabolism did not indicate the presence of horizontal pleiotropy (Supplementary Tables 3 and 4). To address the potential effects of population stratification, we undertook a MR sensitivity analysis excluding rs12785878, as this SNP has been associated with non-European status10 (Supplementary Table 5). Excluding rs12785878, the inverse relationship between 25(OH)D levels and risk of the GBM subtype identified by the MLE method remains significant (OR = 0.51, 95% CI = 0.33–0.80, P = 0.003), thereby providing no evidence that this association is a result of population stratification.
Discussion
To our knowledge, this is the first MR study evaluating the effect of vitamin D on glioma risk undertaken. Overall our results do not provide evidence for an effect of vitamin D on risk of all forms of glioma. They do however raise the possibility for a protective role of vitamin D in GBM. While vitamin D and its metabolites have been shown to induce death of glioblastoma cells20,21,22, only one epidemiological study has investigated the relationship between pre-diagnostic levels of 25(OH)D and glioma risk23. Researchers found that higher levels of 25(OH)D were protective against high-grade glioma in men over the age of 56 (OR = 0.59), although the reverse trend was shown in men under the age of 56, albeit at a borderline-significant level23. Excluding the possibility of post hoc data mining, such paradoxical findings would support distinct aetiologies between the GBM and non-GBM subtypes, as has been suggested previously9.
Vital to the method of statistical analysis used herein is that none of the MR assumptions are violated. This requires that the variants chosen as IVs are (i) strongly associated with the exposure, (ii) are not associated with any confounding effects between exposure and outcome and (iii) are only associated with the outcome via the exposure. With regard to this study, the instruments chosen were associated with 25(OH)D levels at genome-wide significance levels. The MR-Egger test provided no evidence of horizontal pleiotropy, which we deemed sufficient to satisfy the third assumption. Furthermore, none of the four SNPs were in linkage disequilibrium (i.e. r2 ≥ 0.001) with any of the variants identified by Melin et al.9 as being in the risk region for glioma. With regard to confounding factors, few risk factors are known for glioma, so it was not possible to entirely rule out the possibility of unknown confounding factors causing statistical bias. However it should also be noted that all four SNPs lie either within or near genetic loci whose function in vitamin D physiology is well understood7, although a lack of knowledge of possible confounding factors means it was not possible to entirely rule out the possibility of confounding by unknown factors.
We acknowledge that a weakness of our study was in the small percentage of variability (around 2%) in 25(OH)D levels explained by the IV. Such a low value means any interpretation of these results as true indicators of the effect of total 25(OH)D levels on glioma risk are limited. This is quantified by the high ORs required for sufficient study power. Furthermore the study only accounts for circulating 25(OH)D levels and not for the action of 25(OH)D at the cellular level11. The genetic variants used as IVs in this MR analysis associate with 25(OH)D levels, rather than levels of the biologically active 1,25-dihydroxyvitamin D (1,25(OH)2D) and we therefore cannot explicitly comment on the relationship between 1,25(OH)2D and glioma. The low OR found in the GBM analysis should be noted however, given the fairly consistent indications of protective effects of 25(OH)D across all three methods. As is generally the case with MR, any findings should be viewed as a compliment to other future epidemiological studies, which test more robustly for associations between vitamin D and glioma and its subtypes.
In conclusion our MR analysis provides no evidence for an association between vitamin D and glioma, though findings raise the possibility of a potential association between vitamin D and GBM warranting further investigation.
Data Availability
Genotype data from the GICC GWAS are available from the database of Genotypes and Phenotypes (dbGaP; accession phs001319.v1.p1). Genotype data from the GliomaScan GWAS can also be accessed through dbGaP (accession phs000652.v1.p1). Data from the other studies are available upon request.
Change history
23 May 2019
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Dolecek, T. A., Propp, J. M., Stroup, N. E. & Kruchko, C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro Oncol 14(Suppl 5), v1–49 (2012).
Ostrom, Q. T. et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol 16, 896–913 (2014).
Wang, S. Epidemiology of vitamin D in health and disease. Nutr Res Rev 22, 188–203 (2009).
Feldman, D., Krishnan, A. V., Swami, S., Giovannucci, E. & Feldman, B. J. The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer 14, 342–57 (2014).
Toner, C. D., Davis, C. D. & Milner, J. A. The vitamin D and cancer conundrum: aiming at a moving target. J Am Diet Assoc 110, 1492–500 (2010).
Sheehan, N. A., Didelez, V., Burton, P. R. & Tobin, M. D. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med 5, e177 (2008).
Wang, T. J. et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet 376, 180–8 (2010).
Langsetmo, L. et al. Calcium and vitamin D intake and mortality: results from the Canadian Multicentre Osteoporosis Study (CaMos). J Clin Endocrinol Metab 98, 3010–8 (2013).
Melin, B. S. et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat Genet 49, 789–794 (2017).
Mokry, L. E. et al. Vitamin D and Risk of Multiple Sclerosis: A Mendelian Randomization Study. PLoS Med 12, e1001866 (2015).
Manousaki, D. et al. Vitamin D levels and susceptibility to asthma, elevated immunoglobulin E levels, and atopic dermatitis: A Mendelian randomization study. PLoS Med 14, e1002294 (2017).
Pierce, B. L., Ahsan, H. & Vanderweele, T. J. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol 40, 740–52 (2011).
Louis, D. N. et al. The2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131, 803–20 (2016).
Burgess, S. et al. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 30, 543–52 (2015).
Burgess, S. & Thompson, S. G. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol 181, 251–60 (2015).
de Bakker, P. I. et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet 17, R122–8 (2008).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44, 512–25 (2015).
Dimitrakopoulou, V. I. et al. Circulating vitamin D concentration and risk of seven cancers: Mendelian randomisation study. BMJ 359, j4761 (2017).
Brion, M. J., Shakhbazov, K. & Visscher, P. M. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol 42, 1497–501 (2013).
Magrassi, L., Butti, G., Pezzotta, S., Infuso, L. & Milanesi, G. Effects of vitamin D and retinoic acid on human glioblastoma cell lines. Acta Neurochir (Wien) 133, 184–90 (1995).
Magrassi, L. et al. Vitamin D metabolites activate the sphingomyelin pathway and induce death of glioblastoma cells. Acta Neurochir (Wien) 140, 707–13; discussion 713–4 (1998).
Garcion, E., Wion-Barbot, N., Montero-Menei, C. N., Berger, F. & Wion, D. New clues about vitamin D functions in the nervous system. Trends Endocrinol Metab 13, 100–5 (2002).
Zigmont, V. et al. Association Between Prediagnostic Serum 25-Hydroxyvitamin D Concentration and Glioma. Nutr Cancer 67, 1120–30 (2015).
Acknowledgements
HT was supported by a Wellcome Trust Summer Student bursary. AS is supported by a Cancer Research UK clinical Fellowship. In the UK, funding was provided by Cancer Research UK (C1298/A8362) supported by the Bobby Moore Fund. The GICC was supported by grants from the National Institutes of Health, Bethesda, Maryland (R01CA139020, R01CA52689, P50097257, P30CA125123). The UK Interphone Study was supported by the European Commission Fifth Framework Program “Quality of Life and Management of Living Resources” and the UK Mobile Telecommunications and Health Programme. The Mobile Manufacturers Forum and the GSM Association provided funding for the study through the scientifically independent International Union against Cancer (UICC).
Author information
Authors and Affiliations
Contributions
R.S.H. and A.J.C. managed the project. H.T., A.J.C., A.S., P.J.L. and R.S.H. drafted the manuscript. H.T. and A.J.C. performed statistical analyses. B.K., K.L., M.J.S. and R.H.S. provided U.K. data. M. Simon, P.H., M.M.N. and K.-H.J. provided German data. Q.T.O., J.E.E.-P., G.N.A., E.B.C., D.I., J.S., J.S.B.-S., S.H.O., J.L.B., R.K.L., C.J., R.B.J., B.S.M., M.R.W., M.L.B. and R.S.H. provided GICC data. S.C. and P.R. provided National Cancer Institute (NCI) data. M. Sanson provided French data. All authors reviewed the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Takahashi, H., Cornish, A.J., Sud, A. et al. Mendelian randomisation study of the relationship between vitamin D and risk of glioma. Sci Rep 8, 2339 (2018). https://doi.org/10.1038/s41598-018-20844-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20844-w
This article is cited by
-
Vitamin D and human health: evidence from Mendelian randomization studies
European Journal of Epidemiology (2024)
-
Systematic review of Mendelian randomization studies on risk of cancer
BMC Medicine (2022)
-
Testing for causality between systematically identified risk factors and glioma: a Mendelian randomization study
BMC Cancer (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
