
Abstract
Dominant optic atrophy (DOA) is a rare progressive and irreversible blinding disease which is one of the most frequent forms of hereditary optic neuropathy. DOA is mainly caused by dominant mutation in the OPA1 gene encoding a large mitochondrial GTPase with crucial roles in membrane dynamics and cell survival. Hereditary optic neuropathies are commonly characterized by the degeneration of retinal ganglion cells, leading to the optic nerve atrophy and the progressive loss of visual acuity. Up to now, despite increasing advances in the understanding of the pathological mechanisms, DOA remains intractable. Here, we tested the efficiency of gene therapy on a genetically-modified mouse model reproducing DOA vision loss. We performed intravitreal injections of an Adeno-Associated Virus carrying the human OPA1 cDNA under the control of the cytomegalovirus promotor. Our results provide the first evidence that gene therapy is efficient on a mouse model of DOA as the wild-type OPA1 expression is able to alleviate the OPA1-induced retinal ganglion cell degeneration, the hallmark of the disease. These results displayed encouraging effects of gene therapy for Dominant Optic Atrophy, fostering future investigations aiming at clinical trials in patients.
Similar content being viewed by others

Introduction
Hereditary optic neuropathies define a group of blinding diseases characterized by a progressive degeneration of the retinal ganglion cells (RGC)1. They are mainly due to modifications in genes encoding ubiquitous mitochondrial proteins and their pathological mechanisms responsible for the specific RGC degeneration remain poorly understood2. Dominant optic atrophy (DOA) is one of the most frequent forms of these diseases, and is mainly related to dominant OPA1 mutations3. Importantly, in the majority of DOA cases, haplo-insufficiency of the OPA1 protein represents the princeps mechanism responsible for the disease4 supporting that gene supplementation should be beneficial. Up to now, while gene therapy trials are under process in patients with the maternally-inherited Leber’s optic neuropathy, the other major hereditary optic neuropathy, no gene therapy approach has been reported for the treatment of DOA5. In the last years, we generated and characterized a DOA mouse model carrying the most frequent OPA1 mutation that faithfully reproduces the syndromic form of DOA6,7. Based on this model, we have appraised the efficiency of gene therapy involving the transduction of adeno-associated virus carrying the human wild-type OPA1 cDNA under the control of the Cytomegalovirus (CMV) promotor. Our results show that CMV-induced OPA1 expression prevents RGC loss in Opa1+/− treated mice.
Results
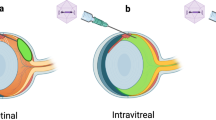
We designed a strategy based on the injection of an AAV2 serotype 2 known to transduce specifically retinal ganglion cells8 (Fig. 1). In this vector, the human isoform #1 OPA1 full length cDNA was incorporated and its expression was driven by the CMV promotor. To control the level of the human OPA1 (HsOPA1) gene expression, we transfected murine NIH3T3 cells with the pGG2 vector carrying the pCMV-HsOPA1 construction (Supplementary Figure S1). While the endogenous murine Opa1 gene was found expressed in untransfected, empty-pGG2 and pGG2-pCMV-HsOPA1 transfected cells, we found that HsOPA1 transcripts were quantified only in pGG2-pCMV-HsOPA1 transfected cells (Figure S1). HsOPA1 expression was found larger than the endogenous Opa1 gene (Figure S1A), which has been confirmed with the OPA1 protein expression monitored by western blot (Figure S1B). Thus, these in vitro experiments validated that the CMV promotor is able to efficiently enhance the HsOPA1 gene production.

Gene therapy strategy. pCMV: CMV promotor; HsOPA1:Homo sapiens OPA1; VEP: Visual Evoked Potentials; RGC: retinal ganglion cells. This strategy included 4 female Opa1+/+ controls, 4 untreated and 8 treated female Opa1+/− mice.
Then, a preliminary in vivo analysis using the AAV2/2-CMV-GFP construction was used to follow the transduction efficiency and long-lasting expression of this vector after injection (Fig. 2). We first quantified in control mice the GFP and the endogenous Opa1 gene expressions. Thus, using real-time quantitative PCR we monitored these gene expressions in retina as well as in optic nerve as negative control, 2 months after intravitreal injections (Fig. 2A). We found the expression of the murine Opa1 gene in both retina and optic nerve. By contrast, GFP transcripts were found expressed in retina but not in optic nerve. Then, we observed by in vivo funduscopy the presence of GFP positive retinal areas (Fig. 2B) and found that GFP positive cells are homogeneously distributed on whole mounted retinas. Co-immunolabelling with the Brn3a RGC specific marker showed that 42.9% ±2.03 of Brn3a positive cells were also GFP positive. Moreover, we found that 83% of the total number of GFP positive cells were Brn3a positive (Fig. 2C, Supplementary Figure S2).

Assessment of the vector transduction efficiency. (A) Quantification of murine endogenous Opa1 transcripts as well as GFP transcripts. MmOpa1: Mus musculus Opa1 (B) In vivo funduscopy was performed on untreated and treated Opa1+/− mice in brightfield (upper panel) and using a specific green filter to observe GFP fluorescence (lower panel). (C) Post-mortem immunohistochemistry was done on whole mount retinas using a RGC specific Brn3a labelling (Upper panel). Zoom-in (lower panel) was done to observe Brn3a/GFP double-positive RGCs (double arrow), Brn3a positive (arrow) and GFP negative RGCs.
We further performed intravitreal injections of the AAV2/2-pCMV-HsOPA1, carrying the human variant #1 OPA1 cDNA full length transcript on 3-month-old Opa1 mice, before the onset of progressive degenerative processes in retina6,7. Eight months following injection, we assessed the quality of the visual pathways from retinal ganglion cells to cortical areas by recording visual evoked potentials. As previously reported, we found a significant increase of the N-wave latency in untreated mice compared to controls (Fig. 3A). In consequence to treatment, Opa1+/− mice harbored similar increase of the N-wave latency as found in untreated mice. In addition, we analysed the scotopic threshold responses (STRs) recorded using flash-electroretinograms, that assess the activity of inner retina neurons9. We found a significant increase in the negative STR (nSTR) latencies of heterozygous untreated mice compared to controls (Fig. 3B). Interestingly, the nSTR latency values obtained after treatment were drastically decrease reaching those recorded in control animals (Fig. 3B). In parallel, we examined in vivo the outcome of the AAV2/2 injection on mice visual acuity. In agreement with our previous data7, Opa1+/− untreated mice developed a loss of visual acuity, revealed by the decrease of the frequency thresholds and the increase of the contrast perception. Interestingly, in treated Opa1 mice we found an increase of the frequency threshold comparable to that of controls (Fig. 3C). In the same way, contrast perception threshold was slightly decreased in treated Opa1+/− mice compared to untreated animals reaching level observed in control mice. Following the in vivo exploration, mice were sacrificed and we performed the RGC counting on whole mount retinas using the specific Brn3a RGC labelling. As previously described at 11 months, Opa1+/− untreated mice harbored a loss of RGCs compared to controls. In consequence to treatment, the number of RGC in Opa1+/− mice was significantly increased compared to untreated mice and reached control values (Fig. 3D).

Efficiency of the AAV2/2 injection on visual function and retinal ganglion cell number. (A and B) In vivo electrophysiological recordings. (A) visual evoked potential (VEP) N-wave latencies, (B) negative scotopic threshold response (nSTR). (C) Assessment of visual acuity using an optokinetic drum. Measurements of frequency thresholds (left) and contrast perception (right). NS: not significant. (D) post-fixed RGC counting using a specific Brn3a immuno-labelling in whole mount retinas. White rectangles: enlargement of whole-mount retinas. Brn3a positive dots were quantified using Imaris and MetaMorph® softwares. Data were expressed as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 using one-way ANOVA.
Discussion
Our results showed the efficiency of the AAV2 construction to transduce and maintain OPA1 expression in the mouse retinal ganglion cells. Moreover, injection of the construction carrying the human variant #1 OPA1 cDNA, which gives rise to both long and short OPA1 isoforms, driven by the CMV promotor prevents RGC progressive degeneration, which is the hallmark of the disease.
Beyond the prevention of retinal ganglion cell degeneration, we showed the benefit of OPA1 gene therapy on the electrophysiological measurements specific of retinal ganglion cell activity. Indeed, although visual evoked potentials (VEPs) recorded did not show any difference 9 months after injection, the analysis of STRs revealed a clear benefit of the treatment on retinal ganglion cell activity. VEPs are basically used to assess whether RGC axons are capable to carrying retinal signals along the geniculo-cortical pathway. On the other hand, STRs remain a more sensitive protocol to investigate inner retinal circuits and thus retinal ganglion cell activity. All together, these two electrophysiological approaches emphasized a dysfunction of retinal ganglion cells in Opa1 untreated mice. But interestingly, the effect of gene therapy improves more the retinal ganglion cell intra-retinal functioning rather than their capability to carry the electric signal to the cortical visual areas. These results are supported by those provided by the assessment of visual acuity in vivo. Indeed, visual acuity, which is the major parameter to appreciate visual capacities, remains slightly improved in Opa1+/− treated mice. Thus, our results show that OPA1 gene therapy may be sufficient to prevent retinal ganglion cell degeneration but not enough to efficiently maintain the conduction of the electric information to the visual perception zones. Interestingly, a discrepancy between cell loss prevention and electrophysiological function has been already described in the case of RPE65 gene therapy trials in both animals and patients with the Leber’s congenital amaurosis10. We can consider that the number of transfected retinal ganglion cells remains insufficient to trigger appreciable improvement of the electric activity along the optic path. Indeed, the amount of Brn3a positive RGCs without GFP expression suggests the existence of a cell reservoir that could be transduced with higher doses of viral genomes. Indeed, this could also explain why the level of GFP protein found in retina is significantly lower than the endogenous Opa1 expression. Nevertheless, this level of transduction remained efficient to prevent retinal ganglion cell dysfunction and degeneration.
These first gene therapy results established the hints that OPA1 expression in retinal ganglion cells can be an effective solution to treat dominant optic atrophy. This provides challenging perspectives to go further on the improvement of trials to efficiently develop pre-clinical trials for this blinding disease that nowadays remains without any treatment.
Material and Method
Opa1delTTAG mouse model
The Opa1 knock-in mice carrying the c.2708_2711delTTAG mutation was described in Sarzi et al.6. Mice were kept in the animal facility of the Institute for Neurosciences of Montpellier (B 3417236, 11/03 2010). All protocols were carried out on Opa1+/− mice and their Opa1+/+ littermates.
Intravitreous injections
Mice were anesthetized by intra-peritoneal injections of a mixture of Ketamine (120 mg/kg) and Rompun (10 mg/kg). The viral constructs used in this study were produced at Laboratoire d’Amplification de Vecteurs – CHU de Nantes, based on the provided sequences, and stocked at −80 °C until use. We introduced the human variant #1 OPA1 cDNA full length under the control of the CMV promotor. Two µl of the AAV2/2-pCMV-HsOPA1 or AAV2/2-pCMV-GFP vector were injected intra-vitreally at a titration of 1.6 1011 vg/ml or 3.2 108 per injection. The injected eye was covered with ophthalmic gel (Lacryvisc; Alcon®, Novartis; Fort Worth, USA). After installing a circular coverslip, the eye was pierced with a 34-gauge needle (Hamilton MicroSyringe; Reno, USA) for pressure equalization. Then, a Hamilton 5 μl syringe (MicroliterTM #65; Hamilton MicroSyringe; Reno, USA) with a blunt 34-gauge needle was used for gene delivery. Vector injection was done on the opposite side of the first hole, and the needle was kept inside several minutes until the eye coloring returned to normal. Finally, the needle was slowly removed. The mouse was kept on a heating plate at 37 °C, with the eye covered by the ophthalmic gel, and was kept in a separate cage until full recovery. Experiments were developed in accordance with the ARVO (Association for Research in Vision and Ophthalmology) Statement for the Use of Animals in Ophthalmic and Visual Research. Injections of the AAV2/2-pCMV-HsOPA1 or the AAV2/2-pCMV-GFP vectors were carried out on Opa1 mice in this proportion: 4 Opa1+/+ female controls, 4 untreated and 8 treated Opa1+/− female mice. Five control mice were injected with the AAV2/2-pCMV-GFP construction to quantify GFP expression in retina and optic nerve.
Quantitative real-time PCR
Quantitative real-time polymerase chain reactions were performed on total RNA extracted from retinas and optic nerves using the RiboPureTM Kit (Ambion). One microgram of RNA product was reverse-transcribed with the VersoTM cDNA Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The endogenous murine Opa1 transcripts as well as the human transduced OPA1 transcripts were quantified using a specific pair of primers with SYBR_ Green/LightCycler_ technology (Roche). Primers for murine Opa1 transcripts quantification: F: CAGGAGAAGTAGACTGTGTC, R: TGTGACTTTATTTTGCACGG. Primers for GFP transcripts: F: AGTGCTTCAGCCGCTACCC and R: CAGCTCGATGCGGTTCACC. Primers for human OPA1 transcripts: F: GCAATTGAAAACATGGTGGGT and R: CTGGGTGCTCCTCATTACAT. Primers for the ribosomal protein L27 gene: F: ACGCAAAGCCGTCATCGTGAAG and R: CTTGGCGATCTTCTTCTTGCC. Gene expressions were reported to the L27 gene expression of each sample.
Visual electrophysiology
Electrophysiology was performed in a darkroom, in the morning after overnight dark-adaptation, as advised11. Visual Evoked Potentials (VEPs) were performed on anesthetized mice, and recorded using three phases of 60 flashes as previously described12. Flash duration was 5 milliseconds with a frequency of 1 Hz and intensity of 159 cd.s−1.m−2. Amplitudes and latencies obtained during each phase were averaged. A cut-off filter was set at 35 Hz. For flash-electroretinogram acquisition mice were exposed to flashes of light lasting 5 ms at 0.1 Hz frequency each, with growing intensities (0.159, 0.3, 0.5, 1.59, 5, 15.9 cd∙s/m2); for the scotopic ERG. That included the scotopic threshold responses (STRs) of retinal ganglion cells9, as the second of six recordings. Electrophysiological data were analyzed with Visiosystem software (SIEM BioMedicale; Nîmes, France).
Virtual-reality optokinetic system
Mouse visual acuity was assessed as described by Prusky et al.13. The apparatus included a virtual optomotor constituted by four computer monitors arranged in a quadrangle arena generating a virtual rotating cylinder with drifting vertical sine wave grating. The OptoMotry (Cerebral Mechanics, Lethbride, Alberta, Canada) was used to control grating parameters and video recordings. Mice were placed on a central platform allowing them to track the grating with reflexive head and neck movements. The increasing of the grating frequency (from 0.042 to 0.442 cyc/deg.) until the optomotor reflex was not detectable anymore, defined the visual acuity. Contrast sensitivity was given at the 0.042 cyc/deg frequency. Mice were tested for both eyes in the morning at the beginning of their daylight cycle. Experiments were done blind to the genotype.
Funduscopy
In vivo funduscopy was performed using a Micron III retinal-imaging microscope (Phoenix Research Laboratories, Pleasanton, CA). Mice were anesthetized by intra-peritoneal injections of a mixture of Ketamine (120 mg/kg) and Rompun (10 mg/kg) and pupils were dilated with 1% tropicamide. The Lacryvisc® corneal ophthalmic gel (Alcon, Fort Worth, TX) was added before starting. Bright field and a specific green filter were used to observe retinal fundus and GFP expression, respectively. Still-frame images and video-rate sequences were acquired with Streampix III image acquisition software (NorPix, Montreal, Canada).
Retinal ganglion cell counting
Mice were euthanized by cervical dislocation. Eyes were collected and neuroretinas were isolated and fixed in paraformaldehyde 4%. For RGC counting, whole neuroretinas were immunolabelled using a Brn3a primary antibody (1:500, Santa Cruz Biotechnology) along with an Alexa 594 conjugated anti-goat secondary antibody (1:800, Invitrogen, Oregon, USA). Four lateral sections were done on retinas to allow their correct unfolding and mounting between slide and coverslip. Whole-mount retina images were obtained using the right Axioimager Z1 microscope with ApoTome module (Zeiss). Retinal ganglion cell number was calculated by the Imaris software and related to the retina surface calculated by the MetaMorph® Offline software.
Statistical analysis
Mann-Withney U-test and one-way ANOVA followed by the Student-Newman-Keuls multiple comparison test were used. Statistical significance thresholds were determined at P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***) values.
Study approval
All protocols carried out on animals were approved by the Animal Care and Use Committee Languedoc-Roussillon and recorded under the reference: CEEA-LR-11058, in agreement with the ARRIVE guidelines. All efforts were made to minimize the number of used animals and their suffering, according to the European directive 2010/63/UE. The authors confirm that they are in compliance with their Institutional Review Boards (IRBs).
References
Yu-Wai-Man, P., Griffiths, P. G., Hudson, G. & Chinnery, P. F. Inherited mitochondrial optic neuropathies. Journal of medical genetics 46, 145–158, https://doi.org/10.1136/jmg.2007.054270 (2009).
Yu-Wai-Man, P. & Newman, N. J. Inherited eye-related disorders due to mitochondrial dysfunction. Human molecular genetics 26, R12–R20, https://doi.org/10.1093/hmg/ddx182 (2017).
Delettre, C. et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26, 207–210, https://doi.org/10.1038/79936 (2000).
Yu-Wai-Man, P., Griffiths, P. G. & Chinnery, P. F. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Progress in retinal and eye research 30, 81–114, https://doi.org/10.1016/j.preteyeres.2010.11.002 (2011).
Yu-Wai-Man, P. Therapeutic Approaches to Inherited Optic Neuropathies. Semin Neurol 35, 578–586, https://doi.org/10.1055/s-0035-1563574 (2015).
Sarzi, E. et al. The human OPA1delTTAG mutation induces premature age-related systemic neurodegeneration in mouse. Brain: a journal of neurology 135, 3599–3613, https://doi.org/10.1093/brain/aws303 (2012).
Sarzi, E. et al. Increased steroidogenesis promotes early-onset and severe vision loss in females with OPA1 dominant optic atrophy. Human molecular genetics 25, 2539–2551, https://doi.org/10.1093/hmg/ddw117 (2016).
Harvey, A. R., Hellstrom, M. & Rodger, J. Gene therapy and transplantation in the retinofugal pathway. Progress in brain research 175, 151–161, https://doi.org/10.1016/S0079-6123(09)17510-6 (2009).
Porciatti, V. Electrophysiological assessment of retinal ganglion cell function. Experimental eye research 141, 164–170, https://doi.org/10.1016/j.exer.2015.05.008 (2015).
Petit, L., Khanna, H. & Punzo, C. Advances in Gene Therapy for Diseases of the Eye. Human gene therapy 27, 563–579, https://doi.org/10.1089/hum.2016.040 (2016).
Hankins, M. W., Jones, R. J. & Ruddock, K. H. Diurnal variation in the b-wave implicit time of the human electroretinogram. Vis Neurosci 15, 55–67 (1998).
Chekroud, K. et al. Simple and efficient: validation of a cotton wick electrode for animal electroretinography. Ophthalmic Res 45, 174–179, https://doi.org/10.1159/000321118 (2011).
Prusky, G. T., Alam, N. M., Beekman, S. & Douglas, R. M. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Investigative ophthalmology & visual science 45, 4611–4616, https://doi.org/10.1167/iovs.04-0541 (2004).
Acknowledgements
The authors gratefully acknowledge the Association Rétina France, the Association Française contre les Myopathies (AFM), the Association Kjer France, the Association Ouvrir Les Yeux and the Fondation de France for supporting this work. Authors also thank Dr Vasiliki Kalatzis for critical advices and the University of Montpellier, the Montpellier Rio Imaging (MRI) platform and the Réseau des Animaleries Montpelliéraines (RAM).
Author information
Authors and Affiliations
Contributions
E.S., C.L. and G.L. designed and supervised experiments. E.S. wrote the main manuscript and, with M.S., performed experiments. E.S. analyzed data and statistics. C.P.-M., L.E., M.Q. and M.P. contributed to the intravitreal injections. E.S., M.P., A.M., C.P.H., G.L. and C.D. reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sarzi, E., Seveno, M., Piro-Mégy, C. et al. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci Rep 8, 2468 (2018). https://doi.org/10.1038/s41598-018-20838-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20838-8
This article is cited by
-
Current advances in gene therapy of mitochondrial diseases
Journal of Translational Medicine (2022)
-
Autosomal dominant optic atrophy caused by six novel pathogenic OPA1 variants and genotype–phenotype correlation analysis
BMC Ophthalmology (2022)
-
Sustained intracellular calcium rise mediates neuronal mitophagy in models of autosomal dominant optic atrophy
Cell Death & Differentiation (2022)
-
Clinical electrophysiology of the optic nerve and retinal ganglion cells
Eye (2021)
-
Therapeutic Options in Hereditary Optic Neuropathies
Drugs (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
