
Abstract
Extra-Mediterranean glacial refugia of thermophilic biota, in particular in northern latitudes, are controversial. In the present study we provide genetic evidence for extra-Mediterranean refugia in two species of grass snake. The refuge of a widely distributed western European lineage of the barred grass snake (Natrix helvetica) was most likely located in southern France, outside the classical refuges in the southern European peninsulas. One genetic lineage of the common grass snake (N. natrix), distributed in Scandinavia, Central Europe and the Balkan Peninsula, had two distinct glacial refuges. We show that one was located in the southern Balkan Peninsula. However, Central Europe and Scandinavia were not colonized from there, but from a second refuge in Central Europe. This refuge was located in between the northern ice sheet and the Alpine glaciers of the last glaciation and most likely in a permafrost region. Another co-distributed genetic lineage of N. natrix, now massively hybridizing with the aforementioned lineage, survived the last glaciation in a structured refuge in the southern Balkan Peninsula, according to the idea of ‘refugia-within-refugia’. It reached Central Europe only very recently. This study reports for the first time the glacial survival of a thermophilic egg-laying reptile species in Central Europe.
Similar content being viewed by others


Introduction
Climatic oscillations in the Pleistocene induced large-scale range shifts of many animal and plant species all over the world1. The extant phylogeographic structure of thermophilic European biota is often explained by their retreat to refugia in southern peninsulas (Iberian, Apennine, Balkan) during the last glacial and postglacial recolonization of northern regions1,2,3,4,5. However, there is increasing evidence for extra-Mediterranean refugia6, and this is also true for thermophilic species like reptiles7,8. Grass snakes (Natrix astreptophora, N. helvetica, N. natrix; formerly lumped together under N. natrix9,10,11) are widely distributed across the western Palaearctic9,12. The range of the barred grass snake (N. helvetica) extends from the Pyrenees to the Rhine region, and it includes the Italian Peninsula, Sicily, Corsica and Sardinia. In the north, the range reaches to Northumberland, close to the Scottish border9. The common or eastern grass snake (N. natrix sensu stricto) occurs from the Rhine region eastwards to Lake Baikal in Siberia9,12. Its northern range extension is controversial, with unambiguous records up to central Sweden and southern Finland and debated records close to the Arctic Circle further north in Fennoscandia9,13,14. In any case, these two grass snake species are cold-tolerant compared to many other egg-laying reptile species9. This suggests that extra-Mediterranean refugia might have existed, like recently shown for the more thermophilic wall lizard (Podarcis muralis). For this species, extra-Mediterranean refugia were inferred for southern France, northern Italy, the eastern Alps and the Central Balkans8.
A previous study15 revealed glacial refugia for grass snakes in each of the southern European peninsulas, Corso-Sardinia, North Africa, Anatolia and the neighbouring Near and Middle East, with multiple microrefugia in continental Italy plus Sicily, the Balkan Peninsula, Anatolia and the Near and Middle East. In continental Italy, one refugium was inferred for the Padan Plain, south of the Alps, which qualifies for an extra-Mediterranean refugium. However, the location of the glacial refugia of some of the most widely distributed mitochondrial lineages of N. helvetica and N. natrix remain unclear. In N. helvetica, the concerned lineage is distributed from the Pyrenees across France, the Benelux countries, Switzerland and westernmost Germany to the Rhine region. Its range also includes Britain up to Northumberland, but not Ireland (Fig. 1: blue lineage). The Apennine Peninsula can be excluded as potential location of its glacial refuge because there are related, but distinct mitochondrial lineages of N. helvetica occurring15 (Supplementary Fig. S1). Thus, the refuge of the blue lineage could have been either in southern France or further northwest, perhaps in one of the areas emerged from sea during glacial low sea level stands (Fig. 2). If a southern refuge existed, genetic diversity should conform to the classical paradigm of ‘southern richness and northern purity’1,2,3, with high diversity in the south and low diversity in the north (Fig. 2a). If a northern refugium existed (Fig. 2b), a similar, but inversed, pattern is expected, with high genetic diversity in the north and low diversity in the south because ‘the rapid postglacial colonization and founder events inevitably reduce within-lineage genetic diversity’16.

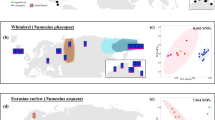
Sampling sites for mitochondrial lineages of the barred grass snake (Natrix helvetica, blue triangles) and the common grass snake (N. natrix, yellow circles and red stars). Insets: N. helvetica (left, Linz am Rhein, Germany; photo: Wolfgang Böhme) and N. natrix (right, Moritzburg, Germany; photo: Melita Vamberger). Map was created using arcgis 10.2 (www.esri.com/arcgis) and adobe illustrator CS6 (www.adobe.com/products/illustrator.html).

Hypotheses for the location of the glacial refuge and Holocene range expansion of the blue lineage of the barred grass snake (Natrix helvetica). Schematic representation of landmass, ice sheet and glacier areas during the Last Glacial Maximum (approx. 20,000 years ago) according to Diercke International Atlas 2010 (ISBN 978-3-14-100790-9). Light blue areas correspond to the possible southern (a) or northern refuge (b) of the blue lineage of N. helvetica. ‘High’ and ‘low’ indicate expected genetic diversities; location of putative refugia is speculative and arbitrary. Map was created using arcgis 10.2 (www.esri.com/arcgis) and adobe illustrator CS6 (www.adobe.com/products/illustrator.html).
In N. natrix, the two concerned lineages are distributed from the southern Balkan Peninsula across Central Europe to the Baltic region (Fig. 1: red lineage) or Scandinavia (Fig. 1: yellow lineage), with sympatric records in many regions. However, the red lineage has not been found in the northernmost parts of the species’ range, supporting that the yellow lineage arrived first14, either from a southeastern glacial refuge or from a northern extra-Mediterranean refuge. Indeed, we speculated in a pilot study that a refuge north of the Alps could have existed for the yellow lineage17 because then no southern records were known. However, using larger and geographically more comprehensive sampling, we found the yellow lineage also widely distributed in the southern Balkans15, suggesting rather multiple refuges for the yellow and red lineages and other lineages in the southern Balkan Peninsula. Yet, the southern Balkan records of the yellow lineage are separated by an enigmatic distribution gap from the northern records, whereas the red lineage is continuously distributed from the southern Balkans to Central Europe (Fig. 1)11,15.
This distribution pattern could result either from range expansions from two distinct refugia in the southern Balkan Peninsula (Fig. 3a) or from antidromic range expansions from a northern refugium of the yellow lineage and from a southern refugium of the red lineage (Fig. 3b). Alternatively, it could be speculated that both mitochondrial lineages existed together in one and the same southern refugium (ancestral polymorphism; Fig. 3c), what could explain their co-occurrence in many regions.

Hypotheses for the location of glacial refuges and Holocene range expansions of the yellow and red lineages of the common grass snake (Natrix natrix). (a) Range expansions from two distinct southern Balkan refuges (yellow and red circles) leading to secondary admixture in Central Europe and in the Balkan Peninsula, (b) antidromic range expansions from a northern (yellow circle) and southern refuge (red circle) leading to secondary admixture in Central Europe and in the Balkan Peninsula, (c) range expansion from one Balkan refuge harbouring two distinct mtDNA lineages (ancestral polymorphism). ‘High’ and ‘low’ indicate expected genetic diversities; location of putative refugia is speculative and arbitrary. Maps were created using arcgis 10.2 (www.esri.com/arcgis) and adobe illustrator CS6 (www.adobe.com/products/illustrator.html).
If only southern refugia would have existed (Fig. 3a and c), genetic diversity should be high in the south and low in the north1,2,3. If for the yellow lineage a northern refugium existed (Fig. 3b), the pattern is expected to be inversed, though, with genetic diversity being high in the north and low in the south.
Recent phylogeographic studies10,11,14,15 provided a wealth of mitochondrial DNA (mtDNA) and microsatellite data for grass snakes. However, nuclear gene pools turned out to be highly admixed, especially in the south of the range of N. natrix11, with several genetic lineages involved (Supplementary Fig. S1). Thus, microsatellite data are not feasible for unravelling the location of glacial refugia because high genetic diversity could be the result from admixture and not ‘southern richness’ of an individual genetic lineage. In contrast, maternally inherited mtDNA is not affected by admixture so that data for nearly 1,400 grass snakes are expected to be informative for inferring the location of former glacial refugia, even though nuclear DNA of the same grass snakes may be impacted by hybridization.
Materials and Methods
Sampling and used marker system
Geographically broad sampling of 1,372 grass snakes covered the distribution ranges of the blue lineage of Natrix helvetica and the yellow and red lineages of N. natrix11,15. In our previous studies11,15, we used 13 polymorphic microsatellite loci and two mitochondrial DNA blocks, the cytochrome b gene and the partial ND4 gene plus adjacent DNA coding for tRNAs = ND4 + tRNAs (866 bp). Here we focus on the latter mitochondrial marker because more data are available and differentiation patterns were found to be the same11,15. Microsatellite data could not be used for examining the location of glacial refuges because in the south massive admixture of distinct genetic lineages11 causes high diversity indices that have nothing to do with the original diversity in former refuges. The same is true for the Rhine region, where a narrow hybrid belt of the two grass snake species results in high nuclear genomic diversity11.
Among the used mtDNA sequences of grass snakes, 403 corresponded to the blue lineage of N. helvetica11,15, 526 to the yellow lineage and 443 to the red lineage of N. natrix. An additional sequence from a N. helvetica from northwestern Italy, generated according to laboratory procedures described before15, was added to our previously published data set11. Sequences of obviously non-native grass snakes from the study regions11 were excluded from calculations. All samples studied are listed in Supplementary Table S1.
The new sample (from a roadkill) was obtained and methods were carried out according to relevant guidelines, regulations and best ethical and experimental practice of the Senckenberg Nature Research Society.
Haplotype network analyses and PCAs
Using popart (www.popart.otago.ac.nz) and the implemented parsimony network algorithm of tcs18, a network was built for the 1,372 mtDNA sequences for displaying relationships of haplotypes. In addition, Principal Component Analyses (PCAs) were calculated using the same data set and the R package adegenet19.
Genetic diversity indices and demographic analyses of mtDNA
Genetic diversity in glacial refugia should be much higher than in recently colonized regions1,2,16. To examine this expectation, samples were arbitrarily assigned to northern and southern groups. For the blue lineage of N. helvetica, the divide was the Massif Central in southeastern France (Fig. 4). For the yellow and red lineages of N. natrix, the Carpathians served as primary divide (Figs 5 and 6, left). Because the yellow lineage has a wide distribution gap in the northern Balkan Peninsula11,15, no data were available for the Carpathian Basin. Thus, the southern group of the yellow lineage corresponds only to the southern Balkan. For fine scale analysis of genetic diversities, the data sets were further subdivided (Figs 5 and 6, right). For the northern yellow group, genetic diversity was examined for two subgroups north and south of a line running approximately from the Ore Mountains to the Hunsrück. The southern red group was approximately subdivided along the Balkan Mountains.

Northern and southern groups for samples of Natrix helvetica (n = 403). Map was created using arcgis 10.2 (www.esri.com/arcgis) and adobe illustrator CS6 (www.adobe.com/products/illustrator.html).

Northern and southern groups of the yellow lineage (n = 499) of Natrix natrix (left) and subdivision of the northern group (right). Maps were created using arcgis 10.2 (www.esri.com/arcgis) and adobe illustrator CS6 (www.adobe.com/products/illustrator.html).

Northern and southern groups of the red lineage (n = 443) of Natrix natrix (left) and subdivision of the southern group (right). Maps were created using arcgis 10.2 (www.esri.com/arcgis) and adobe illustrator CS6 (www.adobe.com/products/illustrator.html).
To assess genetic diversity of each group, the following diversity indices were calculated using dnasp 5.10.0120: number of segregating sites S, nucleotide diversity π, number of haplotypes h, and haplotype diversity Hd. The number of private haplotypes hP was determined in arlequin 3.5.1.221. dnasp was also used to examine for signals for population size changes by generating pairwise mismatch distributions. Unimodal mismatch distributions are expected when range or demographic expansions lead to an excess of recent, and thus rare, mutations. In contrast, multimodal mismatch distributions indicate stationary population sizes22,23,24,25. Moreover, a mismatch distribution test was run in arlequin and parameters of sudden demographic expansion (τ, θ0 and θ1) were calculated. To verify the validity of the expansion model, a parametric bootstrap approach was used, comparing the fit of the observed and 100 simulated mismatch distributions. The fit to the expected mismatch distribution was quantified by the sum of square deviations (SSD). SSD values are non-significant when the data do not deviate from the expectation of population expansion. Raggedness indices (rg)26,27 were computed quantifying the smoothness of the observed mismatch distribution. Non-significant results indicate an expanding population27. Finally, changes in population size were examined by calculating Tajima’s D and Fu’s FS values in dnasp. An increasing population size (or purifying selection) is indicated by significantly negative D and FS values, whereas positive values suggest a recent bottleneck28,29.
Sample sizes differed considerably among populations. In order to eliminate the positive bias of sample size on inferred diversity, a rarefaction procedure was employed. For each sample larger than the smallest one with size N, a subsample of N individuals was drawn randomly (without replacement) and diversity indices were calculated. The procedure was repeated five times for each sample, and then the mean subsample diversity for each sample was calculated.
For comparing specific hypotheses for the demographic history of the three studied lineages (blue, yellow and red), we utilized the Approximate Bayesian Computation approach implemented in diyabc 2.0.330. This software allows for rapid testing of different scenarios by calculating summary statistics rather than exact likelihoods31. For each lineage, three alternative scenarios were examined that represented either gradual diversification from refuge areas or sudden expansion (Fig. 7). Three groups were defined for each lineage and a rarefaction procedure (as described above) was applied to obtain even sample sizes. For the blue lineage of N. helvetica, the groups corresponded to samples from Britain, France, and the Rhine region (East). For N. natrix, the yellow lineage was divided into the groups North (Scandinavia), Centre (Central Europe), and Balkan. The red lineage was divided into the groups North, Carpathian Basin, and southern Balkans. Priors were set to follow uniform distributions ranging from 10 to 10,000 for effective population sizes (Ne) and from 10 to 10,000 for diversification times (t1 and t2, in generations). For each lineage 300,000 simulations were run, after which summary statistics were drawn for: (i) number of haplotypes, (ii) number of segregating sites, (iii) mean of pairwise differences, (iv) private segregating sites, (v) mean of pairwise differences (W), (vi) mean of pairwise differences (B), and (vii) FST. According to previous studies32,33, the priors for the mutation rate were set to a minimum of 10−8 and a maximum of 10−6. All other settings were left as suggested in the diyabc 2.0 handbook. Posterior probabilities for scenarios were calculated by logistic regression considering the 3,000 simulated datasets that were closest to the observed values. To convert the obtained estimates of divergence times from generations to years, a generation time of 10 years was used, based on life history data (sexual maturity, maximum age)9.

Phylogeographic hypotheses for genetic lineages of Natrix helvetica and N. natrix explicitly tested using diyabc. Scenarios 1 and 2 correspond to gradual diversification from an ancient population, while scenario 3 corresponds to sudden expansion.
Data availability
All data analysed during this study are included in this published article and its Supplementary Information files. DNA sequences have been uploaded to the European Nucleotide Archive (ENA) and are available under the accession numbers listed in Supplementary Table S1.
Results
Haplotype networks and PCAs
The haplotypes of each grass snake lineage corresponded to a highly distinct cluster in parsimony network analyses (Fig. 8; for geographic distribution of individual haplotypes, see Supplementary Figs S2–S5). Haplotypes of Natrix helvetica (blue lineage) were separated by a minimum of 40 mutation steps from the haplotype cluster of the yellow lineage of N. natrix, and by a minimum of 54 steps from the haplotypes of the red lineage of N. natrix. Between the two clusters of N. natrix, a minimum of 40 mutations occurred. Haplotypes of all lineages had star-like genealogies with a common internal haplotype surrounded by rare haplotypes differing only in one or a few mutations from the central haplotype. However, only for N. helvetica (blue lineages) this topology was unambiguous, with the internal haplotype h1 and eleven rare haplotypes (h2–h11) differing in only one mutation step from h1. There was no obvious pattern of a geographically correlated distribution of any haplotypes.

Parsimony networks of 1,372 ND4+tRNA sequences of barred and common grass snakes. Symbol sizes reflect haplotype frequencies. Small black circles are missing node haplotypes; each line connecting two haplotypes corresponds to one mutation step, if not otherwise indicated by numbers. Haplotype colours represent mitochondrial lineages. Lighter or darker nuances show the occurrence of haplotypes in the northern or southern distribution area according to Figs 4, 5 (left) and 6 (left). Two-coloured haplotypes occurred in the respective northern and southern groups; colours do not indicate percentages of occurrence.
The haplotype cluster of the yellow lineage of N. natrix consisted also of a common central haplotype (y1) surrounded by many rare haplotypes that differed in one or two mutations from y1. Yet, there were some network reticulations, and the more distinct haplotypes y15-y17 differed by four to five mutations from y1. Notably, these haplotypes y15-y17 and the rare haplotypes y12, y21 and y23 occurred only in the southern Balkan region, whereas the vast majority of other haplotypes was found only north of the Carpathians. There was only one shared haplotype (y20) between the two geographical groups. However, some unique southern haplotypes (y12, y18, y21, y23) differed in only one or two mutations from the most common central northern haplotype y1.
The haplotype cluster of the red lineage of N. natrix corresponded not to one, but to two star-like subclusters, with tip haplotypes differing in up to three mutations from the two frequent central haplotypes r3 and r4. The latter two haplotypes differed by two mutation steps from one another. There was no clear pattern in geographical distribution of haplotypes of the red lineage, with haplotypes of both subclusters occurring north and south of the Carpathians.
In accordance with the network analyses, the PCAs revealed three highly distinct clusters corresponding to the blue, yellow and red lineages (Supplementary Fig. S6). When PCAs were calculated for the sequences of each lineage alone, only for the yellow lineage the northern and southern groups were clearly differentiated, with largely non-overlapping 95% confidential intervals. The confidential intervals of the northern and southern groups of the blue and red lineages were massively overlapping, reflecting that these lineages comprise less differentiated haplotypes or haplotype clusters (Supplementary Fig. S7).
Genetic diversity indices and demographic analyses of mtDNA
Diversity indices increased with sample size, suggestive of a positive sampling bias. Therefore, we applied our rarefaction procedure to obtain balanced sample sizes (Table 1, Supplementary Tables S2–S5). However, with respect to mismatch distributions, no differences were observed for total and rarefied samples (Supplementary Tables S6–S11).
For the blue lineage of Natrix helvetica, diversity in the south was higher than in the north when the much larger northern sample was rarefied (Table 1). Mismatch distributions were unimodal (Fig. 9) and Tajima’s D and Fu’s FS values were significantly negative for both groups considering all data, suggestive of population expansion. This is supported by non-significant values of the sum of square deviations (SSD) and the raggedness index (rg) indicating that the data do not deviate from the expectation of population expansion (Supplementary Table S6).

Pairwise mismatch distributions using all samples of the respective groups. Rarefied sampling resulted consistently in the same patterns. Broken green lines are expected frequencies of pairwise differences under demographic expansion. Solid red lines and circles, observed values. Symbols correspond to Figs 4–6.
For the yellow lineage of N. natrix, diversities of the southern Balkan group were higher than for the northern group using a rarefied northern sample (Table 1). For the northern group, there was a signal for recent expansion (unimodal mismatch distribution, Fig. 9), which is in line with significantly negative Tajima’s D and Fu’s FS values and non-significant SSD and rg values (Supplementary Table S6). However, the southern Balkan group was characterized by a multimodal mismatch distribution (Fig. 9), with non-significant positive Tajima’s D and Fu’s FS values, arguing for a stationary, non-expanding population. Contrary to expectation, SSD and rg values were also non-significant (Supplementary Table S6).
To examine genetic diversity on both sides of the distribution gap in the northern Balkans (Fig. 5 left), the northern group was subdivided along a line running from the Ore Mountains to the Hunsrück (Fig. 5, right). Genetic diversity in the northern Central European and Scandinavian subgroup was higher than in the southern subgroup (Table 1). For both subgroups, mismatch graphs were unimodal and Tajima’s D and Fu’s FS values were significantly negative and SSD and rg values were not significant, indicating population expansion (Fig. 9; Supplementary Table S6).
Sample sizes of the red lineage north and south of the Carpathians (Fig. 6, left) were nearly even, so that no rarefication was required. Genetic diversity was distinctly higher south of the Carpathians (Table 1). Mismatch distributions were multimodal for the northern and southern groups (Fig. 9), suggestive of stationary population sizes. However, this was not supported by mostly significant negative Tajima’s D and Fu’s FS values and non-significant SSD and rg values, suggesting rather expansion (Supplementary Table S6). When the southern group was subdivided into the two subgroups Carpathian Basin and southern Balkan Peninsula (Fig. 6, right), highly uneven sample sizes were obtained because only 11 samples were available for the southern Balkan Peninsula. Using the rarefication approach described above to achieve similar sample sizes, diversities in both southern subgroups were similar and higher than in the northern group (Table 1). Mismatch distributions were multimodal for both southern subgroups (Fig. 9). However, Tajima’s D and Fu’s FS values were significantly negative for the Carpathian Basin, complemented by non-significant SSD and rg values (Supplementary Table S6). The Tajima’s D value for the southern Balkan subgroup was negative, the Fu’s FS value was positive, but both were not significant. Significant SSD and rg values support stationary population sizes indicated by the multimodal mismatch distribution graph for the southern Balkans (Supplementary Table S6).
The haplotypes of the red lineage corresponded to two distinct star-like clusters (Fig. 8), which were also analysed separately. Genetic diversity of the cluster with the central haplotype “r4” was much higher than in the cluster having the central haplotype “r3” (Supplementary Table S12). For both clusters, mismatch distributions were unimodal (Fig. 9), SSD and rg values were not significant. Tajima’s D and Fu’s FS values were for both clusters significantly negative (Supplementary Table S13), suggesting expansion.
The models preferred by our ABC analyses (Fig. 7; Table 2; Supplementary Figs S8 and S9) supported the results of the other demographic analyses. If the northern populations are derived from Holocene range expansions from a southern refuge, either scenario 1 (stepwise range expansion from south to north) or scenario 3 (sudden expansion) were expected. The stepwise range expansion of scenario 1 could also correspond, however, to two distinct glacial refugia if the time elapsed between the branching events would conflict with Holocene expansion. Conversely, scenario 2 would indicate a stepwise southward range expansion from the north. For the blue lineage of N. helvetica and the red lineage of N. natrix, scenario 3 was best supported (Table 2). The sudden expansion for the blue lineage was dated to 12,000 years before present (BP), while the expansion was dated only to approximately 3,000 BP for the red lineage. For the yellow lineage of N. natrix scenario 1, representing a stepwise range expansion from the Balkan, was supported. The divergence of the Balkan group and the common ancestor of the central and northern groups was dated to approximately 39,000 BP, whereas the divergence of the Central and northern European groups was dated to 7,700 BP (Table 2).
Discussion
Our analyses of genetic diversities of mtDNA suggest for the blue lineage of the barred grass snake (Natrix helvetica) and the red lineage of the common grass snake (N. natrix) glacial refugia in the south, close to the Mediterranean Sea or inland of the southern Balkan Peninsula. This is indicated by higher genetic diversities in the former refuges and supported by our diyabc analyses. Typically, such refugia are thought to be located in the southern peninsulas1,4,5,34,35,36,37. This was not the case for the blue lineage of N. helvetica, because the two peninsulas adjacent to its range, the Iberian and Italian peninsulas, are occupied by genetically distinct grass snakes (N. astreptophora in the Iberian Peninsula, several distinct lineages of N. helvetica in the Italian Peninsula15 (Supplementary Fig. S1). Our new sample from northwestern Italy constitutes the southeastern most record of N. helvetica. Thus, our results are in line with the previous suggestion15 that the refuge of the blue lineage of N. helvetica was in southern France (Fig. 10). This region has also been inferred as a possible glacial refuge for other reptile species (Podarcis muralis8, Zootoca vivipara38, Vipera aspis39, V. berus40). From there, the blue lineage seems to have rapidly expanded its range approximately 12,000 BP (Table 2). This date coincides with a rapid warming event evinced from ice core and climate records from northern Europe that has triggered also range shifts in other species41,42,43 and explains the colonization of Britain by N. helvetica via what is now the English Channel.

Putative glacial refuges and schematic range expansions of genetic lineages of Natrix helvetica (blue lineage) and N. natrix (yellow and red lineages). Expansion times from diyabc indicated. Exact location of refugia is arbitrary. Map was created using arcgis 10.2 (www.esri.com/arcgis) and adobe illustrator CS6 (www.adobe.com/products/illustrator.html).
For the red lineage of N. natrix, the higher southern genetic diversity indicates that its glacial refuge was located in the southern Balkan Peninsula, conforming to one of the classical refuges4,5,34,44,45,46. However, the structured, bipartite haplotype network and the multimodal mismatch distributions of the red lineage of N. natrix support a different demographic history compared to the blue lineage of N. helvetica. The network for the blue lineage is perfectly star-like, as typical for demographic expansions, coupled or not with range expansions. Such networks are characterized by a presumably ancestral common central haplotype. Expansion is also supported by an excess of rare mutations corresponding to rare tip haplotypes, and unimodal mismatch distributions. In contrast, intricate network topologies with multiple internal haplotypes and multimodal mismatch distributions as in the red lineage argue normally for stationary and geographically structured populations22,23,25,47. However, for the red lineage, some demographic analyses were conflicting. When the samples corresponding to two star-like haplotype clusters were examined separately for the red lineage, signal for expansion was obtained. This suggests that the refuge of the red lineage was structured into two microrefugia (or ‘refugia within refugia’48). These putative microrefuges, and the occurrence of the yellow and other mitochondrial lineages in the southern Balkan Peninsula, are in line with the idea of multiple glacial refuges of grass snakes15 and other reptiles8,49,50,51 there. The microrefuges of the red lineage must have been in very close proximity, so that haplotypes of each microrefuge came rapidly into contact, spread together and are now co-distributed across the entire range (Fig. 10). Also our diyabc analyses support a range expansion from the south, which is, however, very recent with only approximately 3,300 BP.
Haplotypes of the red lineage are occurring together with haplotypes of the yellow lineage in many parts of their distribution ranges. Theoretically, such a pattern could also originate from ancestral polymorphism, i.e. from the survival of the two mitochondrial lineages in one and the same refugial population. However, based on our previously published analyses of 13 microsatellite loci11, this hypothesis can be firmly rejected, because then only one nuclear gene pool should correspond to grass snakes harbouring haplotypes of the yellow and red mtDNA lineages. This is not the case. The yellow and the red mtDNA lineages clearly match with two distinct nuclear clusters (Supplementary Fig. S10), which is why for both distinct refuges have to be inferred.
Consequently, the question remains whether the yellow lineage of N. natrix had another refuge in the southern Balkan Peninsula, in close proximity of those of the red lineage, or whether the yellow lineage might have survived the last glaciation somewhere in a northern extra-Mediterranean refuge17 (Fig. 3). At first glance our data support a southern refugium because genetic diversity in the south is much higher (Table 1). However, our PCA analyses indicate that the northern and southern groups of the yellow lineage are genetically distinct, a sharp contrast to the blue and red lineages (Supplementary Fig. S7). Moreover, demographic analyses suggest no Holocene expansion for the southern group, in contrast to the northern one. There is only one shared haplotype between the northern and the southern groups (Fig. 8), and the central haplotype of the northern group has not been found in the southern Balkans. If the northern grass snakes spread from a southern Balkan refuge, it should be expected that more haplotypes of the southern group occur also in the north and that a southern haplotype is ancestral to northern tip haplotypes. Also when the southern group were derived from the northern one, we should expect a similar, but inversed pattern (Fig. 3b), with high diversity in the north and low diversity in the south. Then, the central haplotype should occur in the northern group. This scenario of an exclusively northern refuge for the yellow lineage is rejected by our diyabc calculations (Table 2) and not supported by any demographic analyses.
In the face of the wide distribution gap in the northern Balkans, yet another possibility comes into mind, namely two glacial refugia, one north of the Alps and one in the southern Balkan Peninsula. It seems plausible that the last glaciation interrupted an originally continuous population of the yellow lineage, with survivors in two distinct refuges, one in the north and one in the south. This hypothesis is supported by the high genetic diversity indices of northern Central European and Scandinavian grass snakes compared southern Central European ones (Table 1) and by our diyabc analyses (Table 2). The diyabc analyses suggest that the Balkan group separated from the more northerly populations of the yellow lineage approximately 39,000 years ago. This predates the Last Glacial Maximum (LGM) and coincides with the time when the ice sheets first reached their local last glacial maxima and the sea level dropped first to glacial lowstand52, suggesting that the range interruption was caused by corollary environmental changes. The divergence of the Scandinavian and Central European populations was dated to only 7,700 BP, arguing for a colonization of Scandinavia from the Central European source populations during the Holocene climatic optimum.
The absence of the red mitochondrial lineage in northern Europe and its rareness in northern Central Europe (Fig. 1) is in agreement with late invasion14 and our ABC modelling, which suggests that the red lineage expanded its range into Central Europe only 3,300 BP (Table 2). Late invasion is also supported by the largely pure nuclear gene pool of grass snakes of the yellow lineage in northern Germany and southern Scandinavia, without admixture with the red nuclear cluster11. This pattern perfectly matches with the idea of a northern refuge of the yellow lineage because grass snakes of the yellow lineage had to cover much lower distances to reach Scandinavia than grass snakes of the red lineage coming from the southern Balkans.
Also for other taxa, there is growing evidence for extra-Mediterranean glacial refugia, in particular in the Alpine region, the Carpathian and the Caucasian regions6,7,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71. However, our data (Tables 1 and 2) suggest an even more northerly location for the refuge of the yellow lineage of N. natrix, namely within Central Europe, i.e. in between the northern ice sheet and the Alpine glaciers of the last glaciation (Fig. 10). In this region, refuges are expected mainly, but not exclusively, for artic or alpine species6,53,54,55, but not necessarily for an egg-laying thermophilic reptile. Such northern refugia are controversially debated55,72,73,74, especially with respect to their location in permafrost regions. For instance, the glacial survival of the adder (Vipera berus), a species widely co-distributed with grass snakes, has been doubted for permafrost regions40. Yet, even today, grass snakes occur in regions with discontinuous and continuous permafrost, for example near Lake Baikal in Siberia9,12,75,76, and the same is true for the adder12,76. Based on fossil and genetic evidence, Bhagwat & Willis72 examined glacial refuges of more than 50 European vertebrate species and concluded that there is a positive correlation between present-day distribution (northern range limits beyond 60°N) and persistence in northern glacial refugia. Species with northern range limits beyond 60°N are likely to have survived in northern refuges, and N. natrix qualifies for this category. Even if the northernmost questionable records9,13,14 are disregarded, there are unambiguous records of grass snakes beyond 60°N (for instance in Finland at 61.9°N14).
With respect to refugia in permafrost regions it has to be kept in mind that this does not necessarily imply life on permanently frozen ground. The surface of permafrost soils thaws during the warm season, and permanent frost remains only in different depths beneath. Moreover, it has been suggested that thermophilic biota have persisted in sheltered valleys with suitable microclimates, and there is hard evidence for some northern refugia up to the Norwegian coast55,77. For instance, forest land snails have been recorded from the late glacial of the Bohemian Massif, providing evidence for the persistence of small patches of broadleaf forest in Central Europe78.
In conclusion (Fig. 10), our study supports the existence of extra-Mediterranean refugia in grass snakes. The refugium of the blue lineage of N. helvetica was most likely located in southern France, outside the Mediterranean peninsulas. The red lineage of N. natrix seems to have survived the last glacial in a classical southern refugium in the Balkan Peninsula. Haplotype networks, mismatch distributions and demographic analyses of mtDNA sequences suggest that its refuge was geographically structured. From the Balkan, the red lineage has spread late to Central Europe and the Carpathian Basin and admixed en route with the yellow lineage of N. natrix11. For the yellow lineage, there is strong evidence for two distinct refugia during the LGM, one in the southern Balkan Peninsula and another one in Central Europe. This is supported (1) by the wide distribution gap in the northern Balkan Peninsula, (2) largely distinct mitochondrial haplotypes of grass snakes from the disjunct southern and northern parts of the distribution range, (3) by the higher genetic diversity of grass snakes from northern Germany and Scandinavia compared to snakes from southern Central Europe, (4) the lack of the red lineage in Scandinavia and northern Central Europe, and (5) by modelling using Approximate Bayesian Computation. From the Central European refugium, the yellow lineage colonized Scandinavia during the Holocene climatic optimum, long before the red lineage arrived in Central Europe.
References
Hewitt, G. M. The genetic legacy of the Quaternary ice ages. Nature 405, 907–913 (2000).
Hewitt, G. M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 58, 247–276 (1996).
Hewitt, G. M. Quaternary phylogeography: the roots of hybrid zones. Genetica 139, 617–638 (2011).
Taberlet, P., Fumagalli, L., Wust-Saucy, A.-G. & Cosson, J.-F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 7, 453–464 (1998).
Schmitt, T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front. Zool. 4, 11 (2007).
Schmitt, T. & Varga, Z. Extra-Mediterranean refugia: The rule and not the exception? Front. Zool. 9, 22 (2012).
Ursenbacher, S., Carlsson, M., Helfer, V., Tegelström, H. & Fumagalli, L. Phylogeography and Pleistocene refugia of the adder (Vipera berus) as inferred from mitochondrial DNA sequence data. Mol. Ecol. 15, 3425–3437 (2006).
Salvi, D., Harris, D. J., Kaliontzopoulou, A., Carretero, M. A. & Pinho, C. Persistence across Pleistocene ice ages in Mediterranean and extra-Mediterranean refugia: phylogeographic insights from the common wall lizard. BMC Evol. Biol. 13, 147 (2013).
Kabisch, K. Natrix natrix (Linnaeus, 1758) – Ringelnatter in Handbuch der Reptilien und Amphibien Europas. Band 3/II A – Schlangen II (ed Böhme, W.), 513–580 (Aula-Verlag, 1999).
Pokrant, F. et al. Integrative taxonomy provides evidence for the species status of the Ibero-Maghrebian grass snake Natrix astreptophora. Biol. J. Linn. Soc. 118, 873–888 (2016).
Kindler, C. et al. Hybridization patterns in two contact zones of grass snakes reveal a new Central European snake species. Sci. Rep. 7, 7378 (2017).
Bannikov, A. G., Darevskii, I. S., Ishchenko, V. G., Rustamov, A. K. & Shcherbak, N. N. Opredelitel’ zemnovodnykh i presmykayushchikhsya fauny SSSR (Prosveshchenie, 1977).
Fog, K., Schmedes, A. & Rosenørn de Lasson, D. Nordens padder og krybdyr (G.E.C. Gad, 1997).
Kindler, C., Bringsøe, H. & Fritz, U. Phylogeography of grass snakes (Natrix natrix) all around the Baltic Sea: implications for the Holocene colonization of Fennoscandia. Amphibia-Reptilia 35, 413–424 (2014).
Kindler, C. et al. Mitochondrial phylogeography, contact zones and taxonomy of grass snakes (Natrix natrix, N. megalocephala). Zool. Scr. 42, 458–472 (2013).
Waters, J., Fraser, C. I. & Hewitt, G. M. Founder takes all: density-dependent processes structure biodiversity. Trends Ecol. Evol. 28, 78–85 (2013).
Fritz, U., Corti, C. & Päckert, M. Mitochondrial DNA sequences suggest unexpected phylogenetic position of Corso-Sardinian grass snakes (Natrix cetti) and do not support their species status, with notes on phylogeography and subspecies delineation of grass snakes. Org. Divers. Evol. 12, 71–80 (2012).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Jombart, T. ADEGENET: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Librado, P. & Rozas, J. DNASPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Excoffier, L. & Lischer, H. E. L. ARLEQUIN suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Slatkin, M. & Hudson, R. R. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129, 555–562 (1991).
Rogers, A. R. & Harpending, H. Population growth makes waves in the distribution of pairwise genetic divergences. Mol. Biol. Evol. 9, 552–569 (1992).
Ray, N., Currat, M. & Excoffier, L. Intra-deme molecular diversity in spatially expanding populations. Mol. Biol. Evol. 20, 76–86 (2003).
Excoffier, L. Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol. Ecol. 13, 853–864 (2004).
Harpending, H. C., Sherry, S. T., Rogers, A. R. & Stoneking, M. Genetic structure of ancient human populations. Curr. Anthropol. 34, 483–496 (1993).
Harpending, H. C. Signature of ancient population growth in a low resolution mitochondrial DNA mismatch distribution. Hum. Biol. 66, 591–600 (1994).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Fu, Y.-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Cornuet, J. M. et al. DIYABCv2. 0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism. DNA sequence and microsatellite data. Bioinformatics 30, 1187–1189 (2014).
Csilléry, K., Blum, M. G., Gaggiotti, O. E. & François, O. Approximate Bayesian computation (ABC) in practice. Trends Ecol. Evol. 25, 410–418 (2010).
Guicking, D., Lawson, R., Joger, U. & Wink, M. Evolution and phylogeny of the genus Natrix (Serpentes: Colubridae). Biol. J. Linn. Soc. 87, 127–143 (2006).
Kindler, C. et al. Phylogeography of the Ibero-Maghrebian red-eyed grass snake (Natrix astreptophora). Org. Divers. Evol., https://doi.org/10.1007/s13127-017-0354-2 (2018).
Hewitt, G. M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 68, 87–112 (1999).
Lenk, P., Fritz, U., Joger, U. & Wink, M. Mitochondrial phylogeography of the European pond turtle, Emys orbicularis (Linnaeus 1758). Mol. Ecol. 8, 1911–1922 (1999).
Brito, P. H. The influence of Pleistocene glacial refugia on tawny owl genetic diversity and phylogeography in western Europe. Mol. Ecol. 14, 3077–3094 (2005).
Dapporto, L. Speciation in Mediterranean refugia and post-glacial expansion of Zerynthia polyxena (Lepidoptera, Papilionidae). J. Zool. Syst. Evol. Res. 48, 229–237 (2010).
Guillaume, C.-P., Heulin, B., Arrayago, M. J., Bea, A. & Braña, F. Refuge areas and suture zones in the Pyrenean and Cantabrian regions: geographic variation of the female MPI sex-linked alleles among oviparous populations of the lizard. Lacerta (Zootoca) vivipara. Ecography 23, 3–10 (2000).
Ursenbacher, S. et al. Phylogeography of the asp viper (Vipera aspis) inferred from mitochondrial DNA sequence data: Evidence for multiple Mediterranean refugial areas. Mol. Phylogenet. Evol. 38, 546–552 (2006).
Ursenbacher, S. et al. Postglacial recolonization in a cold climate specialist in western Europe: patterns of genetic diversity in the adder (Vipera berus) support the central-marginal hypothesis. Mol. Ecol. 24, 3639–3651 (2015).
Steffensen, J. P. et al. High-resolution Greenland ice core data show abrupt climate change happens in few years. Science 321, 680–684 (2008).
Vinther, B. M. et al. Holocene thinning of the Greenland ice sheet. Nature 461, 385–388 (2009).
Sommer, R. S. et al. When the pond turtle followed the reindeer: effect of the last extreme global warming event on the timing of faunal change in Northern Europe. Glob. Change Biol. 17, 2049–2053 (2011).
Böhme, M. U. et al. Phylogeography and cryptic variation within the Lacerta viridis complex (Lacertidae, Reptilia). Zool. Scr. 36, 119–131 (2007).
Skog, A. et al. Phylogeography of red deer (Cervus elaphus) in Europe. J. Biogeogr. 36, 66–77 (2009).
Vilaça, S. T. et al. Mitochondrial phylogeography of the European wild boar: the effect of climate on genetic diversity and spatial lineage sorting across Europe. J. Biogeogr. 41, 987–998 (2014).
Harpending, H. & Rogers, A. Genetic perspectives on human origins and differentiation. PNAS 96, 10597–10602 (2000).
Gómez, A. & Lunt, D. H. Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula in Phylogeography of Southern European Refugia (eds Weiss, S. & Ferrand, N.), 155–188 (Springer, 2007).
Fritz, U. et al. Mitochondrial phylogeography of European pond turtles (Emys orbicularis, Emys trinacris) – an update. Amphibia-Reptilia 28, 418–426 (2007).
Jablonski, D. et al. Contrasting evolutionary histories of the legless lizards slow worms (Anguis) shaped by the topography of the Balkan Peninsula. BMC Evol. Biol. 16, 99 (2016).
Marzahn, E. et al. Phylogeography of the Lacerta viridis complex: mitochondrial and nuclear markers provide taxonomic insights. J. Zool. Syst. Evol. Res. 54, 85–105 (2016).
Clark, P. U. et al. The Last Glacial Maximum. Science 325, 710–714 (2009).
de Lattin, G. Die Ausbreitungszentren der holarktischen Landtierwelt in Verhandlungen der Deutschen Zoologischen Gesellschaft vom 21. bis 26. Mai 1956 in Hamburg (ed. Pflugfelder, O.), 380–410 (Geest & Portig, 1957).
de Lattin, G. Grundriß der Zoogeographie. (Gustav Fischer, 1967).
Stewart, J. R. & Lister, A. M. Cryptic northern refugia and the origins of the modern biota. Trends Ecol. Evol. 16, 608–613 (2001).
Jaarola, M. & Searle, J. B. Phylogeography of field voles (Microtus agrestis) in Eurasia inferred from mitochondrial DNA sequences. Mol. Ecol. 11, 2613–2621 (2002).
Babik, W. et al. Mitochondrial phylogeography of the moor frog, Rana arvalis. Mol. Ecol. 13, 1469–1480 (2004).
Schönswetter, P., Stehlik, I., Holderegger, R. & Tribsch, A. Molecular evidence for glacial refugia of mountain plants in the European Alps. Mol. Ecol. 14, 3547–3555 (2005).
Kotlík, P. et al. A northern glacial refugium for bank voles (Clethrionomys glareolus). PNAS 103, 14860–14864 (2006).
Magri, D. et al. A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol. 171, 199–221 (2006).
Sommer, R. S. & Nadachowski, A. Glacial refugia of mammals in Europe: evidence from fossil records. Mammal Rev. 36, 251–265 (2006).
Gratton, P., Konopinski, M. K. & Sbordoni, V. Pleistocene evolutionary history of the Clouded Apollo (Parnassius mnemosyne): genetic signatures of climate cycles and a ‘time-dependent’ mitochondrial substitution rate. Mol. Ecol. 17, 4248–4262 (2008).
Varga, Z. Extra-Mediterranean refugia, post-glacial vegetation history and area dynamics in eastern Central Europe in Relict Species. Phylogeography and Conservation Biology (eds Habel, J. C. & Assmann, T.), 57–87 (Springer, 2010).
Homburg, K. et al. Multiple glacial refugia of the low-dispersal ground beetle Carabus irregularis: molecular data support predictions of species distribution models. PLoS One 8, e61185 (2013).
Boston, E. S. M., Puechmaille, S. J., Clissmann, F. & Teeling, E. C. Further evidence for cryptic north-western refugia in Europe? Mitochondrial phylogeography of the sibling species Pipistrellus pipistrellus and Pipistrellus pygmaeus. Acta Chiropterol. 16, 263–277 (2014).
Wielstra, B., Babik, W. & Arntzen, J. W. The crested newt Triturus cristatus recolonized temperate Eurasia from an extra-Mediterranean glacial refugium. Biol. J. Linn. Soc. 114, 574–587 (2015).
Wielstra, B., Zieliński, P. & Babik, W. The Carpathians hosted extra-Mediterranean refugia-within-refugia during the Pleistocene Ice Age: genomic evidence from two newt genera. Biol. J. Linn. Soc. 122, 605–613 (2017).
Ghielmi, S., Menegon, M., Marsden, S. J., Laddaga, L. & Ursenbacher, S. A new vertebrate for Europe: the discovery of a range-restricted relict viper in the western Italian Alps. J. Zool. Syst. Evol. Res. 54, 161–173 (2016).
Stojak, J. et al. Between the Balkans and the Baltic: phylogeography of a common vole – Mitochondrial DNA lineage limited to Central Europe. PLoS One 11, e0168621 (2016).
Kühne, G., Kosuch, J., Hochkirch, A. & Schmitt, T. Extra-Mediterranean glacial refugia in a Mediterranean faunal element: the phylogeography of the chalkhill blue Polyommatus coridon (Lepidoptera, Lycaenidae). Sci. Rep. 7, 43533 (2017).
Pabijan, M., Zieliński, P., Dudek, K., Stuglik, M. & Babik, W. Isolation and gene flow in a speciation continuum in newts. Mol. Phylogenet. Evol. 116, 1–12 (2017).
Bhagwat, S. A. & Willis, K. J. Species persistence in northerly glacial refugia of Europe: a matter of chance or biogeographical traits? J. Biogeogr. 35, 464–482 (2008).
Tzedakis, P. C., Emerson, B. C. & Hewitt, G. M. Cryptic or mystic? Glacial tree refugia in northern Europe. Trends Ecol. Evol. 28, 696–704 (2013).
de Lafontaine, G., Guerra, C. A. A., Ducousso, A., Sánchez-Goñi, M.-F. & Petit, R. J. Beyond skepticism: uncovering cryptic refugia using multiple lines of evidence. New Phytol. 204, 450–454 (2014).
Tyszkowski, S. et al. Geology, permafrost, and lake level changes as factors initiating landslides on Olkhon Island (Lake Baikal, Siberia). Landslides 12, 573–583 (2015).
Shiklomanov, N. I., Streletskiy, D. A., Swales, T. B. & Kokorev, V. A. Climate change and stability of urban infrastructure in Russian permafrost regions: prognostic assessment based on GCM climate projections. Geogr. Rev. 107, 125–142 (2017).
Stewart, J. R., Lister, A. M., Barnes, I. & Dalén, L. Refugia revisited: individualistic responses of species in space and time. Proc. R. Soc. B 277, 661–671 (2010).
Juřičková, L., Horáčková, J. & Ložek, V. Direct evidence of central European forest refugia during the last glacial period based on mollusc fossils. Quat. Res. 82, 222–228 (2014).
Acknowledgements
This study was funded by the German Research Foundation (DFG; FR 1435/11-1 and FR 1435/11-2). In addition, Eva Graciá was supported by the Spanish Ministry of Economy, Industry and Competitiveness and the European Regional Development Fund (Project CGL2015-64144; MINECO/FEDER, UE). Many thanks go to Birgit Blosat for donating a grass snake sample, to Wolfgang Böhme for the photo of the Natrix helvetica and to Melita Vamberger for discussing the data analyses strategy and supplying the photo of the Natrix natrix. We thank Robert Sommer, an anonymous reviewer, and the editor for helpful comments.
Author information
Authors and Affiliations
Contributions
C.K. performed the lab work, raw data editing, most analyses, created the figures and wrote most of the draft. E.G. performed the DIYABC analyses and contributed to the manuscript. U.F. conceived and designed the study, discussed the data and text, and revised the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kindler, C., Graciá, E. & Fritz, U. Extra-Mediterranean glacial refuges in barred and common grass snakes (Natrix helvetica, N. natrix). Sci Rep 8, 1821 (2018). https://doi.org/10.1038/s41598-018-20218-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20218-2
This article is cited by
-
The last interglacial-glacial cycle in the Meuse Valley (southern Belgium) inferred from the amphibian and reptile assemblages: implications for Neanderthals and anatomically modern humans
Archaeological and Anthropological Sciences (2022)
-
Rate of change for the thermal adapted inversions in Drosophila subobscura
Genetica (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
