
Abstract
Necrotic enteritis (NE) is a severe intestinal disease, which can change gut microbiota and result in a high cost for the poultry industry worldwide. However, little is known regarding how the gut microbiota of NE chicken ileum are changed by Bacillus licheniformis. This study was conducted to investigate how ileum microbiota structure was changed by B. licheniformis in broiler chickens challenged with Clostridium perfringens-induced NE through Illumina MiSeq sequencing. The broilers were randomly separated into four groups: the negative control group (NC), the positive control group (PC), the fishmeal and coccidia group (FC), and the PC group supplied with feed containing B. licheniformis (BL). Compared to the PC and FC, alpha diversity, beta diversity, and the bacterial taxa of the ileum microbiota were more similar in BL and NC. Some genera, which were related to the NE control, became insignificant in BL with NC, such as Lactobacillus, Lactococcus, Bacteroides, Ruminococcus and Helicobacter. The PICRUSt analysis revealed that a tumour suppressor gene, p53, which was negatively correlated with Helicobacter, was enriched in the BL group. Our findings showed that the ileum microbiota disorder caused by NE in chickens was normalized by dietary B. licheniformis supplementation.
Similar content being viewed by others


Introduction
Necrotic enteritis (NE) in chickens, which was first reported by Parish in 19611, is a common illness caused by Clostridium perfringens2. There is increasing evidence that NE outbreaks in broiler chickens have a severe economic impact and globally cost over $2 billion annually in losses and disease control because of the high mortality rates and reduced growth performance3. NE not only causes economic losses but also results in illnesses in humans. NE in chickens can cause a threat to public health through the food chain by C. perfringens4. C. perfringens is a strictly anaerobic gram-positive bacterium, which can form spores. C. perfringens commonly presents in the intestinal tract of chickens but is not pathogenic and causes enterotoxaemia only under certain conditions. An experimental disease challenge trial showed that it is necessary to introduce induction factors, such as Eimeria co-infection and high protein feed supplementation (like fishmeal), to cause diseases5,6. Research on NE in chickens has been conducted for decades, but it is still one of the major challenges in the poultry industry. This is especially true since in-feed antibiotics have been banned in more and more countries7, and it is increasingly important to search for alternatives for the treatment of NE in chickens.
Probiotics are “friendly” bacteria that help to maintain a normal balance in the intestinal tract by aiding normal digestion, supporting the immune system and promoting overall health8. They can likely prevent and treat disease effectively by mainly modulating mucosal immune activity and epithelial barrier function as a biological antagonist, which has already been proven through clinical trials for maintaining disease treatment9. In addition, probiotics are characterized as safe, free from pollution and have no remaining compounds. Probiotics have become the ideal alternative for antibiotics. Bacillus subtilis and Lactic acid bacteria are common probiotics, and Bacillus licheniformis, which is “generally recognized as safe”, has been extensively used for a long time in the poultry industry3. B. licheniformis has promising affects in many industries, such as poultry and fishing, as a supplement. Research showed that supplementing feed water with B. licheniformis could enhance the growth performance of chickens10. This mechanism may explain how B. licheniformis could produce different enzymes, such as lipase, protease and amylase, which could increase the digestion and absorption ability of chickens11. Meanwhile, the metabolite of B. licheniformis could also help prevent some diseases, such as piglet diarrhoea and NE in chickens12.
The chicken’s ileum is an important site for digestion and nutrient absorption13 and is home to a large and varied microbiota community14. Several studies have shown that NE could induce disease of the intestines and change the intestinal microbiota structure of patients, especially for the ileum15,16. Chicken intestinal physiology and health were influenced by these changes. Therefore, the objective of this study was to investigate how the ileum microbiota structure was changed by B. licheniformis in broiler chickens challenged with C. perfringens-induced NE through high-throughput next-generation sequencing.
Results
Metadata and sequencing
A total of 60 chicken ileum samples (15 NC, 15 PC, 15 FC and 15 BL) were collected and sent for sequencing. After selecting the OTU and checking for chimaera, a total of 1,532,759 reads were assigned to 4,883 non-singleton OTUs from 60 samples. Each sample had 25,546 ± 9,653 reads and 428 ± 172 OTUs on average (Table S1). We used a Venn diagram, which is based on the OTUs, to compare the similarities and differences between the microbiota community of the different groups. Compared with NC, the FC, PC and BL communities had 1483, 1359 and 1205 shared OTUs (Figure S1), with the OTUs comprising 50.43%, 43.87% and 49.32% of the sequences in the FC, PC and BL communities, respectively.
The microbiota diversity analysis
The alpha diversity analysis was included for the observed species and the Shannon index, which was intended to be representative of the community richness and community diversity. Figure 1 shows the quartile deviation of observed species and the Shannon index. Noticeably, the community richness and community diversity in chickens with NC and BL were lower than those of PC and FC by the observed species (Fig. 1a) and the Shannon index (Fig. 1b). We found that BL and NC were significantly different from FC (P < 0.01) and were significantly different from PC (P < 0.05) in the observed species by boxplot. For the community diversity comparison, samples of NC were significantly different from those of FC (P < 0.01), and those of the BL group were also significantly different from those of FC (P < 0.05) in the Shannon index. However, no significant difference in Alpha diversity analysis was observed between the samples of NC and BL (P > 0.05).

Alpha diversity analysis of the ileum gut microbiota of NC, FC, PC and BL. Panel a represents differences in bacterial community richness (Observed species) between the four groups. Panel b represents differences in bacterial community diversity (Shannon) between the four groups. Asterisk shows significant differences between groups (**P < 0.01, *P < 0.05, Mann-Whitney U test).
The principal coordinates analysis (PCoA) and the gut microbiota trees, which revealed the similarity measure of bacterial communities based on the phylogenetic distance, were performed based on the weighted Unifrac distance matrixes (Fig. 2). The results of PCoA showed a degree of diversity discrepancy between each group, but the samples from NC, BL and FC clustered together, which was consistent with the results of the gut microbiota tree.

Principal Coordinate Analysis (PCoA) and Gut microbiota trees of Unifrac distances of the ileum gut microbiota of NC, FC, PC and BL. Panel a represents a 2 dimensional weighted PCoA plot by sample type. Panel b represents the Gut microbiota trees clustering on weighted Unifrac distances. Sequences were normalized to the depth of 9,718 sequences with 10 times of subsampling to minimize the effect of sequencing depth.
Comparison of microbiota in the ileum in each group
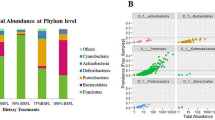
At the phylum level, the abundance, whose relative abundance was more than 0.1% in all of the groups, were classified into 7 phyla, and 6 were identified as a phylum, and the average relative abundances of them were over 99.50% of the overall bacteria community. Firmicutes, Proteobacteria and Bacteroidetes were significantly different in NC and PC. Proteobacteria and Bacteroidetes increased with the severity of necrotic enteritis (NC, FC and PC), whereas Firmicutes decreased. However, this phenomenon we called microbiota disorder was alleviated in BL group (Fig. 3).

Relative abundance of the broilers’ ileum microbiota in level phylum. The abundances of each groups are more than 0.01%.
Figure 4 shows the structures of the bacterial community of four groups in the chicken ileum samples. At the genus level, 422 genera were detected. The top 20 genuses with higher relative abundance, of which the average relative abundance was over 95.31% of the overall bacteria community, are illustrated. We performed LEfSe (LDA score = 3) to identify significant differences in the bacterial taxa. This threshold could guarantee that the meaningful taxa would be compared and eliminate most rare taxa. In NC, 7 bacterial taxa were significantly more abundant (P < 0.05), whereas 14 taxa were overrepresented in PC (P < 0.05) (Fig. 5a), and 2 bacterial taxa were significantly more abundant in NC (P < 0.05), whereas 38 taxa were overrepresented in FC (P < 0.05) (Figure S2a). No bacterial taxa were significantly more abundant in NC (P < 0.05), whereas only 2 taxa were overrepresented in BL (P < 0.05) (Fig. 5b). In BL, 2 bacterial taxa were significantly more abundant in BL (P < 0.05), whereas 10 taxa were overrepresented in PC (P < 0.05) (Figure S2b), and only one bacterial taxa was significantly more abundant in BL (P < 0.05), whereas 6 taxa were overrepresented in FC (P < 0.05) (Figure S2c). The results show that Lactobacillus and Lactococcus were significantly more abundant in NC than PC and FC (P < 0.05, Fig. 6a,b). However, Bacteroides and Ruminococcus were significantly more abundant in PC than in NC and BL (P < 0.05, Fig. 6c,d). The results also showed the abundance of Streptomyces and Helicobacter in four groups with a bar graph (Fig. 6e,f). The abundance of Streptomyces were more abundant in BL than in the other groups. In addition, the abundance of Helicobacter were more abundant in PC than in the other groups.

Microbiota composition of NC, FC, PC and BL. Each bar represents the average relative abundance of each bacterial taxon within a group. The top 20 abundant taxa are shown.

Taxa that were significantly differentially represented between groups were examined by linear discriminant analysis coupled with effect size (LEfSe) using the default parameters (LDA score = 3). Panel a show different taxa between NC and PC. Panel b show different taxa between NC and BL. The mean and median relative abundance of these bacterial taxa are indicated with straight and dotted lines, respectively.

The relative abundance of 6 bacterial taxa in 4 groups (NC, FC, PC and BL). Panel a show relative abundance of Lactobacillus. Panel b show relative abundance of Lactococcus. Panel c show relative abundance of Bacteroides. Panel d show relative abundance of Ruminococcus. Panel e show relative abundance of Streptomyces. Panel f show relative abundance of Helicobacter.
Bacterial function prediction
A computational tool, the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt), was used to predict the functional changes of the gut microbiota genomes in different treated groups from the Kyoto Encyclopedia of Genes and Genomes pathways. A total of 328 functions were detected in FC, PC and BL grounds, and 21 functions had a significant difference (bootstrap Mann-Whitney U-test, P < 0.01), which consisted of 6.40% of the overall functions, such as N−glycan biosynthesis, lipopolysaccharide biosynthesis and the P53 signalling pathway (Fig. 7).

Predicted function in the gut micorbiota of FC, PC and BL. The gene copy numbers of samples within the same sample group were pooled. Values of each functional gene (row) were log10 transformed. The third level of the Kyoto Encyclopedia of Genes and Genomes pathway was shown in the heatmap. The significant test of the gene distribution between groups were performed using bootstrap Mann-Whitney u-test with cutoffs of P < 0.01, FDR < 0.1.
Discussion
Necrotic enteritis is a common disease of chickens. There are many predisposing factors of NE, but fishmeal and coccidium are the 2 most used factors. As reported, fishmeal feed can increase the amount of mucus secreted by the intestine and make the intestinal contents very viscous. Coccidia infection can directly damage the intestinal mucosa, induce the proliferation of pathogens and finally lead to NE infection. Weakened immunity also increase the probability that chickens are infected with NE6. The animal experiment of our published paper had used the same flocks of chickens, which found that the growth performance of the FC group was better than that of the PC group and none of the chickens in FC group were dead compared with the PC group16, which agreed with our previous study. Therefore, we speculated that chickens in the FC group were healthier than those of the PC group. This was because of the function of C. perfringens, which could produce toxins and enzymes such as α toxin17. This kind of toxin is a main A type toxin of C. perfringens, and it has the enzymatic activity of Phospholipase C and Sphingomyelinase. The alpha toxin can also hydrolyse Phosphatidylcholine and Sphingomyelin and destroy the cell membrane structure. It can induce the neutrophil granulocyte and macrophage to release TNF-α, which results in the haemolysis and death of the cell18. And that paper also showed that B. licheniformis supplement could improve the health status of chickens suffering from NE according to the results of mortality and growth performance16. The reason may be that B. licheniformis could enhance the gut barrier ability, which could stop the endotoxin and pathogens from passing through the intestinal mucosa into the blood. This can enhance the growth of immune organs, activate the lymphocyte, increase the level of immune globulin, and improve the cellular immunity and humoral immunity19. Simultaneously, B. licheniformis can also adjust the gut microbiota within animals and reduce the number of pathogens. It can also produce many bioactivators, such as lysozyme, bacteriocin, antifungal protein and varieties of antibiotics (including phospholipids, amino acids and polyenoid)20.
The gut microbiota community was relatively stable, which guarantees the healthy status of the host. The microbiota of the host plays an important role in the health of mankind by absorbing nutrients, enhancing growth and metabolism, protecting against harmful bacteria and modulating the immune system such that the function is irreplaceable. There have been studies showing that an intestinal flora disturbance could cause many diseases, and the health status of the same species has similar microbiota communities21. NE is a severe intestinal disease, and it can change the microbiota community structure5. We can observe that the gut microbiota communities can reflect the healthy status of the NE in chickens indirectly.
This study found that NC and BL were more similar than PC or FC in community richness and community diversity. The cause of the difference to NC may be explained by the damage of the ileum mucous membrane, which stimulates the growth of the invading organisms in PC and FC. For BL, B. licheniformis could probably prevent a flora disorder by mainly modulating mucosal immune activity and epithelial barrier function as a biological antagonist, which would reduce the succession of the invading organisms and indigenous microbiota. Using PCoA and gut microbiota tree analysis, we found that BL were more similar to NC and FC. This also might be because of the regulation by B. licheniformis.
Results of LEfSe showed that 21 bacterial taxa were significantly different between NC and PC, but only 2 bacterial taxa were significantly different between NC and BL. Therefore, we presume that NC and BL were more similar than PC in the ileum microbiota community.
Many gut microbiota species, especially Lactobacillus, Lactococcus and Bacteroides, have an intimate relationship with the health of animals. In addition, Lactobacillus, Lactococcus and Bacteroides were significantly different taxa based on the LEfSe analysis.
Lactobacillus spp., which is one of the most important bacteria in the intestinal tract of humans and animals, functions by maintaining the bacterial community in the intestinal tract, improving immunity, and facilitating the absorption of nutrients. Some studies show that Lactobacillus inhibited the inflammatory responses of the gut and had antagonistic effects against intestinal and food-borne pathogens by enhancing the immune functions in the intestinal mucosa22. Lactobacillus in the gut of animals can regulate the expression of the toll-like receptor gene, activate the immune response of DC/NK interaction, and balance the immune response of Th1/Th222. In other words, the stability of Lactobacillus is very important to health. We found that the abundance of Lactobacillus in NC was significantly higher in PC but not significantly higher in BL. Therefore, we presume that BL may have stronger immune responses than PC. At the same time, we also found that the change in the abundances of Lactobacillus in chicken ileum were different than in chicken caecal pouches. Previous research showed that Lactobacillus of NE in chickens’ caecal pouches were changed in the composition of the species without changing the total abundance5. However, our study showed that the total abundances of Lactobacillus in PC and FC were significantly less than NC.
Lactococcus spp., a model LAB lactic acid bacteria, is one of the well-known lactate producing probiotics and is often used to improve the efficiency of animal digestion. The leading areas of study are the food-industry, biopharmaceuticals, and vaccines. The members of Lactococcus can produce bacteriocin (e.g., Nisin). Nisin is a member of the lantibiotic family of antimicrobial peptides that exhibit potent antibacterial activity against many gram-positive bacteria, including human and animal pathogens such as Staphylococcus, Bacillus, Listeria, and Clostridium23. Lactococcus lactis, which is a member of Lactococcus spp., can reduce the bacterial abundance of breeding ground for corruption, especially for Leuconostoc24. It is very important that the abundance of Lactococcus remain relatively stable. The results showed that the abundance of Lactococcus in BL were not significantly different from NC.
The LEfSe results showed that Bacteroides spp. were over-represented in PC. Bacteroides is a normal intestinal flora but can evolve into a pathogenic form, and the amounts could increase when the gut is pathologically changed or impaired25. Previous research shows that a member of Bacteroides could produce cell surface molecules. Cell surface molecules produced by this organism likely play important roles in colonization, communication with other microbes, and pathogenicity26, or even induce carcinoma of the intestine27. Human intestinal disease study analysis demonstrated that Bacteroides could cause diseases such as intra-abdominal infections28 and anaerobic bacteremia29. It could also enhance the resistance to antimicrobial agents such as antibiotics30,31 (e.g., carbapenems). The reason why Bacteroides were highest in PC may be explained by the fact that the disease of PC was the most serious. In BL, Bacteroides did not change, which may be due to the B. licheniformis, i.e., B. licheniformis can adjust the imbalance of normal intestinal flora and have a protective effect on the intestine.
Ruminococcus was the natural flora from the gut of chickens. In human studies, some Ruminococcus flora help cells to absorb sugars, which might contribute to weight gain32. The study showed that Ruminococcus in PC was higher than that of other groups. The relative results are the same as Willing et al.33 and Joossen et al.34, who studied on changes of intestinal microbiota of enteritis host, and the highly significant increase of the abundance of Ruminococcus was related closely to enteritis. There was another interesting report of increasing Ruminococcus in enteritis patients. It has been reported previously that Ruminococcus could produce lantibiotics, which could enhance sterilization activity against some clostridia and bifidobacteria species35. Thus, it was not desirable that the abundance of the Ruminococcus of the chicken ileum significant increased. Previous research showed that the increase in the number of Ruminococcus was related to mucin glycans36. Mucin, which is secreted by the gut epithelial cell, plays a key role in the gastrointestinal mucosal barrier, and it usually provides many adherens junctions and dietary requirements for bacteria. Some research showed its alterations were associated with numerous diseases, including carcinomas and inflammation37. The results of our study may be explained by the fact that the chicken ileum physiological environment was damaged, and the increased mucin was caused by damage, which finally led to an increasing abundance of Ruminococcus.
We found that the abundance of Streptomyces in BL was higher than in other groups. It also had an obvious dissimilarity between BL and the other groups, although LEfSe was not shown. Streptomyces spp., which have complex and large secondary metabolism regulatory networks, can produce many beneficial bioactive substances through secondary metabolism. Streptomycin, one of secondary metabolites of Streptomyces, has powerful antibacterial activity against mycobacterium tuberculosis and antimicrobial efficiency against a number of gram-negative bacteria (e.g., Escherichia coli, Hepatitis bacillus, Enterobacter, Salmonella and Brucella). In addition to antibiotics, the secondary metabolism of Streptomyces is varied, including anti-tumour agents, immune inhibitors, insect resistance agents and exocellular enzymes38 (e.g., pectinase, cellulase, and chitinase). In addition, recent research suggests that Streptomyces spp. could have benefits that extend beyond the gut, such as curing salmonella pullorum disease and Pasteurella multocida in infected chickens, but there are few studies on treating NE in chickens. It is also an anti-coccidial drug used in the poultry industry39, which is an important aetiological factor. Therefore, even Streptomyces spp. poorly exist in the chicken ileum by LEfSe, and it might be interesting to study the effects of Streptomyces in broiler chickens challenged with C. perfringens-induced NE.
We also found that a part of the different bacterial taxa were shared between the NC group and the other 2 groups (PC and FC), such as Lactobacillus. Lactobacillus was significantly more abundant in the NC group than the PC and FC groups in the LEfSe analysis, and the abundance of Lactobacillus in PC was less than FC. This may be related to the worsening of the illness. We found an interesting result, which showed that although chickens in the FC and PC group were infected with necrotic enteritis, Lactococcus was significantly more abundant in the FC group than the NC group, but it was significantly less in the PC group compared with the NC group. The healthy gut microbiota maintains relative stability or keeps the dynamic equilibrium status. Significantly increasing or decreasing the gut microbiota is bad for the host, especially the high relative abundance of a microorganism such as Lactococcus. This result might be similar to the impact on the host of changing the abundance of Bacteroides25.
The gut microbiota community was one of the factors affecting normal physiological functions. In human studies, abnormalities of the gut microbiota community can lead to allergies, obesity, diabetes and even cirrhosis of the liver40,41. The PICRUSt aims to predict the unobserved character states in a community of organisms from phylogenetic information regarding the organisms in the community. And they are commonly used to study animal intestinal function42. With respect to PICRUSt functional profiles, the enrichment of “Glycan Biosynthesis and Metabolism” (e.g., N−Glycan biosynthesis and lipopolysaccharide biosynthesis) pathways in the PC group is remarkable. This result was construable due to its high Ruminococcus abundance32. This was similar to our results of Ruminococcus abundance. We found that the P53 signalling pathway in the BL group is highly enriched. P53 is recognized as one of the important genes that participates in apoptosis control and is closely related to the incidence of a variety of human tumuors43. Inactivation of p53 by mutation occurs in over 50% of all human cancers. Some studies have shown that p53 inactivation is related to Helicobacter pylor infection44. Our study showed that the abundance of Helicobacter in PC was higher than the other groups. This was why the p53 signalling pathway in PC is not enriched. Chickens in PC was the most likely to develop bowel cancer, beacuse of the decrease of p53.
Our previous research showed that the supplement of B. licheniformis could significantly enhance the growth of immune organs45, number of white blood cells, mRNA expression levels of cytokines related to enteritis by the real-time PCR and villous histology of ileum was similar with those of the healthy chickens compared with chickens infected with NE46. The recent publication of ours also showed dietary B. licheniformis supplementation could adjust the expression levels of certain key genes related to lipid metabolism3. Our former paper also showed that probiotics could prevent NE by enhancing the immune function of ileum47,48. As everyone knows, gut microbiome can affect the immune function of host, the reason of which might be microbiome of ileum turns to be more balanced after probiotics treatment. At the same time, chickens of the BL group effectively alleviate the negative effects of NE infection and can also reduce antioxidant stress and enhance growth performance5. Therefore, we presume that chickens of the BL group was more health and hold more stable intestinal flora than chickens of the PC group.
In summary, the results of our study showed that the ileum microbiota of necrotic enteritis in chickens was normalized by dietary B. licheniformis supplementation. Furthermore, the data of this study may provide a new insight into the prevention and treatment of NE in broilers.
Materials and Methods
Test strain information
B. licheniformis H2 (CCTCC NO: M2011133) that was isolated from the ileum of healthy chickens was provided by the Animal Microecological Research Center (College of Veterinary Medicine, Sichuan Agricultural University, Chengdu, China).
The C. perfringens type-A (CVCC2030) strain isolated from a chicken clinically diagnosed with NE was obtained from the China Veterinary Culture Collection Center.
The DLV coccidium vaccine was developed by Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences.
Experimental design and sampling
The experiments were approved before animal testing by the Sichuan Agricultural University Committee on Ethics in the Care and Use of Laboratory Animals. According to the Administration of Affairs Concerning Experimental Animals standard, the animals were subject to a carefully managed plan. The flowchart of this study is shown in Figure S3. Simply stated, 240 chickens were born on the same day with similar body masses and were purchased from a local commercial hatchery. All the samples were randomly assigned to four groups, five replicates per treatment, and twelve chickens per replicate in a pen. The four groups were (1) a negative control group fed with a corn-soybean meal diet (NC, negative control); (2) an NE experimental model group (PC, positive control); (3) a group that was fed a diet supplemented with 30% of fishmeal from day 14 onwards and challenged with the DLV coccidiosis vaccine (FC, fishmeal and coccidia); and (4) an infected group given a diet supplemented with B. licheniformis (BL, B. licheniformis at a dose of 1.0 × 106 CFU/g). During this experiment, all the chickens were fed with free access to water and food in a temperature controlled room, and light was provided 24 hours a day. Table S2 shows the compositions of an un-medicated corn-soybean meal diet and a high fishmeal diet. The diets were formulated according to NRC (1994) (NRC. Nutrient Requirements of Poultry. 9th ed. Washington: The National Academies Press; 1994).
The birds were fed with a basal diet from day 1 to day 13. From day 14 onward, the diets of all the birds were changed to the basal diets supplemented with 30% fishmeal (w/w), except for those of the NC group. On day 15, all the birds, with the exception of those in the NC group, were inoculated with a 10-fold coccidiosis vaccine by oral gavage. In contrast, the birds in the NC group received sterile phosphate buffered saline. On days 18, 19, and 20, the birds in the PC and BL groups were individually infected with 1 mL of C. perfringens through a plastic tube containing approximately 2.2 × 108 CFU/mL. The feed of the BL group was dosed with 1.0 × 106 CFU/g B. licheniformis throughout the experiment.
On day 22, three chickens per pen were sacrificed randomly, and the contents in the ileum were collected. Sixty samples were immediately frozen in liquid nitrogen containers. The samples were then stored at −80 °C for further analysis.
All the experiments were performed in accordance with the approved guidelines and regulations.
DNA extraction and pyrosequencing
We extracted the total bacteria DNA from the contents in the ileum (100 ± 10 mg each) by the EZNA Stool DNA Kit(Omega Bio-tek) according to the manufacturer’s instructions, and DNA samples were stored at −80 °C before further analysis. The sequencing was performed at the Chengdu Institute of Biology, Chinese Academy of Sciences. Basically, 16S rRNA genes of 16sV4 were amplified using 515 F/806 R (515 F: 5′-GTGCCAGCMGCCGCGGTAA-3′, 806 R: 5′-GGACTACVSGGG TATCTAAT-3′) with the barcode. The PCR amplification system is 100 μL, including 0.75 units Ex Taq DNA polymerase (TaKaRa, Dalian, China), 1 × Ex Taq loading buffer (TaKaRa, Dalian, China), 0.2 mM dNTP mix (TaKaRa, Dalian, China), 0.2 µM of each primer, and 100 ng template DNA. The conditions of the PCR amplification were 95 °C for 3 min (1 cycle), 94 °C for 100 s/50 °C for 60 s/72 °C for 60 s (35 cycles), and a last step of 72 °C for 10 min. The PCR-amplification products were purified by 2% agarose gel electrophoresis, and the EZNA Gel Extraction Kit (Omega Bio-tek) was used for the recovery of DNA from the gels.
Bioinformatics and statistical analysis
The sequence reads were merged using mothur v1.31.249. The sequences were assembled using Qiime 1.8.0 after the chimeric sequences were removed by Usearch 7.0.1001 and based on the Unweighted Pair-group Method with Arithmetic Means algorithm50. Operational taxonomic units (OTUs) were generated at a 97% similarity threshold, and sequences less than two were removed from all samples. The microbiota diversity analysis included an Alpha diversity analysis (Observed species and Shannon index) and Jackknifed beta diversity (weighted Unifrac distances calculated with 10 times of subsampling) in different samples, and weighted Unifrac distances were visualized by PCoA. The gut microbiota trees were generated using the weighted Pair Group Method with an Arithmetic Mean algorithm based on the different distance metrics generated by Qiime. The significant difference test of alpha diversity (Mann-Whitney U-test) and significance test of beta diversity (two-sided Student’s t-test) were analysed using QIIME. The significant difference test of the phylum level was analysed using SPSS 19.0. Linear discriminant analysis coupled with effect size (LEfSe) was performed to identify the differential expression in the genus level between the groups of bacterial taxa and compared with the relative contents of the differential expression in the genus level of the bacterial taxa. The function of the ileum microbiota community was predicted using PICRUSt51. The R packages “Biom”, “Phyloseq”, and “Pheatmap” were used for the data analysis and plotting. The raw read sequences of our 60 samples have been deposited at the Sequence Read Archive of the National Center for Biotechnology Information, with the study accession number SRP128297.
References
Parish, W. E. Necrotic enteritis in the fowl (Gallus gallus domesticus). I. Histopathology of the disease and isolation of a strain of Clostridium welchii. Journal of Comparative Pathology. 71, 377–393 (1961).
Prescott, J. F., Smyth, J. A., Shojadoost, B. & Vince, A. Experimental reproduction of necrotic enteritis in chickens: A review. Avian Pathology. 45, 317–322 (2016).
Zhou, M. et al. Effects of Bacillus licheniformis on the growth performance and expression of lipid metabolism-related genes in broiler chickens challenged with Clostridium perfringens -induced necrotic enteritis. Lipids in Health & Disease. 15, 48, https://doi.org/10.1186/s12944-016-0219-2 (2016).
Van Immerseel, F. et al. Clostridium perfringens in poultry: an emerging threat for animal and public health. Avian Pathology. 33, 537–549 (2004).
Stanley, D., Wu, S. B., Rodgers, N., Swick, R. A. & Moore, R. J. Differential responses of cecal microbiota to fishmeal, Eimeria and Clostridium perfringens in a necrotic enteritis challenge model in chickens. PLoS One. 9, e104739, https://doi.org/10.1371/journal.pone.0104739 (2014).
Shojadoost, B., Vince, A. R. & Prescott, J. F. The successful experimental induction of necrotic enteritis in chickens by Clostridium perfringens: a critical review. Veterinary Research. 43, 74, https://doi.org/10.1186/1297-9716-43-74 (2012).
M’Sadeq, S. A., Wu, S., Swick, R. A. & Choct, M. Towards the control of necrotic enteritis in broiler chickens with in-feed antibiotics phasing-out worldwide. Animal Nutrition. 1, 1–11 (2015).
Pourabedin, M. & Zhao, X. Prebiotics and gut microbiota in chickens. FEMS Microbiology Letters. 362, v122, https://doi.org/10.1093/femsle/fnv122 (2015).
Jones, R. M. et al. Lactobacilli modulate epithelial cytoprotection through the Nrf2 pathway. Cell Reports. 12, 1217–1225 (2015).
Liu, X. et al. Growth performance and meat quality of broiler chickens supplemented with bacillus licheniformis in drinking water. British Poultry Science. 25, 682–689 (2014).
Rozs, M., Manczinger, L., Vágvölgyi, C. & Kevei, F. Secretion of a trypsin-like thiol protease by a new keratinolytic strain of Bacillus licheniformis. FEMS Microbiology Letters. 205, 221–224 (2001).
Knap, I., Lund, B., Kehlet, A. B., Hofacre, C. & Mathis, G. Bacillus licheniformis prevents necrotic enteritis in broiler chickens. Avian Diseases. 54, 931–935 (2010).
Shaufi, M. A. M., Sieo, C. C., Chong, C. W., Gan, H. M. & Yin, W. H. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathogens. 7, 4, https://doi.org/10.1186/s13099-015-0051-7 (2015).
Stanley, D., Hughes, R. J. & Moore, R. J. Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Applied Microbiology and Biotechnology. 98, 4301–4310 (2014).
Kim, J. E. et al. Dietary Capsicum and Curcuma longa oleoresins increase intestinal microbiome and necrotic enteritis in three commercial broiler breeds. Research in Veterinary Science. 102, 150–158 (2015).
Lin, Y. et al. Disruption in the cecal microbiota of chickens challenged with Clostridium perfringens and other factors was alleviated by Bacillus licheniformis supplementation. Plos One. 12, e0182426, https://doi.org/10.1371/journal.pone.0182426 (2017).
Masaya, T. et al. Clostridium perfringens α-toxin impairs innate immunity via inhibition of neutrophil differentiation. Scientific Reports. 6, 28192, https://doi.org/10.1038/srep28192 (2016).
Dahiya, J. P., Wilkie, D. C., Van Kessel, A. G. & Drew, M. D. Potential strategies for controlling necrotic enteritis in broiler chickens in post-antibiotic era. Animal Feed Science & Technology. 129, 60–88 (2006).
Deng, W., Dong, X. F., Tong, J. M. & Zhang, Q. The probiotic Bacillus licheniformis ameliorates heat stress-induced impairment of egg production, gut morphology, and intestinal mucosal immunity in laying hens. Poultry Science. 91, 575–582 (2012).
Kim, Y. et al. Identification and antimicrobial activity of phenylacetic acid produced by Bacillus licheniformis isolated from fermented soybean, chungkook-jang. Current Microbiology. 48, 312–317 (2004).
Karlsson, F. H. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 498, 99–103 (2013).
Tsai, Y. T., Cheng, P. C. & Pan, T. M. The immunomodulatory effects of lactic acid bacteria for improving immune functions and benefits. Applied Microbiology and Biotechnology. 96, 853–862 (2012).
Field, D. et al. A Bioengineered nisin derivative to control biofilms of Staphylococcus pseudintermedius. PLoS One. 10, e119684, https://doi.org/10.1371/journal.pone.0119684 (2014).
Comi, G., Andyanto, D., Manzano, M. & Iacumin, L. Lactococcus lactis and Lactobacillus sakei as bio-protective culture to eliminate Leuconostoc mesenteroides spoilage and improve the shelf life and sensorial characteristics of commercial cooked bacon. Food Microbiology. 58, 16–22 (2016).
Rocha, E. R. & Smith, C. J. Ferritin-like family proteins in the anaerobe Bacteroides fragilis: when an oxygen storm is coming, take your iron to the shelter. Biometals. 26, 577–591 (2013).
Wilson, M. M., Anderson, D. E. & Bernstein, H. D. Analysis of the outer membrane proteome and secretome of bacteroides fragilis reveals a multiplicity of secretion mechanisms. PLoS One. 10, e117732, https://doi.org/10.1371/journal.pone.0117732 (2015).
Kelly, D. et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nature Immunology. 5, 104–112 (2003).
Brook, I. & Frazier, E. H. Aerobic and anaerobic microbiology in intra-abdominal infections associated with diverticulitis. Journal of Medical Microbiology. 49, 827–830 (2000).
Lassmann, B., Gustafson, D. R., Wood, C. M. & Rosenblatt, J. E. Reemergence of anaerobic bacteremia. Clinical Infectious Diseases. 44, 895–900 (2007).
Treviño, M. et al. Susceptibility trends of Bacteroides fragilis group and characterisation of carbapenemase-producing strains by automated REP-PCR and MALDI TOF. Anaerobe. 18, 37–43 (2012).
Fernándezcanigia, L. et al. First national survey of antibiotic susceptibility of the bacteroides fragilis group: emerging resistance to carbapenems in Argentina. Antimicrob Agents Chemother. 56, 1309–1314 (2012).
Arumugam, M. et al. Enterotypes of the human gut microbiome. Nature. 473, 174–180 (2011).
Willing, B. P. et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 139, 1844–1854 (2010).
Joossens, M. et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 60, 631–637 (2011).
Dabard, J. et al. Ruminococcin A, a new lantibiotic produced by a Ruminococcus gnavus strain isolated from human feces. Applied & Environmental Microbiology. 67, 4111–4118 (2001).
Crost, E. H. et al. Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS One. 8, e76341, https://doi.org/10.1371/journal.pone.0076341 (2013).
Zhang, H. Y. et al. Expression of adhesion molecules and mucins in human and rhesus macaque gastrointestinal epithelial cells. Histology and Histopathology. 26, 1405–1413 (2011).
Saratale, G. D., Saratale, R. G. & Sangeun, O. Production and characterization of multiple cellulolytic enzymes by isolated Streptomyces sp. MDS. Biomass & Bioenergy. 47, 302–315 (2012).
Kant, V. et al. Anticoccidial drugs used in the Poultry: an overview. Science Internationa. 1, 261–265 (2013).
Wang, B. et al. Altered fecal microbiota correlates with liver biochemistry in nonobese patients with non-alcoholic fatty liver disease. Scientific Reports. 6, 32002, https://doi.org/10.1038/srep32002 (2016).
Qin, N. E. A. Alterations of the human gut microbiome in liver cirrhosis. Nature. 7516, 59–64 (2014).
Zeng, B. et al. The bacterial communities associated with fecal types and body weight of rex rabbits. Scientific Reports. 5, 9342, https://doi.org/10.1038/srep09342 (2015).
Gillotin, S. IASPP, a potential drug target in cancer therapy. Leukemia Research. 33, 1175–1177 (2009).
Bhardwaj, V. et al. Helicobacter pylori bacteria alter the p53 stress response via ERK-HDM2 pathway. Oncotarget. 6, 1531–1543 (2015).
Li, J. Genetic diversity of Clostridium perfringens and effects of Bacillus licheniformis on the ileum flora of chicken necrotizing enterocolitis: Sichuan Agricultural University (2010).
Liu, L. Influences of Bacillus licheniformis on ileum histology of chickling necroticans enteritis and Cytokine dynamic variation: Sichuan Agricultural University (2010).
Wang, H. et al. Probiotic Enhanced Intestinal Immunity in Broilers against Subclinical Necrotic Enteritis[J]. Frontiers in Immunology. 10, https://doi.org/10.3389/fmmu.2017.01592 (2017)
Qing, X. et al. Preventing subclinical necrotic enteritis through Lactobacillus johnsonii, BS15 by ameliorating lipid metabolism and intestinal microflora in broiler chickens[J]. Amb Express. 7, https://doi.org/10.1186/s13568-017-0439-5 (2017).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 75, 7537–7541 (2009).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 27, 2194–2200 (2011).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology. 31, 814–821 (2013).
Acknowledgements
International Cooperative Project of Science and Technology Bureau of Sichuan Province (2013HH0055, 2014FZ0076). Agricultural science and technology research and development project of Chengdu science and Technology Bureau (2015-NY02-00295-NC).
Author information
Authors and Affiliations
Contributions
S.X., Y.L., M.Z., D.Z. and X.N. designed the experiment. S.X., Y.L., M.Z. and H.W., performed the experiments. S.X., H.Z. and Y.L. analyzed the experimental data. S.X., Y.Z., Y.Z., K.P., and B.J. contributed reagents/materials/analysis tools. S.X., Y.L., D.Z. and X.N. discussed the results and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, S., Lin, Y., Zeng, D. et al. Bacillus licheniformis normalize the ileum microbiota of chickens infected with necrotic enteritis. Sci Rep 8, 1744 (2018). https://doi.org/10.1038/s41598-018-20059-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20059-z
This article is cited by
-
Compound Probiotics Improve Body Growth Performance by Enhancing Intestinal Development of Broilers with Subclinical Necrotic Enteritis
Probiotics and Antimicrobial Proteins (2023)
-
Effects of Bacillus methylotrophicus SY200 Supplementation on Growth Performance, Antioxidant Status, Intestinal Morphology, and Immune Function in Broiler Chickens
Probiotics and Antimicrobial Proteins (2023)
-
Bacillus licheniformis PF9 improves barrier function and alleviates inflammatory responses against enterotoxigenic Escherichia coli F4 infection in the porcine intestinal epithelial cells
Journal of Animal Science and Biotechnology (2022)
-
Choice of 16S ribosomal RNA primers affects the microbiome analysis in chicken ceca
Scientific Reports (2021)
-
A study on fungal defensin against multidrug-resistant Clostridium perfringens and its treatment on infected poultry
Applied Microbiology and Biotechnology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
