
Abstract
Identifying single nucleotide polymorphisms (SNPs) that influence chemotherapy disposition may help to personalize cancer treatment and limit toxicity. Genome-wide approaches are unbiased, compared with candidate gene studies, but usually require large cohorts. As most chemotherapy is given cyclically multiple blood sampling is required to adequately define drug disposition, limiting patient recruitment. We found that carboplatin and paclitaxel disposition are stable phenotypes in ovarian cancer patients and tested a genome-wide association study (GWAS) design to identify SNPs associated with chemotherapy disposition. We found highly significant SNPs in ABCC2, a known carboplatin transporter, associated with carboplatin clearance (asymptotic P = 5.2 × 106, empirical P = 1.4 × 10−5), indicating biological plausibility. We also identified novel SNPs associated with paclitaxel disposition, including rs17130142 with genome-wide significance (asymptotic P = 2.0 × 10−9, empirical P = 1.3 × 10−7). Although requiring further validation, our work demonstrated that GWAS of chemotherapeutic drug disposition can be effective, even in relatively small cohorts, and can be adopted in drug development and treatment programs.
Similar content being viewed by others


Introduction
Large inter-individual variation in drug disposition has been reported for most chemotherapeutic drugs at standard dosing schedules1, including paclitaxel and carboplatin2,3. For women with advanced epithelial ovarian cancer (EOC), the standard of care is cytoreductive surgery and six cycles of paclitaxel and carboplatin chemotherapy4,5. Despite a high initial response rate (~75%), the median progression-free survival is between 16–21 months and there is a wide range in median overall survival reported, between 31–57 months6. There is also significant variability in chemotherapy side effects, some with a genetic component. For example, more haematological and neurological toxicities have been reported in Asian ovarian cancer patients compared to European patients5,7,8.
It may be hypothesized that variation in drug disposition is a contributing factor in variable treatment effects in EOC. There have been several well-studied associations between germline genetic variants and chemotherapeutic drug toxicity, leading to the inclusion of pharmacogenomic information in the US Food and Drug Administration (FDA) advice to prescribers for drugs including Irinotecan and Capecitabine9,10,11. In examples to date, these genetic variants were identified using a candidate gene approach, focusing on genes involved in drug metabolism. However, this approach requires a priori biological knowledge and will not reveal novel biomarkers or associations. The aim of this study was to identify novel germline genetic variants that influence carboplatin and paclitaxel disposition in ovarian cancer patients using a genome-wide association study (GWAS) approach.
Results
Patient selection and characteristics
The clinical characteristics of the two patient cohorts in this study, the Australian and Dutch cohorts, were similar, except that Dutch patients had higher body weight (due in part to increased height), resulting in higher paclitaxel doses (Table 1). The median creatinine level was lower (P = 0.002) in the Australian cohort (although both were within normal range), resulting in higher estimated glomerular filtration rate (GFR) (P = 0.003).
A significant inter-individual, but small intra-individual variability in paclitaxel and carboplatin disposition
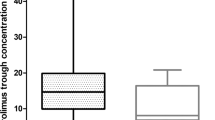
The paclitaxel and carboplatin pharmacokinetic (PK) parameters were summarized in Tables 2 and 3, respectively. These results were consistent with published data12,13. Significant inter-individual variability was observed in paclitaxel TC>0.05 (Time of paclitaxel concentration > 0.05 µmol/L) (>2-fold) and carboplatin clearance (~5-fold). In contrast, there were no statistically significant differences of paclitaxel TC>0.05 (P = 0.595) (Fig. 1a) and carboplatin clearance (P = 0.682) (Fig. 1b) between two treatments in the seven Australian patients with PK sampling performed at both cycle one and three. These results demonstrated that chemotherapeutic drug disposition was a stable phenotype in individuals (at least over a six-week period), and supported the hypothesis that there is a genetic component underlying drug disposition.

Pharmacokinetic parameter intra-individual variability. (a) Time of paclitaxel concentration >0.05 µmol/L (TC>0.05) and (b) carboplatin clearance from cycle one and three of chemotherapy in seven Australian patients, analysed by paired sample t test. The patients are ordered according to increasing carboplatin clearance rate.
Correlations between drug disposition and hematological toxicity
Hematological assessment after the first cycle of chemotherapy was performed in 49 Australian patients receiving combination chemotherapy. Paclitaxel TC>0.05 was associated with relative neutropenia (Fig. 2a) (R = 0.436, P = 0.002) but not with relative thrombocytopenia (Fig. 2b) (R = 0.247, P = 0.087). On the other hand, the correlation between carboplatin clearance and relative neutropenia was not statistically significant (Fig. 2c) (R = −0.144, P = 0.324), but there was a statistically significant inverse correlation between carboplatin clearance and relative thrombocytopenia (Fig. 2d) (R = −0.336, P = 0.018) by simple linear regression analysis. These findings are in accordance with previous publications14,15,16 and confirmed paclitaxel TC>0.05 is a good predictive marker for severe neutropenia, whereas carboplatin clearance predicts thrombocytopenia, using the population pharmacokinetic modelling we applied.

Associations between paclitaxel and carboplatin disposition and haematological toxicities. (a) Association between time of paclitaxel concentration >0.05 µmol/L (TC>0.05) and relative neutropenia; (b) association between TC > 0.05 and relative thrombocytopenia; (c) association between carboplatin clearance and relative neutropenia and (d) association between carboplatin clearance and relative thrombocytopenia, analysed by simple linear regression analysis in 49 Australian patients receiving paclitaxel and carboplatin combination chemotherapy.
GWAS identified novel polymorphisms associated with paclitaxel exposure
We found that ~27% variability in paclitaxel TC>0.05 was accounted for by paclitaxel dose, GFR and AST (aspartate amino transaminase), therefore tested each SNP for its association with paclitaxel TC>0.05 without and with adjustment for these three covariates. Thirty-seven patients of European ethnicity and 14 of Asian ethnicity from the Australian cohort, and 35 patients of European ethnicity from the Dutch cohort were available for analysis. Six Dutch patients were missing AST values therefore 29 were included in the adjusted analysis.
Manhattan plots, showing SNP association P-values from the meta-analysis (-log10(p)) against genomic position, in unadjusted and adjusted analyses are presented in Fig. 3a and b. The genomic control parameters were 1.01 and 1.02 respectively, suggesting that there was little population substructure or other artefacts. In the unadjusted GWAS (Fig. 3a), 25 SNPs were found to be associated with paclitaxel TC>0.05 with P < 1 × 10−5 (Supplementary Table S1). The SNP with the highest statistical significance was rs17130142. The P value from meta-analysis was 1.8 × 10−7. We ran 107 permutations to correct for the asymptotically approximated results and empirical p-value remained unchanged. In the adjusted analysis (Fig. 3b), 19 SNPs were associated with paclitaxel TC>0.05 with P < 1 × 10−5 (Supplementary Table S2). Rs17130142 remained the most statistically significant polymorphism.

Paclitaxel GWAS. Manhattan plots of paclitaxel disposition GWAS (a) unadjusted and (b) adjusted for paclitaxel dose, glomerular filtration rate (GFR) and aspartate amino transaminase (AST); (c) LocusZoom plot for rs17130142 from the adjusted paclitaxel GWAS. The color shading indicates the strength of LD (r2) from the reference of 1000 Genome Project (CEU samples) between the corresponding SNP and the top SNP, rs17130142. The red horizontal line indicates the genome-wide significance threshold of P = 5 × 10−8; the blue line is the threshold for putative association (P = 10−5).
Rs17130142 is an intergenic polymorphism on chromosome 1 (Fig. 3c). It has a minor allele frequency (MAF) ~5% in European patients (7% and 3% in the Australian and Dutch cohort respectively), but was monomorphic in the 14 Asian patients. The minor allele A was associated with increased paclitaxel TC>0.05 in European patients from both the Australian cohort (effect = 8.58, P = 6.5 × 10−6) and the Dutch cohort (effect = 18.24, P = 7.7 × 10−5). In the meta-analysis, it met the criteria for genome-wide statistical significance (P = 2.0 × 10−9). Due to the small number of heterozygous patients (5 from 37 Australian patients and 2 from 29 Rotterdam patients), the empirical p-value after 107 permutations dropped to 1.3 × 10−7.
ABCC2 polymorphisms associated with carboplatin clearance
Consistent with published data17, we found GFR accounted for >90% variability in carboplatin clearance. Therefore, we tested association between each SNP and carboplatin clearance unadjusted and adjusted for GFR. 42 European patients (1 removed due to low genotyping call rate) and 14 Asian patients in the Australian cohort were included in meta-analysis weighted by sample size.
The genomic control parameters for the unadjusted and adjusted analysis were 1.00 and 1.03 respectively. In the unadjusted GWAS (Fig. 4a), four SNPs were found to be associated with carboplatin clearance with P < 1 × 10−5 (Supplementary Table S3). After adjusting for GFR, these associations were not statistically significant (P > 0.05), suggesting the putative associations found between these SNPs and carboplatin clearance were due to associations with GFR.

Carboplatin GWAS. Manhattan plots of carboplatin clearance GWAS (a) unadjusted and (b) adjusted for glomerular filtration rate; (c) LocusZoom plot for ABCC2 loci from adjusted carboplatin GWAS. The colour shading indicates the strength of LD (r2) from the reference of 1000 Genome Project (CEU samples) between the corresponding SNP and the labelled SNP, rs8187710. The red horizontal line indicates the genome-wide significance threshold of P = 5 × 10−8; the blue line is the threshold for putative association (P = 10−5).
However, in the adjusted GWAS (Fig. 4b), 17 SNPs were found to be associated with carboplatin clearance with P < 1 × 10−5 (Supplementary Table S4). Of highest statistical significance was rs7230264 (P = 8.7 × 107), a SNP that lies within an intron of RP11-739N10, a long intergenic non-coding RNA (lincRNA) of unknown function on chromosome 18. The minor allele A was associated with decreased carboplatin clearance and has a frequency of 12% in European patients and 25% in Asian patients.
Interestingly, a cluster of SNPs in complete linkage disequilibrium (r2 = 1) around ABCC2 were found to be putatively associated with carboplatin clearance in 42 patients with European ancestry (P = 5.2 × 10−6) (Fig. 4c). They are monomorphic in 14 patients with Asian ancestry (Supplementary Table S4). Located on chromosome 10q24, ABCC2 encodes a carboplatin efflux transporter ABCC2 (also known as multiple resistant protein 2 - MRP2) (see the locusZoom plot18 in Fig. 4c). Due to the small number of heterozygous patients for these SNPs (N = 3), we also ran 107 permutations to correct for the asymptotically approximated results. The empirical p value from the permutation was 1.4 × 10−5. The details of the four SNPs in ABCC2 are summarised in Table 4. Patients carrying the variant alleles at these SNPs had decreased carboplatin clearance (effect = −1.21; Supplementary Table S4), potentially leading to a higher carboplatin systemic exposure.
Discussion
Inherited genetic variation has an impact on drug absorption, metabolism and excretion, and therefore on pharmacodynamics. Identifying SNPs that influence chemotherapy disposition may help to personalize individual treatment. While GWAS has the advantage of being unbiased, they usually require a large sample size. Since most cytotoxic drugs have a short half-life and are given cyclically, multiple blood samples are required to adequately define drug disposition and systemic exposure. This tends to limit patient sample sizes in pharmacokinetic studies. Having demonstrated that carboplatin and paclitaxel disposition is a stable phenotype within an individual, we performed two separate GWAS of paclitaxel and carboplatin disposition and identified novel inherited genetic variants associated with drug disposition in ovarian cancer patients.
In the paclitaxel GWAS, we identified a novel polymorphism associated with paclitaxel TC>0.05 with genome-wide significance. Rs17130142 is located on chromosome 1 between LOC100505768 (distance = 402 kb) and PKN2 (Protein kinase N2) (distance = 910 kb). PKN2 encodes a transmembrane serine/threonine protein kinase. It plays a role in cell cycle progression and cytokinesis19. Being an intergenic polymorphism, the function and effects of rs17130142 are unknown, but it may regulate upstream or downstream genes and affect gene splicing, transcription factor binding, messenger RNA degradation, or the sequence of non-coding RNA20,21. Further fine mapping and functional analysis to determine the target gene and underlying mechanism is warranted.
In the adjusted carboplatin GWAS, four ABCC2 SNPs in complete linkage disequilibrium were associated with carboplatin clearance. ABCC2 is localized in the apical luminal membrane of polarized epithelial cells of several excretory organs including liver, intestine and kidney22. It has an important function in the biliary excretion of endogenous metabolites. ABCC2 transports drug conjugated to glutathione including platinum agents23 and also exports many unconjugated substances24. ABCC2 polymorphism has been found associated with response and survival in patients with gastric25, oesophageal26, ovarian27, non-small cell cancer lung cancer28,29. Among the four SNPs, rs8187710 (4544 G > A) in exon 32 is non-synonymous leading to a cysteine to tyrosine transition at position 1515 in the last cytoplasmic loop of ABCC2. In stably transfected human embryonic kidney cells (HEK293), a 4544 A clone showed higher accumulation of lopinavir, calcein and carboxyfluorescein diacetate (ABCC2 substrates) as well as reduced ABCC2 mediated efflux of lopinavir compared with a 4544G clone30. Increased risks of acute anthracycline-induced cardiac toxicity31 and congestive heart failure32 have also been reported in patients carrying the minor allele at rs8087710, who received doxorubicin chemotherapy, another ABCC2 substrate. As there is no physiological evidence of ABCC2 expression in the heart, increased cardiac toxicity could be caused by a reduced biliary elimination of doxorubicin, which normally accounts for 50% of its disposition33. Further supporting our findings, the major G allele of rs8187710 has been reported to be associated with inferior overall survival in 188 platinum-treated stage IV non-small-cell lung cancer (ASCO annual meeting 2012, abstract number 7586), presumably due to increased platinum clearance. These reports are in agreement with our finding that minor alleles of these ABCC2 SNPs are associated with reduced ABCC2 transporter function, leading to decreased carboplatin clearance. This could be clinically relevant for platinum toxicity and response. Although fine mapping and functional analyses have not been performed, ABCC2 is an obvious candidate and it is reasonable to assume that it is the functionally relevant target of this GWAS finding.
Additional SNPs were found to be associated with carboplatin clearance in the adjusted GWAS. Among the SNPs listed in Supplementary Table S4, rs7230264 located in lincRNA RP11-739N10 on chromosome 18 was of the highest statistical significance. LincRNAs have been implicated in a wide range of cellular processes including gene transcription regulation34, post-transcriptional regulation35 and epigenetic regulation36. The role of lincRNA RP11-739N10 in relation to carboplatin clearance remains to be determined.
GWAS have been used to identify common DNA sequence variants that are associated with susceptibility to over 250 complex disease traits, using very large sample size of thousands to hundreds of thousands of patients. With few exceptions, the odds ratios (ORs) for the effects are typically in the range of 1.05 to 1.15. In contrast, GWAS studies of drug efficacy or safety with small patient cohorts, with some ranging from only 20 to 80 patients37,38,39,40, have revealed a substantial number of unanticipated and often striking signals. The large effects identified in pharmacogenomics studies are likely due to the lack of an evolutionary response to modern exogenous agents. Unlike genes associated with common disease, which are located on haplotype blocks that have been selected to overcome environmental influences and selection pressure over thousands if not millions of years, the genome has had very little time to adapt to exposure to prescription medications41. This could explain our significant findings in a relatively small patient cohort.
In conclusion, our GWAS approach identified SNPs putatively associated with carboplatin and paclitaxel disposition. Examination of the functional characteristics of the SNPs may provide novel pharmacogenomic biomarkers for personalized ovarian cancer treatment. More importantly, our findings support that GWAS of chemotherapeutic drug disposition can be effective even when performed in a relatively small number of samples and can be used to improve the drug candidate pipeline and boost the efficacy while limiting the toxicity of medications already in use. As genotyping and sequencing costs continue to decline, every effort should be made to incorporate comprehensive, unbiased pharmacogenomic analyses in strategies to optimise treatment outcome.
Materials and Methods
Patient population
Patients with EOC receiving paclitaxel and carboplatin (N = 55) or single agent carboplatin (N = 6), were recruited between 2009 and 2012 from Westmead Hospital and Royal Prince Alfred Hospital, Sydney, Australia. Key inclusion criteria were histologically or cytologically confirmed EOC; ECOG ≤ 2; adequate organ function and no concurrent use of moderate or strong cytochrome P450 and ATP-binding cassette transporter (ABC transporter) inhibitors or inducers. An independent paclitaxel pharmacokinetic (PK) cohort of 35 EOC patients was recruited through the Erasmus MC - Cancer Institute, Rotterdam, the Netherlands. This study was approved by the Medical Ethics Review Board Erasmus MC in the Netherlands and the Sydney West Area Health Service Human Research Ethics Committee (now known as the Western Sydney Local Health District Human Research Ethics Committee) in Australia. All experiments were performed in accordance with relevant guideline and regulations. Written informed consent was obtained from each patient.
Cremophor EL formulated paclitaxel (175 mg/m2) was administered intravenously (i.v.) over a planned 3-hour infusion. This was immediately followed by a 1-h infusion of carboplatin (AUC 5-6 mg/mL*min) in patients receiving combination chemotherapy. The carboplatin doses were calculated according to the method described by Calvert42 and glomerular filtration rate (GFR) was estimated using the Cockcroft-Gault formula43.
Pharmacokinetic blood sampling and bioanalysis
Blood samples for PK analyses were taken in 10-mL ETDA tubes during the cycle one treatment for all patients. In consenting participants from the Australian cohort, additional PK blood samples were also collected during cycle three of chemotherapy (N = 7). In the Australian cohort, blood sampling was taken immediately before the start, 1.5 hours after the start, at the end of paclitaxel infusion, at the end of carboplatin infusion, ~2–3 hours after the end of carboplatin infusion and ~ 21 hours after the end of paclitaxel infusion respectively, with the precise time for each blood sampling recorded. In the Dutch cohort, blood sampling was performed before the start, 1.5 hours after the start, at the end and ~2–3 hours after the end of paclitaxel infusion.
Whole blood was centrifuged immediately for 10 minutes at 3,000 rpm (1,500 g) at 4 °C. For paclitaxel PK analysis, plasma was collected and stored at -80 °C. For carboplatin PK analysis, plasma was immediately transferred and centrifuged through a 30-kDa cut-off Amicon® ultrafiltrate filter (Millipore, Billerica, Massachusetts USA) for 15 min at 3,000 rpm at 20 °C. The preparation of ultrafiltrate was done immediately after blood collection to prevent the decrease of free carboplatin level due to ex vivo binding of carboplatin to plasma proteins and erythrocytes. All specimens were stored in cryovials at -80 °C until analysis.
Paclitaxel concentrations were measured using liquid chromatography - tandem mass spectrometry (LC-MS/MS) (Waters Chromatography B.V, Etten-Leur, the Netherlands) at the Laboratory of Translational Pharmacology, Department of Medical Oncology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands44. Carboplatin levels were measured using a Varian 820 inductively coupled plasma mass spectrometry (ICP-MS) connected to a Varian SPS 3 auto-sampler as previously described with minor modification45 at Trace Inorganics Laboratory, Division of Analytical Laboratories, Sydney West Area Health Service, Australia.
Population pharmacokinetic modelling
The paclitaxel and carboplatin concentration-time data were analysed using the non-linear, mixed-effects modelling program (NONMEM) version VI (ICON, Hanover, MD, USA). R (version 2.13 or above, R programming) and PsN (Perl Speaks for NONMEM, Uppsala) were used for data preparation, graphical analysis, model diagnostics. The first-order conditional estimation with interaction (FOCEI) method in NONMEM was employed for model runs.
A stepwise forward selection and backward elimination approach was used for both paclitaxel and carboplatin modelling. Generally, a base model without any covariates was developed. Random effects were treated as lognormal in distribution. Residual error was modelled as additive error, proportional error or a combination of these two. One, two or three compartment models were tested when necessary. After a best base model was found, covariate modeling was then performed. The covariates examined in the model included age, weight, GFR, albumin, bilirubin, alanine aminotransferase (ALT), aspartate aminotransferase (AST), haemoglobin, white blood cell (WBC) count, absolute neutrophil count (ANC) and platelet. Final model was assessed by a visual predictive check, comparing observed concentration vs. time data and the 90% confidence interval generated using 500 simulated concentration–time data sets.
One compartment model with proportional and additive residual error described carboplatin concentration data very well. The final model for paclitaxel is a 3-compartment model with proportional residual error46. Paclitaxel clearance is non-linear and it is not clear which time points are the most indicative of clinically important paclitaxel clearance. Time of paclitaxel concentration >0.05 µmol/L (TC>0.05) has been reported to be associated with both haematological toxicity and chemotherapy response and survival14, simulation was therefore performed to obtain TC>0.05 by introducing more frequent sampling schedules in the dataset.
Haematological toxicity assessment
Baseline full blood counts (FBCs) were measured within 14 days prior to cycle one chemotherapy. Nadir FBCs were measured on day 14 ± 3 after cycle one chemotherapy. Haematological toxicities were assessed by relative neutropenia and thrombocytopenia, defined as the percentage reduction of nadir absolute neutrophil counts and platelet counts compared with baseline.
DNA extraction and genotyping
DNA was extracted from whole blood using DNA Mini Kit (Qiagen, GmbH, Hilden, Germany). A total of 719,665 SNPs was genotyped on the Australian and Dutch samples using Illumina Human OmniExpress arrays (Illumina Inc, San Diego, CA USA). The following quality control steps were applied: (1) removing samples with >5% missing genotypes; (2) excluding any SNP with less than 1% MAF; (3) excluding any SNP that failed the Hardy-Weinberg Equilibrium (HWE) test at the significance level of P = 1 × 10–7; (4) excluding the SNPs with MAF > 5% when the per SNP no-call rate >5%, and SNPs with MAF < 5% when the per SNP no-call rate >1%. One Australian patient was removed from analysis due to low genotyping rate (~75%). A total of 610 255 autosomal SNPs were retained after quality control. In addition, we checked samples for cryptic relatedness using the software PLINK47 and genetic ancestry using EIGENSTRAT48. We confirmed that self-reported ethnicities were concordant with genetic ancestries for all samples.
Statistical analysis
Linear regression was used to test the association between each SNP and the trait of interest, both adjusted and unadjusted for corresponding covariates, using PLINK47. Because meta-analysis has been shown to be more efficient than replication-based analysis49, we performed separate analysis within each population and then ran meta-analysis combining results from different populations weighted by sample sizes using METAL50. P values < 1 × 10−5 were considered as putative associations, and P values < 5 × 10−8 were regarded as genome-wide significance.
References
Mathijssen, R. H., Sparreboom, A. & Verweij, J. Determining the optimal dose in the development of anticancer agents. Nat. Rev. Clin. Oncol. 11, 272–281, https://doi.org/10.1038/nrclinonc.2014.40 (2014).
de Jonge, M. E. et al. Bayesian pharmacokinetically guided dosing of paclitaxel in patients with non-small cell lung cancer. Clin. Cancer Res. 10, 2237–2244 (2004).
Millward, M. J. et al. Carboplatin dosing based on measurement of renal function–experience at the Peter MacCallum Cancer Institute. Aust. N. Z. J. Med. 26, 372–379 (1996).
Ozols, R. F. et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 21, 3194–3200, https://doi.org/10.1200/JCO.2003.02.153 (2003).
du Bois, A. et al. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J. Natl. Cancer Inst. 95, 1320–1329 (2003).
Heintz, A. P. et al. Carcinoma of the ovary. FIGO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int. J. Gynaecol. Obstet. 95(Suppl 1), S161–192, https://doi.org/10.1016/S0020-7292(06)60033-7 (2006).
Takei, Y. et al. A feasibility study of paclitaxel and carboplatin therapy in Japanese patients with epithelial ovarian cancer. Oncol. Rep. 10, 951–955 (2003).
Katsumata, N. et al. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: a phase 3, open-label, randomised controlled trial. Lancet 374, 1331–1338, https://doi.org/10.1016/S0140-6736(09)61157-0 (2009).
Yim, B. T., Sims-McCallum, R. P. & Chong, P. H. Rasburicase for the treatment and prevention of hyperuricemia. Ann. Pharmacother. 37, 1047–1054 (2003).
Relling, M. V. et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharmacol. Ther. 89, 387–391, https://doi.org/10.1038/clpt.2010.320 (2011).
Cote, J. F. et al. UGT1A1 polymorphism can predict hematologic toxicity in patients treated with irinotecan. Clin. Cancer Res. 13, 3269–3275, https://doi.org/10.1158/1078-0432.CCR-06-2290 (2007).
Green, H. et al. Pharmacogenetic studies of Paclitaxel in the treatment of ovarian cancer. Basic Clin Pharmacol Toxicol 104, 130–137, https://doi.org/10.1111/j.1742-7843.2008.00351.x (2009).
Okamoto, H., Nagatomo, A., Kunitoh, H., Kunikane, H. & Watanabe, K. Prediction of carboplatin clearance calculated by patient characteristics or 24-hour creatinine clearance: a comparison of the performance of three formulae. Cancer Chemother. Pharmacol. 42, 307–312 (1998).
Joerger, M. et al. Population pharmacokinetics and pharmacodynamics of paclitaxel and carboplatin in ovarian cancer patients: a study by the European organization for research and treatment of cancer-pharmacology and molecular mechanisms group and new drug development group. Clin Cancer Res 13, 6410–6418, https://doi.org/10.1158/1078-0432.CCR-07-0064 (2007).
Huizing, M. T. et al. Pharmacokinetics of paclitaxel and three major metabolites in patients with advanced breast carcinoma refractory to anthracycline therapy treated with a 3-hour paclitaxel infusion: a European Cancer Centre (ECC) trial. Ann. Oncol. 6, 699–704 (1995).
Gianni, L. et al. Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J. Clin. Oncol. 13, 180–190 (1995).
Calvert, A. H. et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J. Clin. Oncol. 7, 1748–1756 (1989).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337, https://doi.org/10.1093/bioinformatics/btq419 (2010).
Schmidt, A., Durgan, J., Magalhaes, A. & Hall, A. Rho GTPases regulate PRK2/PKN2 to control entry into mitosis and exit from cytokinesis. EMBO J. 26, 1624–1636, https://doi.org/10.1038/sj.emboj.7601637 (2007).
Encode Project Consortium. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816, https://doi.org/10.1038/nature05874 (2007).
Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, https://doi.org/10.1038/nature11247 (2012).
Konig, J., Nies, A. T., Cui, Y., Leier, I. & Keppler, D. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim. Biophys. Acta 1461, 377–394 (1999).
Keppler, D., Leier, I. & Jedlitschky, G. Transport of glutathione conjugates and glucuronides by the multidrug resistance proteins MRP1 and MRP2. Biol. Chem. 378, 787–791 (1997).
Payen, L. et al. The drug efflux pump MRP2: regulation of expression in physiopathological situations and by endogenous and exogenous compounds. Cell Biol. Toxicol. 18, 221–233 (2002).
Li, Z. et al. ABCC2-24C> T polymorphism is associated with the response to platinum/5-Fu-based neoadjuvant chemotherapy and better clinical outcomes in advanced gastric cancer patients. Oncotarget 7, 55449–55457, https://doi.org/10.18632/oncotarget.10961 (2016).
Rumiato, E. et al. A germline predictive signature of response to platinum chemotherapy in esophageal cancer. Transl Res 171(29–37), e21, https://doi.org/10.1016/j.trsl.2015.12.011 (2016).
Lambrechts, S. et al. Genetic variability in drug transport, metabolism or DNA repair affecting toxicity of chemotherapy in ovarian cancer. BMC Pharmacol Toxicol 16, 2, https://doi.org/10.1186/s40360-015-0001-5 (2015).
Han, B. et al. Association of ABCC2 polymorphisms with platinum-based chemotherapy response and severe toxicity in non-small cell lung cancer patients. Lung Cancer 72, 238–243, https://doi.org/10.1016/j.lungcan.2010.09.001 (2011).
Sun, N. et al. MRP2 and GSTP1 polymorphisms and chemotherapy response in advanced non-small cell lung cancer. Cancer Chemother Pharmacol 65, 437–446, https://doi.org/10.1007/s00280-009-1046-1 (2010).
Elens, L. et al. Functional defect caused by the 4544G> A SNP in ABCC2: potential impact for drug cellular disposition. Pharmacogenet. Genomics 21, 884–893, https://doi.org/10.1097/FPC.0b013e32834d672b (2011).
Wojnowski, L. et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation 112, 3754–3762, https://doi.org/10.1161/CIRCULATIONAHA.105.576850 (2005).
Armenian, S. H. et al. Genetic susceptibility to anthracycline-related congestive heart failure in survivors of haematopoietic cell transplantation. Br. J. Haematol. 163, 205–213, https://doi.org/10.1111/bjh.12516 (2013).
Hidemura, K. et al. Shiga-like toxin II impairs hepatobiliary transport of doxorubicin in rats by down-regulation of hepatic P glycoprotein and multidrug resistance-associated protein Mrp2. Antimicrob. Agents Chemother. 47, 1636–1642 (2003).
Goodrich, J. A. & Kugel, J. F. Non-coding-RNA regulators of RNA polymerase II transcription. Nat. Rev. Mol. Cell Biol. 7, 612–616, https://doi.org/10.1038/nrm1946 (2006).
Beltran, M. et al. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 22, 756–769, https://doi.org/10.1101/gad.455708 (2008).
Sanchez-Elsner, T., Gou, D., Kremmer, E. & Sauer, F. Noncoding RNAs of trithorax response elements recruit Drosophila Ash1 to Ultrabithorax. Science 311, 1118–1123, https://doi.org/10.1126/science.1117705 (2006).
Daly, A. K. et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 41, 816–819, https://doi.org/10.1038/ng.379 (2009).
Link, E. et al. SLCO1B1 variants and statin-induced myopathy–a genomewide study. N. Engl. J. Med. 359, 789–799, https://doi.org/10.1056/NEJMoa0801936 (2008).
McCormack, M. et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 364, 1134–1143, https://doi.org/10.1056/NEJMoa1013297 (2011).
Ozeki, T. et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum. Mol. Genet. 20, 1034–1041, https://doi.org/10.1093/hmg/ddq537 (2011).
Harper, A. R. & Topol, E. J. Pharmacogenomics in clinical practice and drug development. Nat. Biotechnol. 30, 1117–1124, https://doi.org/10.1038/nbt.2424 (2012).
Alberts, D. S. & Dorr, R. T. New Perspectives on an Old Friend: Optimizing Carboplatin for the Treatment of Solid Tumors. Oncologist 3, 15–34 (1998).
Cockcroft, D. W. & Gault, M. H. Prediction of creatinine clearance from serum creatinine. Nephron 16, 31–41 (1976).
Sparreboom, A. et al. Determination of paclitaxel in human plasma using single solvent extraction prior to isocratic reversed-phase high-performance liquid chromatography with ultraviolet detection. J. Chromatogr. B Biomed. Sci. Appl. 705, 159–164 (1998).
Louie, H., Wong, C., Huang, Y. J. & Fredrickson, S. A study of techniques for the preservation of mercury and other trace elements in water for analysis by inductively coupled plasma mass spectrometry (ICP-MS). Anal Methods 4, 522–529 (2012).
Callies, S. et al. A population pharmacokinetic model for paclitaxel in the presence of a novel P-gp modulator, Zosuquidar Trihydrochloride (LY335979). Br. J. Clin. Pharmacol. 56, 46–56 (2003).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575, https://doi.org/10.1086/519795 (2007).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909, https://doi.org/10.1038/ng1847 (2006).
Skol, A. D., Scott, L. J., Abecasis, G. R. & Boehnke, M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 38, 209–213, https://doi.org/10.1038/ng1706 (2006).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191, https://doi.org/10.1093/bioinformatics/btq340 (2010).
Acknowledgements
This project was supported by the National Health and Medical Research Council (NHMRC) of Australia (scholarship to BG), Cancer Institute New South Wales (CINSW) through the Sydney West Translational Research Centre and support for BG, Cancer Council New South Wales through the PRIMe consortium, and Cancer Australia (552481). AdeF was funded by the University of Sydney Cancer Research Fund. SM acknowledges support from an NHMRC Fellowship and an Australian Research Council Fellowship. RB was a CINSW Fellow. CL, DC and the genotyping infrastructure were supported by Cancer Research - UK grants [C8197/A16565]. The Gynaecological Oncology Biobank at Westmead is a member of the Australasian Biospecimen Network-Oncology group, funded by the National Health and Medical Research Council Enabling Grants ID 310670 & ID 628903 and the Cancer Institute NSW Grant ID 12/RIG/1-17. This work was presented in part at the 2014 ASCO Annual Meeting (Chicago, IL, May/June 2014, #5563) and the 2015 AACR Annual Meeting (Philadelphia, Pennsylvania, April 2015, #5493).
Author information
Authors and Affiliations
Contributions
B.G. and A.deF. conceived the experiments and led the study. Y.L. and S.M. performed the GWAS analysis, C.L., D.C. and J.B. performed genotyping. H.X. performed pharmacokinetic modelling, B.G., A.N., I.B., A.deG., P.B., M.H., R.M. and P.H. contributed to patient recruitment and data accrual, P.deB., H.G., C.K., Y.C., S.J., R.B. and G.C.T. contributed the concept development, data accrual and data analysis. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, B., Lu, Y., Nieuweboer, A.J.M. et al. Genome-wide association study of paclitaxel and carboplatin disposition in women with epithelial ovarian cancer. Sci Rep 8, 1508 (2018). https://doi.org/10.1038/s41598-018-19590-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19590-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
