
Abstract
Sperm entry in mammalian oocytes triggers intracellular Ca2+ oscillations that initiate resumption of the meiotic cell cycle and subsequent activations. Here, we show that phospholipase C zeta 1 (PLCζ1) is the long-sought sperm-borne oocyte activation factor (SOAF). Plcz1 gene knockout (KO) mouse spermatozoa fail to induce Ca2+ changes in intracytoplasmic sperm injection (ICSI). In contrast to ICSI, Plcz1 KO spermatozoa induced atypical patterns of Ca2+ changes in normal fertilizations, and most of the fertilized oocytes ceased development at the 1–2-cell stage because of oocyte activation failure or polyspermy. We further discovered that both zona pellucida block to polyspermy (ZPBP) and plasma membrane block to polyspermy (PMBP) were delayed in oocytes fertilized with Plcz1 KO spermatozoa. With the observation that polyspermy is rare in astacin-like metalloendopeptidase (Astl) KO female oocytes that lack ZPBP, we conclude that PMPB plays more critical role than ZPBP in vivo. Finally, we obtained healthy pups from male mice carrying human infertile PLCZ1 mutation by single sperm ICSI supplemented with Plcz1 mRNA injection. These results suggest that mammalian spermatozoa have a primitive oocyte activation mechanism and that PLCζ1 is a SOAF that ensures oocyte activation steps for monospermic fertilization in mammals.
Similar content being viewed by others


Introduction
At mammalian fertilization, sperm entry causes Ca2+ oscillations, repetitive acute increases and decreases in cytosolic Ca2+ levels lasting for several hours in human and mouse eggs, which trigger not only resumption of the meiotic cell cycle but also block polyspermy, the entry of multiple sperm heads into the ooplasm1. These events are collectively referred to as oocyte activation and are prerequisite for normal subsequent mitosis and embryonic development2.
The mechanism of how sperm generate the Ca2+ transients in oocytes have been studied for decades. Microinjection studies introducing extracts from rabbit, hamster, or boar spermatozoa have strongly implied that, in mammals, protein(s) in sperm soluble fraction can trigger Ca2+ oscillations comparable to those caused by fertilization3,4. Such proteins are termed as sperm-borne oocyte activation factor (SOAF). So far, multiple candidates for SOAF have been raised by microinjection studies5,6. However, injection studies of purified protein7,8 or analysis of gene knockout (KO) mice9 showed discrepancies in identifying the SOAF.
A sperm-specific phospholipase C zeta 1 (PLCζ1) was identified as a candidate for SOAF10. In somatic cells, phospholipase C (PLC) mediates the digestion of phosphatidylinositol 4,5-bisphosphate (PIP2) to generate inositol triphosphate (IP3). These cause the release of Ca2+ from the endoplasmic reticulum11. PLCζ1 was also shown that it can induce Ca2+ oscillations in oocytes by releasing Ca2+ from the oocyte’s endoplasmic reticulum at levels far above the unstimulated very low basal intracytoplasmic Ca2+ concentrations12. However, Plcz1 KO mouse was reported preliminary at a conference as male infertile because of defective spermatogenesis13.
In this report, we generated Plcz1 KO mice with the CRISPR/Cas9 system and revealed that PLCζ1 plays an essential role in ICSI fertilization but not in normal fertilization. While no Ca2+ spikes were observed in ICSI fertilized oocytes, atypical Ca2+ spikes were observed in all oocytes from normal fertilization. Very recently, Hachem et al., generated Plcz1 KO mice and reported similar results, but there are significant discrepancies between that report and our data regarding Ca2+ spikes14. We further discovered that the PMBP plays a more critical role than ZPBP in vivo. Our findings revealed the existence of a PLCζ1 independent oocyte activation mechanism and clarified the role of PLCζ1 in ensuring monospermic fertilization.
Results
Generation of Plcz1 KO mice and oocyte activation in micromanipulation
Because conventional gene knockout approaches usually replace one to several exons with a drug-resistant gene cassette, there remains a risk of unexpected effects from the truncated open reading frames and the exogenously introduced promoter15. To exclude such possible artifacts, here we utilized a CRISPR/Cas9 approach that deleted all the coding exons (exons 2–14) to generate Plcz1 KO mice (Supplementary Fig. 1). Absence of the PLCζ1 protein was confirmed by western blotting analysis. Examination of our Plcz1 KO mice did not show any defects in spermatogenesis, sperm morphology, motility, or acrosome reaction rates (Supplementary Fig. 1).
To examine the oocyte activation abilities of Plcz1 KO spermatozoa, we performed intracytoplasmic sperm injection (ICSI) using epididymal spermatozoa. All oocytes injected with wild-type (WT) sperm heads formed pronuclei (PN), and most of them developed to 2-cell embryos (100% and 76.8%, respectively, n = 69; Fig. 1a). In contrast, oocytes injected with Plcz1 KO sperm heads failed to form PN or develop to 2-cell stage embryos (1.6% and 0%, respectively, n = 62; Fig. 1a), similarly to sham ICSI treatments (3.3% and 3.3%, respectively, n = 61; Supplementary Fig. 2a). Recordings of intracellular Ca2+ changes in these ICSI oocytes revealed that Plcz1 KO sperm heads lost the ability to induce rises in intracellular Ca2+ (Fig. 1b). No spikes were observed even after up to three sperm heads were injected (Supplementary Fig. 2b). Injection of whole spermatozoa also resulted in quiescent oocytes indicating no SOAF activity remained (Fig. 1b). These data strongly support the idea that PLCζ1 is the SOAF.

PLCζ1 is the essential SOAF in ICSI. (a) Oocytes at 2, 8, and 20 h after ICSI. Nuclei/pronuclei (PN) were visualized with H2B-mCherry (red). Oocytes with PN (WT 69/69 vs. KO 1/62), and 2cell embryos (WT 53/69 vs. KO 0/62) were counted at 8 h and 20 h after ICSI, respectively. Representative images are shown. Arrows, arrowheads, and asterisk indicate maternal, paternal, and fragmented chromosomes, respectively. Scale bar = 20 μm. (b) Intracellular Ca2+ changes in oocytes after ICSI. Ratio of GEM-GECO fluorescence was recorded.
Fecundity of Plcz1 KO male and oocyte activation in physiological conditions
The impact of PLCζ1 deficiency on male fertility was then examined by mating experiments with WT female mice. Copulations and vaginal plug formation were observed at normal rates for Plcz1 KO males. Unexpectedly, Plcz1 KO male mice sired healthy pups consistently, although the number of pups per copulation was significantly reduced (WT 8.9 pups vs. KO 2.3 pups; Fig. 2a). We then collected the oocytes from females after mating with WT or Plcz1 KO males and examined fertilization status (Fig. 2b). With WT males, all oocytes showed monospermy with a distinct male PN at 12 h after coitus. With Plcz1 KO males, more than 80% of oocytes (90/107) were fertilized, but fewer than one-half formed 2PN (41/90) (Fig. 2c). The remaining fertilized oocytes were observed to experience activation failure (no PN but with fertilization cone(s) at 12 h, indicated as 0PN; 15/90), abnormal activation (1PN; 10/90), or polyspermy (≥3PN; 24/90; Fig. 2c). When we recovered and cultured fertilized oocytes after mating with WT or KO males, increases in PN numbers were observed in some oocytes fertilized by KO sperm, implicating delays in fertilization and/or oocyte activation with Plcz1 KO sperm (Supplementary Table 1). When we further cultivated 2PN oocytes and counted blastocysts at 108 h after coitus, preimplantation developmental ability was lower in oocytes fertilized by KO sperm than WT sperm (29/90 = 32.2% and 56/62 = 90.2%, respectively) (Supplementary Table 1). Thus, although Plcz1 KO male mice are not sterile, the combined defects of activation failure and polyspermy may explain the reduced litter sizes, caused by male subfertility in vivo.

PLCζ1 is not essential for in vivo fertilization. (a) Fecundity of Plcz1 heterozygous and KO males. The horizontal line and numerals indicate the mean numbers of pups/vaginal plug noted. (b) Oocytes collected from oviducts of females mated with Plcz1 KO males at 10 h post coitus. Oocytes with various numbers of pronuclei were observed. Arrow and dotted circles represent the fertilizing cone and pronuclei, respectively. Scale bar = 20 μm. (c) The percentages of oocytes with the numbers of PN at 12 h post coitus.
To examine further the sperm fertilizing ability and oocyte activation ability, we performed in vitro fertilization (IVF) experiments at various sperm concentrations (2, 10, and 50 × 103 spermatozoa/ml) (Fig. 3a, Supplementary Table 2). Fertilization rates were similar between WT and KO groups and increased along with sperm concentration. Characteristically, activation failure was evident with Plcz1 KO spermatozoa at lower sperm concentrations (WT 0% vs. KO 12.4% at 2 × 105/ml), and the polyspermy increased up to ~80% at higher sperm concentrations (WT 7.6% vs. KO 82.4% at 50 × 103/ml; Fig. 3a). We next used live imaging to monitor intracellular Ca2+ concentrations. When we focused on monospermic fertilized oocytes, all the IVF fertilized oocytes with Plcz1 KO spermatozoa demonstrated intracellular Ca2+ spikes regardless of PN formation (Fig. 3b, Supplementary Fig. 2c). The Ca2+ spike patterns were aberrant in these oocytes (usually with decreased amplitude), and the number of spikes significantly decreased (WT 12.0 ± 5.68 spikes, 30 eggs vs. KO 2.75 ± 0.65 spikes, 28 eggs, P < 0.05). When we analysed the oocytes fertilized by a single spermatozoon retrospectively, the number of spikes observed in the 0PN oocytes were significantly fewer than those in the oocytes with formed PNs (Fig. 3c), implicating the existence of a threshold of intracellular Ca2+ spikes to trigger PN formation. Consistently, as more spermatozoa fused, the number of Ca2+ spikes increased (Fig. 3d), and more oocytes resumed the cell cycle (Fig. 3e). These findings revealed that mammalian spermatozoa have an oocyte activation ability independent from PLCζ1. However, a single spermatozoon is insufficient and rarely triggers the resumption of the meiotic cell cycle, with incomplete oocyte activation leading to polyspermy.

PLCζ1-independent oocyte activation causes abnormal fertilization. (a) The percentages of oocytes with each number of PN at 12 h after insemination. (b) Intracellular Ca2+ changes in oocytes after fertilization. Ratios of GEM-GECO fluorescence of oocytes with each genotype/number of pronuclei (representatives are shown). The numbers of PN formed (top left), and the mean numbers of Ca2+ spikes observed in oocytes (bottom right) are indicated. The numbers in parentheses indicate the numbers of oocytes observed. (c) Ca2+ spikes for oocytes fertilized by a single spermatozoon from Plcz1 KO males. Numbers of Ca2+ spikes for each number of pronuclei are indicated. Oocytes with one or two PN contained significantly more spikes than 0PN oocytes (*P < 0.005). (d) Ca2+ spikes for oocytes fertilized with differing numbers of Plcz1 KO sperm. Number of Ca2+ spikes for oocytes fertilized with 2 spermatozoa contained significantly more spikes than that fertilized with a single spermatozoon (*P < 0.005). (e) The percentages of oocytes that formed PN. Percentages are based on total numbers of oocytes fertilized with indicated number of Plcz1 KO sperm.
Polyspermy block systems in oocytes fertilized by KO spermatozoa
There are two major mechanisms to prevent polyspermy in mammals, the ZPBP and the PMBP. The process of the ZPBP is well documented. After fertilization, Ca2+ oscillations induce the cortical reaction (CR), an exocytosis event that releases the ASTL from the cortical granules into the extracellular space. This process exposes glycosylated proteins on the cell surface and can be visualized using Lens culinaris agglutinin-fluorescein isothiocyanate complex (LCA–FITC) labelling. The released ASTL cleaves the ZP2 protein and changes the ZP structure to prevent excess sperm entry16. When we visualized intracellular Ca2+ and the CR by LCA–FITC labelling17, a delayed and incomplete CR was observed coincident with a delayed Ca2+ spike by ~1 h that also had a lower amplitude in oocytes fertilized with Plcz1 KO spermatozoa (Fig. 4a). Some oocytes failed to induce the CR during the initial Ca2+ spike. Immunoblotting indicated that ZP2 cleavage was completed by 4 h after insemination with Plcz1 KO spermatozoa, whereas it was completed in 1 h with WT spermatozoa (Fig. 4b).

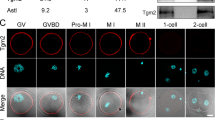
Delayed PMBP establishment by Plcz1 deletion is a cause of polyspermy. (a) Representative fluorescence plots for R-GECO (intracellular Ca2+; gray) and LCA–FITC staining (CR; black) of oocytes after insemination. Black arrowheads highlight Ca2+ spikes that induced the CR, while white arrowheads indicate Ca2+ spikes that failed to initiate the CR. (b) ZP2 immunoblot of oocytes after insemination. Intact ZP2 and the cleaved C-terminal fragments of ZP2 were 120 kD and 90 kD, respectively. A full–length blot is presented in Supplementary Figure 7a. (c) PMBP establishment estimated with the number of fused sperm of in vitro fertilized oocytes. The data reflect the means of separate experiments for each genotype. (d) Oocytes collected from oviduct of Astl heterozygous or KO females mated with WT male. Bright field observation (upper) and nuclear staining (lower) using Hoechst 33342 (blue) at 12 h post coitum are indicated. Oocytes from Astl KO females were denuded to prevent spermatozoa within the perivitelline space (arrowheads) interfering with pronuclei counting. The numbers of 2PN oocytes/total oocytes from five females for each genotype are indicated. No KO oocyte exhibited polyspermy. Scale bar = 50 μm.
Ca2+ is also required for the PMBP, but its mechanism remains unknown18. Here, we assessed the establishment of PMBP by counting the number of spermatozoa fused with ZP-free oocytes (fusion index) at several time points. While the fusion index with WT spermatozoa plateaued at ~2 sperm/oocyte 60 min after insemination as reported previously19 (2.0 ± 0.54 at 60 min and 2.0 ± 0.51 at 80 min, respectively), the fusion indexes with Plcz1 KO spermatozoa were significantly higher than with the WT and continued to increase even after 60 min (4.1 ± 1.14 at 60 min and 4.6 ± 1.12 at 80 min, respectively; Fig. 4c). We also examined PMBP status 12–15 h after the 1st insemination. Oocytes fertilized with WT spermatozoa or with Plcz1 KO spermatozoa rarely fused with WT spermatozoa at the 2nd insemination (Supplementary Fig. 3a, b). Therefore, we conclude that both the ZPBP and PMBP were delayed but eventually established in the oocytes fertilized with PLCζ1-deficient spermatozoa.
The significance of these polyspermy blocking mechanisms was further challenged using Astl KO mice, which lacks the ZP2 cleavage. When Astl KO female mice were mated with WT male mice, the ZP2 protein remained intact after fertilization (Supplementary Fig. 3c), and extra spermatozoa accumulated in the perivitelline space indicating failure of the ZPBP, but the PMBP was still functional and all the oocytes exhibited 2PN (Fig. 4d). This maintenance of monospermic fertilization strongly indicates that the PMBP alone is sufficient to block polyspermy in vivo. Delayed PMBP is the most important reason for the polyspermy found with Plcz1 KO male mice.
Rescue of the infertility of Plcz1 mutants by PLCζ1 supplementation
Human genetic diseases are mainly caused by small mutations rather than gene deletions. There are several PLCZ1 point mutations reported in infertile men20,21. Also, several artificial mutations were shown to influence oocyte activation ability as determined by injecting specific mRNAs into mouse oocytes22 (Supplementary Fig. 4a, b). Here, we phenocopied the human infertility-associated mutations in mice and introduced H435P (an equivalent mutation of human H398P) as well as D210R (an enzymatically dead mutation)10 mutations into mice using CRISPR/Cas9 technology (Supplementary Fig. 4c–e). Both mutant mRNAs were transcribed at WT levels in mutant mouse testes (Fig. 5a). Interestingly, when sperm proteins were analysed by immunoblotting, while the D210R protein was detected at ~74 kDa in similar amounts to WT mice, no H435P protein was detected at ~74 kDa (all 5 males examined for each genotype; Fig. 5b, Supplementary Fig. 5a). It should be noted that we occasionally observed ~20 kDa signal only in H435P sperm (3/5 males; Supplementary Fig. 5a), implicating the instability of H435P protein in vivo. Homozygous male mice carrying D210R or H435P mutations phenocopied Plcz1 KO male mice in spermatogenesis, fertility, IVF, and ICSI outcomes (Fig. 5c,d, Supplementary Fig. 5b–d). By using these point mutant mice as male infertility models, we complemented oocyte activation with WT Plcz1 mRNA injection. The optimized concentrations of human and mouse Plcz1 mRNA for the activation of mouse oocytes23 were adopted in the study. When we injected mouse Plcz1 mRNA (2 ng/μl × 1–3 pl) 1 h after ICSI using mutant spermatozoa, Ca2+ oscillations were followed by successful 2PN formation (Fig. 5d). The injection of 0.2 ng/μl human PLCZ1 mRNA also successfully activated oocytes. Healthy pups were obtained after transplantation of the embryos obtained with Plcz1 mRNA injection (Fig. 5e, Table 1).

Plcz1 mRNA can activate oocytes fertilized with Plcz1 mutant spermatozoa. (a) RT–PCR analysis of testicular RNA. Actb; actin beta. (b) Immunoblot of sperm proteins. Full–length blots are presented in Supplementary Figure 5a and e. (c) The percentages of oocytes with specified numbers of PN 12 h after IVF with Plcz1 point mutant mice. (d) Ratio of GEM-GECO fluorescence of oocytes after ICSI (upper) or 2 ng/µl mouse Plcz1 mRNA injection following ICSI (lower). Images of oocytes 8 h after ICSI or mRNA injection are shown. Nuclei/PN were visualized with H2B-mCherry (red). Scale bar = 20 μm. (e) Representative host mother and pups from ICSI-rescued embryos.
Discussion
Here, we have demonstrated that PLCζ1 is the long-sought SOAF in mice as indicated by the complete deletion of Ca2+ spikes in ICSI (Fig. 1). Further, we found that the existence of a PLCζ1-independent oocyte activation mechanism through normal fertilization where sperm–oocyte membrane interaction occurs. The activity is weak and barely triggers meiotic cell cycle resumption. Thus, this sperm–oocyte membrane interaction is probably a primitive but intrinsic mechanism for oocyte activation.
The results contrast with previous work reported at a conference in which Plcz1 KO male mice were presented to be sterile with defective spermatogenesis13. However, this was likely to be an artifact of the KO strategy that produced a truncated product. Very recently, Hachem et al., reported a similar phenotype of Plcz1 knockout mice14, but there is a notable difference. While we observed atypical but clear Ca2+ oscillations in all the oocytes after normal fertilization, Hachem et al., only found a single spike in one out of forty oocytes. In the present study, by labelling fused sperm chromosomes with Histon H2B-mCherry (Fig. 3b), we only analysed fertilized oocytes. Although the fertilization rate was not indicated, incorporation of unfertilized oocytes in the denominator may explain the difference. The correlation of Ca2+ spikes with the number of fused spermatozoa suggested that spermatozoa have a PLCζ1-independent Ca2+ spike inducing ability (Fig. 3c–e).
In the previous studies with electropermeabilization, more than four Ca2+ spikes were required to induce successful CR and subsequent cell cycle resumption24,25. However, we recently reported that CR completed within a few Ca2+ spikes during normal fertilization17. In this study, we found a significant difference in the number of Ca2+ spikes observed in (0PN) and activated (1 or 2PN) oocytes (2.30 ± 0.48 vs. 2.96 ± 0.56) with Plcz1 KO sperm. Further analysis will be required, however, our data implicates that the mouse oocyte requires an average of three Ca2+ spikes to be activated.
There are several mechanisms proposed for Ca2+ dependent oocyte activation through sperm–oocyte interaction26, such as the Ca2+ conduit model found in Caenorhabditis elegans27, and a membrane receptor model found in Xenopus28. Further study with Plcz1 KO mice will help to elucidate which mechanism(s) are used in mammals. It is interesting to note that newts, birds and insects are known to require polyspermy to ensure oocyte activation but use only a single decondensed sperm head to drive further development and ensure the correct diploid chromosome set in embryogenesis. However, polyspermy compromises further development in mammals. Here, we showed that PLCζ1 plays critical roles in both ZPBP and the PMBP. This ensures monospermic fertilization with successful oocyte activation and the prevention of extra spermatozoa from entering the ooplasm. We also showed that PMBP, rather than ZPBP, plays critical roles in the monospermic fertilization in vivo in mice. This finding will shed light onto development and the roles of these two mechanisms in various organisms.
In infertility clinics, because only a few oocytes are collected during retrieval, conventional IVF protocols have largely been replaced with ICSI protocols to increase the success rate and decrease the superovulatory burden on women29. Although ICSI ensures monospermic fertilization, oocyte activation failures still occur and have been reported as a cause of unexplained infertility30. As we observed polyspermy in Plcz1 mutant mice, such patients might have polyspermy in vivo as well as oocyte activation failure following ICSI. If it is caused by male infertility, this form could possibly be treated by a combination of ICSI and PLCζ1 complementation in vitro.
Methods
Animals
Wild-type mice (C57BL/6 N × DBA/2) F1, also known as B6D2F1, were purchased from CLEA Japan (Tokyo, Japan) or Japan SLC (Shizuoka, Japan). All animal experiments were approved by the Animal Care and Use Committee of the Research Institute for Microbial Diseases, Osaka University, under ethics approval number Biken-AP-H25-02-0, and were performed in accordance with the relevant guidelines and regulations.
Generation of Plcz1 knockout mice with the CRISPR/Cas9 system
Two pX330 plasmids expressing single guide (sg)RNAs targeting the 5′ and 3′ regions of Plcz1 (5′–TAGACGAAGAGCCCTCTATG–3′ and 5′–GTGCGAACCTTGAACCTTCC–3′) were co-transfected with human codon-optimized CAS9 to remove the coding region of Plcz1 in EGR-G101 (C57BL/6) ES cells31. Embryonic stem (ES) cell clones were selected with puromycin. Correctly targeted ES cell clones and germ-line transmission were determined via polymerase chain reaction (PCR) using primers for the KO allele (primer c: 5′–GACCACATCTTTCATGTCC–3′ and primer d: 5′–AGCAACTGAGAATGCAACCC–3′) or the wild-type (WT) allele (primer a: 5′–ATGACTAGGGAGGAGCAGAGAC–3′ and primer b: 5′–ATTCCCATGACCACTCACTACC–3′). A founder mouse with a 50.8 kbp deletion was used to expand the colony.
Intracytoplasmic sperm injection (ICSI)
ICSI was performed as described32. In brief, each sperm head was separated from the tail by applying a few piezo pulses, then injected into a denuded MII oocyte using a piezo manipulator (PrimeTech, Ibaraki, Japan). Whole sperm injection was carried out after immobilization of motile spermatozoa by a piezo pulse.
Fertility testing
Sexually mature male mice of each genotype were caged with B6D2F1 sexually mature female mice. Copulation was confirmed by checking for vaginal plugs every morning and the numbers of pups were counted after caesarean section at 17.5 days post coitum.
In vivo fertilization assay
WT female mice were injected with pregnant mare serum gonadotropin and human chorionic gonadotropin (hCG) at 48 hours intervals. Twelve hours after hCG injection, females were caged with control or mutant male mice. The female mice that copulated with males within 60 min were used in the study. Oocytes were collected from oviducts 7–8 h after coitus with male mice of each genotype and incubated in potassium-supplemented simplex optimized medium (KSOM) medium. The numbers of pronuclei were counted at 12 and 15 h post coitum. Subsequent embryonic development was observed to the blastocyst stage. For the analysis of Astl KO oocytes, heterozygous or homozygous KO female mice17 were superovulated, mated with WT males, and oocytes were collected as described above. To count the number of pronuclei, oocytes were stained with Hoechst 33342 (1 μg/ml, for 10 min) at 12 h after coitus. Zona removal with collagenase (Wako, Osaka, Japan) treatment at 100 μg/ml for 10 min was carried out for KO oocytes to remove sperm fluorescent signals in the perivitelline space.
In vitro fertilization (IVF)
IVF was performed as described33. In brief, mature oocytes were collected and placed in 100 μl of Toyoda, Yokoyama, and Hoshi (TYH) medium. Spermatozoa were collected from the epididymides of sexually mature male mice of each genotype, and incubated in TYH medium for 2 h for capacitation. Capacitated sperm were added to the drop containing oocytes at a final concentration of 2–50 × 103 sperm/ml. After 5 h of co-incubation, oocytes were washed and transferred to KSOM medium.
Sperm motility analysis
Sperm motility was analysed as described34. Cauda epididymal spermatozoa were suspended in TYH medium35,36. Sperm motility was measured using the CEROS sperm analysis system (software version 12.3; Hamilton Thorne Biosciences, Beverly, MA, USA) at 30 min and 3 h after incubation.
Analysis of acrosome reaction
Acrosomal exocytosis was analysed as described36. Spermatozoa expressing enhanced green fluorescent protein (EGFP) in the acrosome were suspended and incubated in TYH medium. At 30 min and 3 h after incubation, an aliquot of the suspension was stained with propidium iodide (final 10 μg/ml) and subjected to fluorescence-activated cell sorting with an EC800 cell analyzer (Sony, Tokyo, Japan). The viability and acrosomal integrity of sperm were determined by propidium iodide staining and the presence of acrosomal EGFP, respectively. A 525 nm and a 595 nm band path filter were used for EGFP and propidium iodide, respectively.
Nuclear/pronuclear imaging
MII oocytes were subjected to microinjection in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid Chatot, Ziomek, Bavister (Hepes-CZB) medium with mRNA for histone H2B-mCherry (5 ng/μL) (a kind gift from Dr. Kazuo Yamagata from Kindai University, Wakayama, Japan)37, followed by incubation in fresh KSOM medium at 37 °C under 5% CO2 in humidified air for 3 h. After ICSI, the oocytes were incubated for at least 10 min to allow membrane sealing, followed by quick transfer into glass-bottomed chambers for observation of pronuclear formation and embryo development using spinning-disk confocal microscopy. The second polar body was distinguishable from the first polar body by its positive mCherry signal. The formation of pronuclei was examined both by bright field observation and by the sized/shape of the mCherry fluorescence.
Ca2+ imaging
Ca2+ imaging was performed as described17. Metaphase (M) stage II oocytes were subjected to microinjection with a mixture of mRNAs for GEM-GECO (60 ng μl) and histone H2B-mCherry (5 ng/μl) in Hepes-CZB medium, followed by incubation in fresh KSOM medium at 37 °C under 5% CO2 in humidified air for 3 h. ICSI was performed as described above within 5 min. IVF was performed as described17 within 10 min using oocytes subjected to partial zona dissection. After fertilization by either method, the oocytes were quickly transferred into glass-bottomed chambers for observation using spinning-disk confocal microscopy. Images of Ca2+ dynamics were taken at 20-s intervals for 5 h. After Ca2+ imaging, images of nuclei/pronuclei with H2B-mCherry were acquired at 15-min intervals for 24 h for evaluation of the number of pronuclei and fused sperm.
Imaging of the cortical reaction
Oocytes were observed as described17. Oocytes microinjected with R-GECO (30 ng/μl) were inseminated with capacitated spermatozoa at a concentration of 1.0 × 106/ml for 10 min. They were observed in CZB medium containing Lens culinaris agglutinin-fluorescein isothiocyanate (LCA–FITC) (5 ng/μl) (J-Oil Mills, Tokyo, Japan).
Zone pellucida protein (ZP2) cleavage assay
ZP2 cleavage assay was performed as described17. Oocytes were collected 0.5, 1, 1.5, 2, 3, and 4 h after IVF and lysed in 3× Tris-glycine sodium dodecyl sulfate (SDS) loading buffer, separated on 5–20% Tris-glycine gels by SDS–polyacrylamide gel electrophoresis (PAGE), transferred to polyvinylidene fluoride membranes (Invitrogen Life Technologies, Carlsbad, CA, USA), blocked in 7.5% nonfat milk in Tris–Bufferd Saline containing 0.05% Tween 20, and probed with primary antibodies for ZP2 (gift from Dr. Jurrien Dean of the National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, USA), followed by secondary antibodies conjugated with horseradish peroxidase38. Chemiluminescence was performed with ECL Plus (GE Healthcare, Little Chalfont, UK), and signals were acquired with the Luminescent Image Analyzer LAS-3000 (Fujifilm, Tokyo, Japan).
Plasma membrane block to polyspermy (PMBP) assay
The PMBP assay was performed using two methods. In the first method, the fusion index was counted as described previously39. The ZP of denuded MII oocytes were removed by incubation in TYH medium containing 100 μg/ml collagenase (Type I-S, Merck KGaA, Darmstadt, Germany) for 5 min. ZP-free oocytes were preloaded with 1 μg/ml Hoechst 33342 for 10 min and washed four times. After 20, 40, 60, or 80 min of insemination with WT or KO spermatozoa at a concentration of 0.5 × 105/ml, the oocytes were fixed in 0.25% glutaraldehyde and observed. Only the fused sperm heads were detected as fluorescent positive and counted. In the second method, oocytes were fertilized by conventional IVF with WT or PLCζ1-deficient spermatozoa at a concentration of 0.5 × 105/ml, as described above. After 12–15 h of incubation in KSOM medium,the ZP of fertilized oocytes were removed by treatment with acidic Tyrode’s solution40 followed by staining with Hoechst 33342 for 10 min and four washes. After 30 min of insemination using WT sperm at a concentration of 0.5 × 105/ml, the oocytes were fixed with 0.25% glutaraldehyde and observed. In this method, all the nuclei are stained and detected as fluorescent positive, thus fused and non-fused sperm nuclei were distinguished by size and shape.
Antibodies
Rabbit polyclonal antibody was produced by immunization with mouse PLCz1 polypeptide (GYRRVPLFSKSGANLEPSS)10. The monoclonal antibody against IZUMO1 (KS64–125) was obtained as described41. The anti-ZP2 antibody used in immunoblotting analysis was a gift from Dr. Jurrien Dean of the National Institute of Diabetes and Digestive and Kidney Diseases.
Data Availability
Mutant Plcz1 mice used in this study are available through Riken BioResource Center, Japan (http://en.brc.riken.jp/). The stock numbers (RBRC numbers) for B6D2-Plcz1 <em1Osb>, B6D2-Plcz1 <em3(D210R)Osb>, and B6D2;B6-Plcz1 <em4(H435P)Osb> are 10014, 10093, and 09988, respectively. All other data are available from the authors on reasonable request.
References
Cuthbertson, K. S. & Cobbold, P. H. Phorbol ester and sperm activate mouse oocytes by inducing sustained oscillations in cell Ca2+. Nature 316, 541–542 (1985).
Ducibella, T. & Fissore, R. The roles of Ca2+, downstream protein kinases, and oscillatory signaling in regulating fertilization and the activation of development. Dev Biol 315, 257–279 (2008).
Swann, K. A cytosolic sperm factor stimulates repetitive calcium increases and mimics fertilization in hamster eggs. Development 110, 1295–1302 (1990).
Stice, S. L. & Robl, J. M. Activation of mammalian oocytes by a factor obtained from rabbit sperm. Mol Reprod Dev 25, 272–280 (1990).
Wu, A. T. et al. PAWP, a sperm-specific WW domain-binding protein, promotes meiotic resumption and pronuclear development during fertilization. J Biol Chem 282, 12164–12175 (2007).
Parrington, J., Swann, K., Shevchenko, V. I., Sesay, A. K. & Lai, F. A. Calcium oscillations in mammalian eggs triggered by a soluble sperm protein. Nature 379, 364–368 (1996).
Nomikos M. et al. Functional disparity between human PAWP and PLCzeta in the generation of Ca2+ oscillations for oocyte activation. Mol Hum Reprod (2015).
Wolny, Y. M. et al. Human glucosamine-6-phosphate isomerase, a homologue of hamster oscillin, does not appear to be involved in Ca2+ release in mammalian oocytes. Mol Reprod Dev 52, 277–287 (1999).
Satouh, Y., Nozawa, K. & Ikawa, M. Sperm Postacrosomal WW Domain-Binding Protein Is Not Required for Mouse Egg Activation. Biol Reprod 93, 94 (2015).
Saunders, C. M. et al. PLC zeta: a sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development 129, 3533–3544 (2002).
Putney, J. W. & Tomita, T. Phospholipase C signaling and calcium influx. Adv Biol Regul 52, 152–164 (2012).
Kouchi, Z. et al. Recombinant phospholipase Czeta has high Ca2+ sensitivity and induces Ca2+ oscillations in mouse eggs. J Biol Chem 279, 10408–10412 (2004).
Ito, M. et al. Arrest of spermatogenesis at round spermatids in PLCZ1-deficient mice. 11th International symposium on Spermatology (abstract) (2010).
Hachem, A. et al. PLCzeta is the physiological trigger of the Ca2+ oscillations that induce embryogenesis in mammals but offspring can be conceived in its absence. Development (2017).
Weissmann, C. & Flechsig, E. PrP knock-out and PrP transgenic mice in prion research. Br Med Bull 66, 43–60 (2003).
Burkart, A. D., Xiong, B., Baibakov, B., Jimenez-Movilla, M. & Dean, J. Ovastacin, a cortical granule protease, cleaves ZP2 in the zona pellucida to prevent polyspermy. J Cell Biol 197, 37–44 (2012).
Satouh, Y., Nozawa, K., Yamagata, K., Fujimoto, T. & Ikawa, M. Viable offspring after imaging of Ca2+ oscillations and visualization of the cortical reaction in mouse eggs. Biol Reprod 96, 563–575 (2017).
McAvey, B. A., Wortzman, G. B., Williams, C. J. & Evans, J. P. Involvement of calcium signaling and the actin cytoskeleton in the membrane block to polyspermy in mouse eggs. Biol Reprod 67, 1342–1352 (2002).
Gardner, A. J. & Evans, J. P. Mammalian membrane block to polyspermy: new insights into how mammalian eggs prevent fertilisation by multiple sperm. Reprod Fertil Dev 18, 53–61 (2006).
Heytens, E. et al. Reduced amounts and abnormal forms of phospholipase C zeta (PLCzeta) in spermatozoa from infertile men. Hum Reprod 24, 2417–2428 (2009).
Yoon, S. Y. et al. Human sperm devoid of PLC, zeta 1 fail to induce Ca(2+) release and are unable to initiate the first step of embryo development. J Clin Invest 118, 3671–3681 (2008).
Nomikos, M. et al. Male infertility-linked point mutation disrupts the Ca2+ oscillation-inducing and PIP(2) hydrolysis activity of sperm PLCzeta. Biochem J 434, 211–217 (2011).
Cox, L. J. et al. Sperm phospholipase Czeta from humans and cynomolgus monkeys triggers Ca2+ oscillations, activation and development of mouse oocytes. Reproduction 124, 611–623 (2002).
Ducibella, T. et al. Egg-to-embryo transition is driven by differential responses to Ca(2+) oscillation number. Dev Biol 250, 280–291 (2002).
Ducibella, T., Schultz, R. M. & Ozil, J. P. Role of calcium signals in early development. Semin Cell Dev Biol 17, 324–332 (2006).
Miyazaki, S. Thirty years of calcium signals at fertilization. Semin Cell Dev Biol 17, 233–243 (2006).
Takayama, J. & Onami, S. The Sperm TRP-3 Channel Mediates the Onset of a Ca(2+) Wave in the Fertilized C. elegans Oocyte. Cell Rep 15, 625–637 (2016).
Sato, K., Tokmakov, A. A., Iwasaki, T. & Fukami, Y. Tyrosine kinase-dependent activation of phospholipase Cgamma is required for calcium transient in Xenopus egg fertilization. Dev Biol 224, 453–469 (2000).
Johnson, L. N., Sasson, I. E., Sammel, M. D. & Dokras, A. Does intracytoplasmic sperm injection improve the fertilization rate and decrease the total fertilization failure rate in couples with well-defined unexplained infertility? A systematic review and meta-analysis. Fertil Steril 100, 704–711 (2013).
Vanden Meerschaut, F. et al. Diagnostic and prognostic value of calcium oscillatory pattern analysis for patients with ICSI fertilization failure. Hum Reprod 28, 87–98 (2013).
Oji, A. et al. CRISPR/Cas9 mediated genome editing in ES cells and its application for chimeric analysis in mice. Sci Rep 6, 31666 (2016).
Kimura, Y. & Yanagimachi, R. Mouse oocytes injected with testicular spermatozoa or round spermatids can develop into normal offspring. Development 121, 2397–2405 (1995).
Tokuhiro, K., Ikawa, M., Benham, A. M. & Okabe, M. Protein disulfide isomerase homolog PDILT is required for quality control of sperm membrane protein ADAM3 and male fertility [corrected]. Proc Natl Acad Sci USA 109, 3850–3855 (2012).
Miyata, H. et al. Sperm calcineurin inhibition prevents mouse fertility with implications for male contraceptive. Science 350, 442–445 (2015).
Toyoda, Y., Yokoyama, M. & Hoshi, T. Studies on the fertilization of mouse egg in vitro. Jpn J Anim Reprod 16, 147–151 (1971).
Nakanishi, T. et al. Real-time observation of acrosomal dispersal from mouse sperm using GFP as a marker protein. FEBS Lett 449, 277–283 (1999).
Yamagata, K. & Suetsugu, R. Wakayama T. Long-term, six-dimensional live-cell imaging for the mouse preimplantation embryo that does not affect full-term development. J Reprod Dev 55, 343–350 (2009).
Gahlay, G., Gauthier, L., Baibakov, B., Epifano, O. & Dean, J. Gamete recognition in mice depends on the cleavage status of an egg’s zona pellucida protein. Science 329, 216–219 (2010).
Inoue, N., Ikawa, M., Isotani, A. & Okabe, M. The immunoglobulin superfamily protein Izumo is required for sperm to fuse with eggs. Nature 434, 234–238 (2005).
Maleszewski, M., Kimura, Y. & Yanagimachi, R. Sperm membrane incorporation into oolemma contributes to the oolemma block to sperm penetration: evidence based on intracytoplasmic sperm injection experiments in the mouse. Mol Reprod Dev 44, 256–259 (1996).
Ikawa, M. et al. Calsperin is a testis-specific chaperone required for sperm fertility. J Biol Chem 286, 5639–5646 (2011).
Acknowledgements
We thank NPO for Biotechnology Research and Development for technical assistance in generating mutant mice; Drs J. Castañeda and G. Pao for critical reading of this manuscript. This work was supported by a KAKENHI JP15J03870 (to KN), JP17K15126 (to YS), JP17H01394, JP 25112007, Takeda Science Foundation, NIH grant P01HD087157, and R01HD088412 (to MI).
Author information
Authors and Affiliations
Contributions
Y.S., K.N., and M.I. designed the study; all authors performed research; all authors analysed data; and Y.S., K.N., and M.I. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nozawa, K., Satouh, Y., Fujimoto, T. et al. Sperm-borne phospholipase C zeta-1 ensures monospermic fertilization in mice. Sci Rep 8, 1315 (2018). https://doi.org/10.1038/s41598-018-19497-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19497-6
This article is cited by
-
Mutations in PLCZ1 induce male infertility associated with polyspermy and fertilization failure
Journal of Assisted Reproduction and Genetics (2023)
-
Advances in the study of genetic factors and clinical interventions for fertilization failure
Journal of Assisted Reproduction and Genetics (2023)
-
A loss-of-function variant in SSFA2 causes male infertility with globozoospermia and failed oocyte activation
Reproductive Biology and Endocrinology (2022)
-
Sperm-oocyte interplay: an overview of spermatozoon’s role in oocyte activation and current perspectives in diagnosis and fertility treatment
Cell & Bioscience (2021)
-
Calcium Oscillatory Patterns and Oocyte Activation During Fertilization: a Possible Mechanism for Total Fertilization Failure (TFF) in Human In Vitro Fertilization?
Reproductive Sciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
