
Abstract
Fungicides are applied intensively to prevent downy mildew infections of grapevines (Vitis vinifera) with high impact on the environment. In order to develop alternative strategies we sequenced the genome of the oomycete pathogen Plasmopara viticola causing this disease. We show that it derives from a Phytophthora-like ancestor that switched to obligate biotrophy by losing genes involved in nitrogen metabolism and γ-Aminobutyric acid catabolism. By combining multiple omics approaches we characterized the pathosystem and identified a RxLR effector that trigger an immune response in the wild species V. riparia. This effector is an ideal marker to screen novel grape resistant varieties. Our study reveals an unprecedented bidirectional noncoding RNA-based mechanism that, in one direction might be fundamental for P. viticola to proficiently infect its host, and in the other might reduce the effects of the infection on the plant.
Similar content being viewed by others


Introduction
Grapevine (Vitis vinifera L.) is an important commodity and comprises varieties for wine production and table grape for human consumption1. Wine production is a very lucrative activity and the world wine trade is worth almost US $30 billion. France, Italy and Spain are the largest European wine producing countries representing altogether half of the world production (http://www.oiv.int/). Grapevines can be infected by a myriad of plant pathogens at all growth stages and in order to secure harvest large quantities of agrochemicals are used to control their spread2. The treatments against powdery and downy mildews, including the oomycete Plasmopara viticola, requires almost two thirds of all synthetic fungicides sprayed in the European Union with adverse effects on the environment3.
Plasmopara viticola (Berk. and Curt.) Berl. and de Toni belongs to the group of oomycetes (Order: Peronosporales, Family: Peronosporaceae) that comprises the most devastating plant pathogens such as Phytophthora infestans responsible for the Irish potato famine in the 19th century4. Unlike the hemibiotroph Phytophthora species, P. viticola is an obligate biotroph and therefore relies entirely on grape as a host to complete its life cycle. P. viticola is endemic in North America where the Vitis species such as V. riparia are naturally resistant, likely as a consequence of a long co-evolutionary process5. Conversely, P. viticola was introduced unintentionally in Europe in the 1870s and immediately spread on V. vinifera cultures causing pandemics all over Europe in the following decades6. Soon after, the extensive use of copper formulations known as the “Bordeaux mixture” restricted the disease spread and later on paved the path to excessive usage of synthetic agrochemicals. As a consequence P. viticola became recently resistant to many fungicides7. Therefore, research on grape resistance mechanisms and the development of alternative strategies to control P. viticola infections are urgently needed to control the environmental burden of grapevine cultures.
Molecular mechanisms occurring during the compatible interaction between P. viticola and V. vinifera are largely unknown. Most of our knowledge about oomycete pathogenicity factors derives from studies focused on Phytophthora species8. During infection oomycetes secrete cytoplasmic and apoplastic effector proteins that usually suppress plant immunity by triggering susceptibility (effector-triggered susceptibility, ETS)9. Certain oomycete effectors with a special protein motif RxLR are recognized by the plant resistance genes and this interaction triggers immunity (effector-triggered immunity, ETI) resulting in localized cell death or hypersensitive response (HR)10. RxLR effectors are found abundantly in all sequenced oomycete genomes and are rapidly evolving11. They even acquired novel functions such as suppressing plant RNA silencing mechanisms12. Their role during the grape downy mildew infection is unclear and their identification requires multi-omics approaches. To this end, we sequenced the DNA extracted from a P. viticola strain isolated in Northern Italy and present its assembled draft genome. By using comparative genomics we discovered the missing metabolic feature in the P. viticola genome that could explain its biotrophic mode of life. We complemented our genome sequencing efforts with genome-wide differential gene expression analyses during the infection process and we identified a protein effector of the RxLR type triggering ETI in the resistant grapevine V. riparia. Small RNA sequencing (sRNA-Seq) combined to a genome-wide degradome study13 revealed a comprehensive interaction network of small RNAs (sRNAs) that target genes during infection allowing us to uncover a potential bi-directional RNA silencing strategy between the pathogen and its host despite species barriers.
Results
Nuclear and mitochondrial genome assemblies, gene annotation and phylogenetic analysis
Assembling the draft genome for P. viticola was a prerequisite for gene and sRNA expression profiling studies during the infection process of V. vinifera. We therefore sequenced the genome of a downy mildew strain, named ‘PvitFEM01’, isolated from a local vineyard in the Trentino region in Italy. DNA extracted from asexual sporangia from in vitro infected plants was used to construct Illumina paired-end libraries (2 × 100 bp) and further sequenced. Although P. viticola genetic material was extracted under sterile conditions, potential contamination with DNA originating from other sources such as the host (grapevine), potential grape endophytic or epiphytic bacteria were addressed. 83 million reads remained for assembly, after filtering against V. vinifera and bacterial sequences (Supplementary Fig. S2 and Supplementary note). In total, 57,890 scaffolds were obtained corresponding to a N50 of 4,645 bp (Table 1, Supplementary Figs S1–S6 and Supplementary note). The total length of the assembly was 83.54 Mb corresponding to 74% of the genome size previously determined by Feulgen staining14. Contigs corresponding to the mitochondrial genome were also identified and provided the first P. viticola mitochondrial reconstruction estimated to be ca. 39.2 kb (Supplementary Fig. S7). P. viticola mitochondrial proteins identity ranged from 80 to 97% with respect to their P. infestans counterparts. A a single amino acid change from Glycine to Alanine at position 143 (G143A) in the mitochondrial apocytochrome b protein suggests that ‘PvitFEM01’ is resistant to Quinone outside Inhibitors (QoI) fungicides (Supplementary Fig. S8). Several polymorphic sites were identified on the P. viticola mitochondrial genome suggesting that our draft assembly might condense several haplotypes (Supplementary Fig. S9).
In total, 38,298 genes were predicted in silico of which 33,982 were annotated using gene ontology terms (Supplementary Table S1). Sequencing of RNA transcripts (RNA-Seq) extracted from P. viticola sporangia and infected leaf material collected at five time points confirmed that 18,335 of these annotated genes were expressed (Table 1, Supplementary Tables S2 and S3, Supplementary Fig. S10 and Supplementary note). Additionally, 320 tRNA-encoding genes encoding representing all 20 isotype classes, as well as the 28 S and 5 S ribosomal genes (rRNAs) were identified in the P. viticola genome (Supplementary Tables S4 to S6 and Supplementary note). The 28 S rRNA is most similar to the one reported for P. viticola f. sp. vinifera5 indicating that ‘PvitFEM01’ isolate belongs to lineage C which is virulent on V. vinifera and some grape hybrids, but not on V. riparia (Supplementary Table S5). To assess the degree of completeness of our draft genome, we defined the oomycete core genome consisting of 1,299 genes of which 81% are also present in P. viticola (Table 1, Supplementary Tables S7 and S8, Supplementary Figs S11–S13 and Supplementary note). This number was corroborated by a BUSCO15 analysis that retrieved a completeness of 87.2% when 303 conserved eukaryotic proteins were considered (Supplementary Table S9 and Supplementary note). The phylogenetic relationship between P. viticola and sequenced oomycetes was established using 312 single-copy core genes. P. viticola is placed, together with Plasmopara halstedii, within the Phytophthora clade with 100% support (Fig. 1, Supplementary Figs S14 and S15 and Supplementary note). Branches in the Plasmopara lineage are longer than the average branch lengths of Phytophthora species. The phylogenetic analysis suggests that P. viticola derives from a necrotrophic Phytophthora-like ancestor.

Phylogenetic relationship between Plasmopara viticola and other oomycetes and abundance of their effectors. The maximum-likelihood phylogenetic tree was built using 312 concatenated proteins selected from single copy genes belonging to the oomycete core genome. The abundance of each class of cytoplasmic and apoplastic effectors effector in biotroph (B), hemibiotroph (H) or necrotroph (N) oomycete species is indicated by a number and a color code. Out of the 87 YxSLK effectors identified in our study only 25 contained a signal peptide and reported in this figure. Darker colors indicate higher abundance.
P. viticola secretes a RxLR effector triggering an immune response in Vitis riparia but not in Vitis vinifera
Oomycetes secrete various types of cytoplasmic effectors to infect their hosts. These effectors were mainly studied in Phytophthora species. We identified that P. viticola encodes 57 RxLR and 68 crinkler (CRN) proteins. In comparison, other oomycetes belonging to the Pythium genus have only five to twelve CRN proteins, suggesting an expansion of this effector type in the ancestor that gave rise to the Hyaloperonospora, Phytophthora and Plasmopara lineages (Fig. 1, Supplementary Tables S10–S14, Supplementary Figs S16–S21 and Supplementary note). The number of genes encoding the recently discovered YxSLK effectors16 is high in P. viticola and P. sojae with, respectively 87 (25 with a signal peptide) and 125 genes, suggesting that they have a special function in these two pathogens (Fig. 1, Supplementary Table S15, Supplementary Fig. S22 and Supplementary note). Most apoplastic effectors previously identified in oomycete genomes are encoded by the P. viticola genome with the largest family being the endo-β-1,3-glucanases inhibitors with a trypsin domain. This family includes 55 members and is the largest among all available oomycete genomes. In contrast to Phytophthora and Pythium species, P. viticola has only 22 elicitins. NPP1 proteins inducing necrosis likely underwent expansion in the Phytophthora lineage whereas only six and 19 members were found in P. viticola and P. halstedii, respectively, which may suggest extensive gene loss as an adaptation to biotrophy. CBEL (cellulose binding elicitor lectin) and PcF (Phytophthora cactorum-Fragaria toxin family) from P. infestans are known to trigger programmed cell death in its host17,18. Their gene families are either strongly reduced or completely absent in Plasmopara species (Fig. 1, Supplementary Table S16 and Supplementary note). In conclusion, except the glucanase inhibitor family (trypsin), all known apoplastic effectors underwent contraction in the Plasmopara lineage from a Phytophthora-like ancestor.
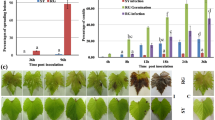
Transcriptional profiling (RNA-Seq) of P. viticola genes during the compatible interaction with V. vinifera revealed differential expression of several annexins, glutathione S-transferases and one glutamic acid decarboxylase (GAD) involved in the production of the non-proteinogenic amino acid γ-Aminobutyric acid (GABA). Furthermore, proteins involved in the hydrolysis of plant material were the first set of apoplastic effectors expressed upon infection (Supplementary Tables S17 and S18). Elicitins, RxLR and YxSLK effectors were among the most highly expressed secreted proteins at later time points (Fig. 2, Supplementary Tables S19–S23, Supplementary Figs S23–S25 and Supplementary note). While the majority of RxLR, Crinkler and YxSLK effectors showed fluctuating levels during infection, the RxLR gene PVITv1008311 was expressed with FPKM values increasing to 9.8 and 17.8 at 96 and 168 hours post-infection (hpi), respectively (Supplementary Table S23). The expression of PVITv1008311 was also measured by qRT-PCR during infection and in sporangia corroborating the RNA-Seq data obtained in our study (Fig. 3a and b). To verify the role of RxLR_PVITv1008311 in pathogenicity, we expressed the coding sequence constructs both with (+sp) or without (Δsp) signal peptide in planta by infiltrating sterile grape leaves grown in vitro. There were no noticeable symptoms visible on V. vinifera leaves even two weeks after infiltration despite strong expression of the effector in planta (Fig. 3c). In contrast, when the same vector constructs were infiltrated in the resistant grape V. riparia a strong necrotic phenotype was observed around the site of infiltration only in leaves infiltrated with RxLR_PVITv1008311Δsp but not with RxLR_PVITv1008311 +sp (Fig. 3d). Trypan blue staining revealed that this halo corresponds to cells that underwent cell death typical of a hypersensitive response (Fig. 3e). Several additional elicitors with or without the signal peptide were tested by infiltrating V. vinifera and V. riparia leaves but the response remained asymptomatic (Supplementary Table S24). Interestingly, the sequence of RxLR_PVITv1008311 was found intact in the European isolate INRA-PV221 but completely fragmented in the Chinese isolate JL-7–2 originally isolated from V riparia (Supplementary Table S25). Collectively, the fact that RxLR_PVITv1008311Δsp elicits a hypersensitive response in V. riparia but not in V. vinifera indicates that grapevine cultivars grown in Europe lost or perhaps did not acquire yet the recognition of P. viticola effectors to initiate a proper immune response.

Distribution of genome-wide expression levels of Plasmopara viticola genes and effectors during the infection time course. Histograms represent the distribution of log10 FPKM values for all P. viticola genes at different hours post-infection (hpi). The values on the y-axis are counts. The gray dots represent all genes classified as cytoplasmic or apoplastic effectors. The colored dots indicate the different classes of effector proteins also possessing a signal peptide for secretion.

Agrobacterium-mediated infiltration assays of RxLR_PVITv1008311 in Vitis vinifera and V. riparia. The relative RxLR PVITv1008311 expression levels were measured during the infection of V. vinifera (a) at different hours post-infection (hpi), in sporangia (b) and one week after infiltration with the RxLR effector PVITv1008311 or the empty vector in V. vinifera (c) and V. riparia (d). The error bars of the relative abundance of the transcript normalized to the P. viticola elongation factor eIF1b in each panel represent the standard deviation of three independent plants or experiments. The necrosis visible on V. riparia leaf in panel (d) is due to dead cells stained in dark blue after trypan blue staining (e). The scale bar represents 5 mm in panels c, d, e and 20 μm in the microscopic pictures in (e).
Nitrogen metabolism and γ-Aminobutyric acid (GABA) catabolism are missing in P. viticola
To better understand P. viticola biotrophic mode of nutrition we identified the metabolic modules that are either completely missing from its genome or differ significantly from the biotroph H. arabidopsidis and the hemibiotroph P. infestans. Similar to the other obligate biotroph H. arabidopsidis19, P. viticola lacks both nitrite and sulfite reductases (Supplementary Figs S26 and S27). However, unlike other obligate biotrophs, P. viticola lacks several enzymes involved in the conversion of L-glutamate to either succinate (KEGG module M00027) or to L-ornithine (M00028). The pathways leading to conversion of leucine to acetoacetate and Acetyl-CoA (M00036), of L-glutamine to uridylic acid (M00051), and of pyruvate to acetyl-CoA (M00307) are also incomplete in P. viticola (Supplementary Fig. S28 and Supplementary Table S26). However, in contrast to the two other oomycetes, the P. viticola genome encodes all genes necessary to convert L-glutamate to L-proline (M00015) and to degrade L-methionine to L-cystathionine (M00035) (Supplementary Fig. S29). Furthermore, the two biotrophs H. arabidopsidis and P. viticola seem to have lost some enzymes required for the biosynthesis of glycosylphosphatidylinositol (GPI)-anchor (M00065), Coenzyme A (M00120), and betaine (M00555). In contrast, the pathways leading to biosynthesis of creatine (M00047), sphingosine (M00099) and N-glycan precursors (M00055, M00073) are conserved in P. viticola and P. infestans but are lost in H. arabidopsidis (Fig. 4a and Supplementary Table S26). The nitrogen metabolism, sulphur assimilation, GABA shunt, ornithine biosynthesis and uridine monophosphate biosynthesis pathways seem also incomplete in the two other P. viticola isolates INRA-PV221 and JL-7–2 (Supplementary Table S27) suggesting that glutamate metabolism and its connection with citric acid (tricarboxylic acid; TCA) and urea cycles, as well as uridylic acid biosynthesis and GABA catabolism, might be impaired in P. viticola (Fig. 4b). Given the role of glutamate in amino acid metabolism and nitrogen utilization, P. viticola infection could have an important impact on grapevine metabolism during the compatible infection. To verify this hypothesis, we performed a differential gene expression analysis using RNA-Seq at multiple time points during infection of grapevine by ‘PvitFEM01’ and characterized the gene sets by functional enrichment analysis using GO annotations (Supplementary Table S28). Interestingly, grapevine genes involved in secondary metabolic processes, cellular amino acid metabolism and derivative metabolic processes were significantly repressed in infected tissues starting at 48 hpi. The genes involved in nitrogen compound metabolic processes and homeostasis start to be expressed later during the infection process at 168 hpi which might suggest that downy mildew stimulates the production of growth nutrients from its host while avoiding triggering cell death (Fig. 4c).

Metabolic pathways missing in Plasmopara viticola and those induced in grapevine during infection. The Venn diagram shows the metabolic pathways specific to P. viticola or shared with two other oomycetes, Phytophthora infestans and Hyaloperonospora arabidopsidis. The KEGG module number M is indicated in brackets (a). A summary of the pathways missing in P. viticola indicated in red. The proline biosynthesis pathway indicated in green is found only in P. viticola but not in the two other oomycetes P. infestans and H. arabidopsidis. (AT: Amino transferase, GAD: glutamic acid decarboxylase, GDH: glutamate dehydrogenase, GOGAT: glutamine oxoglutarate aminotransferase, GS: glutamine synthetase, TCA cycle: tricarboxylic acid cycle) (b). A Venn diagram representing the gene ontology terms of V. vinifera genes enriched at each time point during infection in grapevine. The metabolic pathways indicated in red indicate genes induced whereas those in green refers to genes repressed (c).
A bidirectional cross-species sRNA-mediated gene regulation during the compatible interaction
Plants defense strategies against viral and fungal pathogens rely largely on RNA silencing and the action of sRNAs20. To explore this mechanism of defense during P. viticola-V. vinifera interaction, we sequenced sRNAs from both healthy and infected grapevine plants at 24, 48, 72, 96 and 168 hpi. The sRNA profile of V. vinifera showed enrichment in 21- and 24-nt sRNAs typical of plants, while P. viticola has an almost equal abundance of 21- and 25-nt sRNA classes that are also abundantly expressed in sporangia (Supplementary Fig. S30). In total, two dicer-like proteins (DCLs), two argonaute proteins (AGOs) and one RNA-dependent RNA polymerase (RDR), as well as enzymes known to regulate epigenetic mechanisms were identified confirming the existence a bona fide RNA silencing machinery in P. viticola that is active during its life cycle inside the grapevine host (Supplementary Table S29). Among these proteins two DCL-like enzymes, defined by the presence of a Dicer dimerization domain were found, suggesting that one of them could be dedicated to process the 25-nt sRNA class. P. viticola sRNAs of 21- to 22-nt length were generated from 592 transcripts coding mainly for transporters, transcription factors, methyltransferases, metabolic genes and elicitins. In contrast, the 25- to 26-nt sRNA class derives almost exclusively from genes related to transposition (Supplementary Fig. S31). The 21- to 22-nt sRNAs deriving form coding genes were mapping in sense and antisense orientation suggesting that a dsRNA intermediate is synthesized, most probably, by the unique RDR found in the P. viticola genome (Supplementary Fig. S32). Additionally, a total of 18 CRN (Supplementary Fig. S33), YxSLK (Supplementary Fig. S34) and RxLR (Supplementary Fig. S35a) effector genes produced a high amount of 21/22-nt short interfering RNA (siRNAs) duplexes suggesting a preponderant role in post-transcriptional regulation of these pathogenicity factor during the infection process (Supplementary Table S30). The highly structured PVITv1_T024389 RNA transcript encoded a protein with an unusual LFLAK/RxLR tandem motif and displayed a unique processing pattern among eukaryotes with 21/22-nt siRNA duplexes processed every 60- to 90-nt (Supplementary Figs S35b and S36).
In order to address the potential regulatory role of both grapevine and P. viticola sRNAs during infection, we performed a genome-wide analysis of the RNA degradome or PARE (parallel analysis of RNA ends) of infected material and control plants. In order to support and validate our sRNA target prediction we used SeqTar with stringent parameters and retained only the highly probable sRNA-mRNA interactions based on a mismatch and binding score p-value ≤ 0.001 and a valid peak height of p-value ≤ 10−10. We confirmed that grapevine endogenous microRNAs (miRNAs) regulate genes important for plant growth and development in both infected and control plants (Supplementary Table S31). We also characterized the degradome of P. viticola and identified genes targeted by its endogenous sRNAs such as kinases and a vesicle-associated membrane protein VAC14 only in the degradome-Seq dataset generated from infected plants (Supplementary Table S32). Interestingly, we have identified a potential bidirectional interaction between, on one hand; the sRNAs produced by P. viticola triggering cleavage of grapevine genes and on the other hand, the sRNAs processed from grapevine transcripts and targeting the oomycete messenger RNAs (Fig. 5, Supplementary Tables S33 and S34). Small RNA duplexes processed from grape resistance genes in 21-nt increment and known as phased secondary siRNAs (phasiRNAs), were the most abundant class of grapevine sRNAs and targeted for cleavage P. viticola genes with diverse functions (Fig. 5 and Supplementary Table S34). Other sRNAs processed from grape noncoding RNAs such as the miRNA primary transcripts pri-MIR169, pri-MIR171a, pri-MIR394c, pri-MIR482-like and pri-MIR396a as well as the trans-acting siRNA precursor TAS3 also trigger cleavage of various P. viticola transcripts. Reciprocally, P. viticola sRNAs, including those deriving from the CRN gene PVITv1_T024389, target V. vinifera genes for cleavage at multiple sites (Fig. 5 and Supplementary Table S33). Our results suggest that similarly to mechanisms described for protein-coding gene effectors, noncoding small RNAs potentitally mediate interference between the pathogen and its host in a bi-directional manner in a way not previously known.

Bidirectional cross-species sRNA-mediated gene regulation during the compatible interaction. The hive plot indicates the interactions between P. viticola sRNAs originating from either intergenic (yellow dots) or protein coding genes (gray dots) and V. vinifera genes (blue dots). Reciprocally, V. vinifera sRNAs processed from either noncoding RNA (green dots), intergenic regions (purple dots) or resistance genes (red dots) target P. viticola transcripts (yellow dots). The thickness and color intensity of the yellow and blue lines representing the sRNA-target interactions are proportional to the log transformed p-value calculated for the number of reads from the degradome whose 5′ end corresponds (±1) to the expected sRNA-mediated cleavage site: the larger the edge, the more significant the interaction. The color code is different for regulation starting (from light to dark blue) or arriving at P. viticola (from yellow to red). The size of the dots corresponds to the number of regulations identified for a certain sRNA.
Discussion
The primary goal of this study was the identification of P. viticola pathogenicity factors involved in the infection process of grapevine. To enable the use of transcriptomic approaches we first sequenced the DNA isolated from infected plants and assembled the P. viticola genome. The genome assembly of the Italian P. viticola ‘PvitFEM01’ reached 83.54 Mb, a genome size between the Chinese isolate ‘JL-7–2’ and the French one INRA-PV221 with 101.3 Mb and 74.74 Mb, respectively21,22. The genome of the Chinese isolate reached a higher size of contigs due to the assembly of long reads obtained using the PacBio single-molecule sequencing technology whereas this work relied solely on paired-end Illumina sequencing technology. Nevertheless, the genome obtained in our study reached a completeness that was sufficient enough to identify and annotate 18,335 genes with homology to oomycete proteins for which expression was experimentally verified by RNA-Seq data.
We did not find a massive gene loss that could explain obligate biotrophy in P. viticola as reported for H. arabidopsidis19. On the contrary, our data suggest that P. viticola contain more RxLR, CRN and YxSLK effector genes compared to other biotrophs. The phylogenetic analysis showed that the two downy mildew species P. viticola and P. halstedii share the same clade and are evolutionary close to the Phytophtora species23. In contrast, H. arabidopsidis was placed in a sister group therefore confirming recent findings that downy mildews are not monophyletic24. Our work contribute to the revision of downy mildews phylogenesis and confirms that Plasmopara species derived from a Phytophthora-like ancestor that switched to obligate biotrophy, lost its necrotrophic abilities and became specialized for a defined host species. We provide additional genome sequences and gene annotation that will enable a thorough comparative genomic approach and will help identifying the molecular mechanisms that dictate obligate biotrophy.
The sequenced P. viticola isolate ‘PvitFEM01’ is virulent on V. vinifera but not on the wild North American species V. riparia. Our results also show that this isolate is resistant to Quinone outside Inhibitors (QoI) fungicides since a single amino acid change from Glycine to Alanine at position 143 (G143A) was found in the mitochondrial apocytochrome b protein similarly to other European downy mildew isolates highly resistant to QoI7. Sequencing of the 28 S rRNA showed that P. viticola ‘PvitFEM01’ belongs to the cryptic lineage C and therefore evolved on V. vinifera after introduction of the pathogen in Europe5. Taking this into account, we expected that P. viticola would express effector genes triggering no defense response in V. vinifera. Agrobacterium-mediated infiltration of effectors in V. vinifera leaves resulted in no visible phenotype confirming that the isolate ‘PvitFEM01’ evolved a stealth infection strategy similarly to other obligate biotrophs19. Remarkably, expression of RxLR_PVITv1008311 in leaves of the resistant cultivar V. riparia triggered a hypersensitive response, indicating that this RxLR effector is one of the key evolutionary players in the perpetual arm race between P. viticola and its grapevine host. To successfully infect V. vinifera, P. viticola encodes RxLR effectors with different properties according to the Vitis species with which they have co-evolved. RxLR effectors can suppress plant immunity when P. viticola infects wild Vitis species such as V. amurensis25 or, as in the case of RxLR_PVITv1008311, they have most likely lost the potential to trigger cell death when infecting the domesticated V. vinifera. However, the effector is still recognized by the immune system of V. riparia most probably for the presence in the host genome of the resistance gene recognizing RxLR_PVITv1008311. Our finding opens therefore a new route in grapevine breeding programs since RxLR_PVITv1008311 can be used efficiently in effector-based high-throughput in planta expression assays26. This will help to accelerate the identification of new V. vinifera hybrids or varieties resistant to P. viticola hence reducing the use and the release in the environment of toxic fungicides and chemicals.
Several studies attempted to explain obligate biotrophy by a loss of certain metabolic pathways27. Similarly to other obligate biotrophs P. viticola lost the nitrate and nitrite reductase enzymes suggesting a total dependence on the grape host for acquiring nitrogen in its reduced form19,23. Additionally, our study reveals that not only the Italian P. viticola PvitFEM01 but also the French isolate INRA-PV22121 and the Chinese one JL-7-222,28 lost the genes encoding many enzymes necessary for the conversion of glutamine to uridylic acid and of glutamate to succinate and ornithine. The two latter molecules bridge glutamate metabolism to the TCA and urea cycles. The reconstruction of metabolic pathways by gene annotation provides the evidence for a functional glutamate metabolism in P. viticola, however, relying on the use of ammonia from the host grapevine. This seems a general feature of obligate biotroph pathogens. However, our study also reveals that enzymes involved in amino acid metabolism from glutamate are conserved in P. viticola suggesting that this biotroph does not rely on his host for synthesizing this fundamental amino acid. On the contrary, the GABA shunt pathway was impaired suggesting that succinate is most likely synthesized through the TCA/glyoxylate shunt, but not from GABA in P. viticola. This non-proteinogenic amino acid is synthesized from glutamate by the action of the glutamic acid decarboxylase (GAD) enzyme that is strongly expressed in P. viticola during the course of infection. Taken together our data suggest that enzymatic reactions leading to the production of GABA are strongly activated in P. viticola during infection, however, those associated with GABA catabolism are impaired. This implies an important increase of GABA levels during infection by P. viticola that will not be degraded. Interestingly, GABA reduces H2O2 levels by up-regulating the expression of the catalase VvCAT2 in grapevine29. Whether P. viticola evolved a strategy to increase GABA levels during infection in order to suppress oxidative stress in its host remains to be verified.
Besides the potential role of P. viticola protein-coding genes in the regulation of the infection process, our study unveiled a potential bidirectional gene regulation mediated by noncoding RNAs between P. viticola and its host. Necrotrophic fungi evolved a sRNA-mediated silencing of their host genes to rapidly suppress immunity and successfully infect and destroy the plant tissues30. This post-transcriptional regulation mechanism occurs most probably by a direct uptake of the sRNAs into the cells in the vicinity of the infection site31. In contrast, biotrophic oomycetes must keep their host cells alive until sporulation occurs and exchange of biological material is intensive during the compatible interaction. Taking into account the computational output obtained using SeqTar with stringent parameters, the large number of sRNA-mediated cleavages occurring only in infected tissue but not in control plants are highly probable. This mechanism would implicate an important shuffling of low molecular weight RNA between P. Viticola and its host. This bidirectional exchange could occur either via the haustorium or through simple diffusion between cells in contact. However, additional experimental work is needed to confirm and verify if the pairing of the sRNAs to their cognate target genes occurs randomly in the cytoplasm or if it is evolving towards a type of gene regulation involving specialized protein complexes. It is not excluded that this sRNA-mediated gene regulation is still evolving given that the P. viticola-V. vinifera pathosystem appeared only about 135 years ago32. The winner of this evolving arms race between P. viticola and V. vinifera is difficult to predict.
In conclusion, our work provides new insights on the molecular mechanisms governing pathogenicity of grapevine downy mildew and lays the foundation for future work aiming to develop alternatives to the heavy use of chemical treatments. Based on the results of our work, we propose the development of RNAi-based techniques such as host induced gene silencing (HIGS)33 or spray-induced gene silencing (SIGS)34 to knockdown P. viticola pathogenicity genes as an environmental-friendly alternative of crop protection.
Methods
Methods and any associated references are available in the online version of the paper.
Accession codes
The raw data corresponding to the genome, RNA-Seq, sRNA-Seq and degradome-Seq sequences used in this study has been deposited at GenBank under the project accession PRJNA380033. The mitochondrial genome described in this study has been deposited to GenBank under the accession number KY885002.
Online Methods
Plant material, growth conditions, inoculation and infiltration
V. Vinifera susceptible cultivars Pinot Noir cv. ENTAV115, the near-homozygous Pinot Noir 40024 and Sultanina as well as the resistant V. riparia used in this study were cultivated in vitro in glass tubes on half-strength Murashige Skoog (MS) medium containing 0.6 mg/l thiamine, 100 mg/l myo-inositol, 30 g/l sucrose and 6 g/l agar. The plants were grown at 24 °C under 16 h of light/8 h of dark with an illumination of 70 μmol m−2s−1 light. The isolation from the field of P. viticola “PvitFEM01”, the inoculum preparation and the sterile infection of in vitro grapevine plants were performed as described in Lenzi et al.35. The RxLR and CRN effectors were cloned using the primers described in supplementary Table S35. The Agrobacterium infiltration assays and the gene expression studies by qRT-PCR were performed as described in the supplementary note.
Genome sequencing and gene annotation
P. viticola is a biotroph and therefore cannot be cultivated and grown on synthetic culture medium. The starting DNA for library preparation was isolated from a mix of sporangia, sporangiophores and mycelia emerging from infected grapevine cv. Pinot Noir ENTAV115 grown in vitro. Plasmopara viticola genomic DNA was extracted using the method described in Si-Ammour et al.36. The MicroPlex Library Preparation Kit (Diagenode, www.diagenode.com) was used to build the Illumina library using 60 ng of P. viticola genomic DNA and following the manufacturer’s recommendations. The library was sequenced using a HiSeq. 2500 Illumina platform (Illumina, www.illumina.com) at Fasteris (www.fasteris.ch). DNA fragments were sequenced from both ends to generate 2 × 100 bp paired-end reads. All reads mapping on the grapevine genome cv. PN4002437 and contaminating bacterial sequences were filtered and eliminated to produce a preliminary assembly using Abyss38. Several k-mer lengths were tested and the best N50 (11kbp) value was obtained for a k-mer of 60 nucleotides. To remove sequences not belonging to the P. viticola genome, we followed the flowchart indicated in Supplementary Fig. S2 and explained in details in the supplementary note. The final assembly was then selected among the outputs of Ray39 and Abyss38 ran with different k-mer lengths as described in details in the supplementary note. The P. viticola mitochondrial scaffolds/genes from the Ray assembly were identified on the basis of similarities with P. infestans mitochondrial sequences40. All expected mitochondrial ORFs were found in scaffolds that were subsequently manually assembled. Gene finding and gene training was performed using Augustus41, GlimmerHMM42 and GeneID43. Gene predictions were supported by RNA-Seq data and the genes named as described in the supplementary note. Transfer RNA genes were identified using tRNA-scanSE44 and ribosomal genes annotated using RNAmmer45. The degree of completeness of the assembly was estimated by comparing our final assembly with available data such as the genome size determined by Feulgen staining14, the genome size of P. halstedii, sequences of P. viticola available in different databases, a BUSCO15 analysis and our comparative genomics study, as described in the supplementary note.
Comparative genomics and phylogenetic analyses
The ortholog groups from the 15 oomycete species including P. viticola were used to identify the oomycete core genome. Pairs of genomes were compared using Inparanoid46 and the outputs were integrated using QuickParanoid (http://pl.postech.ac.kr/QuickParanoid/). Phylogenetic analyses of the oomycete dataset were performed using a concatenation of 312 core ortholog proteins containing a single copy per genome and aligned using MAFFT47. The alignment was further filtered with Gblocks48. Phylogenetic trees were built using phyml49 and raxml50. The RxLR, RxLR-like, CRN and YxSLK effectors were identified as described in the supplementary note. Apoplastic effectors were identified by scanning protein sequences with the corresponding HMM models from Pfam51 (http://pfam.xfam.org/).
RNA-Seq, sRNA-Seq and degradome-Seq
Infections of V. vinifera cv. ENTAV115 in vitro plants with P. viticola (isolate ‘PvitFEM01’) were performed as described in Lenzi et al.35. Both infected and non-infected plants were harvested at five time points (0, 24, 48, 72, 96 and 168 hours post-infection, hpi) in duplicates with 20–25 plants in each replicate. The replicates are from independent experiments. Sporangia of P. viticola were collected from infected material at late time points (96 and 168 hpi). Total RNA was extracted using the Spectrum plant total RNA (www.sigmaaldrich.com) and the small RNA fraction recovered from the flowthrough following the manufacturer’s instructions. The RNA-Seq and sRNA-Seq libraries were built using the TruSeq RNA and TruSeq Small RNA Library Prep kits (www.illumina.com), respectively, following the manufacturer’s protocol. The degradome-Seq libraries were constructed from RNA extracted from pooled material of infected and non-infected plants using the parallel analysis of RNA ends (PARE) protocol as described by German et al.13 by Vertis Biotechnologie AG (www.vertis-biotech.com). The RNA-seq and degradome-Seq libraries were sequenced on a HiSeq. 2500 platform (www.illumina.com) at the LaBSSAH facility (www.labssah.eu) and Vertis Biotechnologie AG (www.vertis-biotech.com), respectively. The RNA-seq libraries were processed as described in the supplementary note. Differential gene expression analysis of the P. viticola and V. vinifera transcriptomes were performed by using the Cufflinks pipeline as described in the supplementary note52,53. Targets cleaved by sRNAs were predicted using SeqTar54 and by combining different sets of sRNAs and transcriptomes as described in Šurbanovski et al.55. A set of sRNAs of 21nt that mapped perfectly on P. viticola genome and a set of V. vinifera sRNAs of 21 and 22nt were used to search for mRNA targets in the P. viticola transcriptome from this study and to search for targets in grapevine gene sequences retrieved from Genoscope (www.genoscope.cns.fr) and CRIBI (http://genomes.cribi.unipd.it/). All SeqTar analyses were filtered using a mismatch and binding score p-value ≤ 0.001 and a valid peak height p-value ≤ 10−10.
References
Creasy, G. L. & Creasy, L. L. Grapes (CAB International, Wallingford, Oxfordshire, 2009).
Armijo, G. et al. Grapevine pathogenic microorganisms: understanding infection strategies and host response scenarios. Frontiers in Plant Science 7, 382 (2016).
EUROSTAT. The use of plant protection products in the European Union, Data 1992–2003. (Luxembourg: Office for Official Publications of the European Communities, 2007).
Kamoun, S. et al. The top 10 oomycete pathogens in molecular plant pathology. Molecular Plant Pathology 16, 413–434 (2015).
Rouxel, M. et al. Phylogenetic and experimental evidence for host-specialized cryptic species in a biotrophic oomycete. New Phytologist 197, 251–263 (2013).
Gessler, C., Pertot, I. & Perazzolli, M. Plasmopara viticola: a review of knowledge on downy mildew of grapevine and effective disease management. Phytopathologia Mediterranea 50, 3–44 (2011).
Gisi, U. & Sierotzki, H. Fungicide modes of action and resistance in downy mildews. European Journal of Plant Pathology 122, 157–167 (2008).
Judelson, H. S. Dynamics and innovations within oomycete genomes: insights into biology, pathology, and evolution. Eukaryotic Cell 11, 1304–1312 (2012).
Jones, J. D. G. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
Hein, I., Gilroy, E. M., Armstrong, M. R. & Birch, P. R. J. The zig-zag-zig in oomycete–plant interactions. Molecular Plant Pathology 10, 547–562 (2009).
Win, J. et al. Adaptive evolution has targeted the C-terminal domain of the RxLR effectors of plant pathogenic oomycetes. The Plant Cell 19, 2349–2369 (2007).
Qiao, Y. et al. Oomycete pathogens encode RNA silencing suppressors. Nat Genet 45, 330–333 (2013).
German, M., Luo, S., Schroth, G., Meyers, B. & Green, P. Construction of parallel analysis of RNA ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nature Protocols 4, 356–362 (2009).
Voglmayr, H. & Greilhuber, J. Genome size determination in Peronosporales (Oomycota) by Feulgen image analysis. Fungal Genetics and Biology 25, 181–195 (1998).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Krajaejun, T. et al. Expressed sequence tags reveal genetic diversity and putative virulence factors of the pathogenic oomycete Pythium insidiosum. Fungal Biology 115, 683–696 (2011).
Mateos, F. V., Rickauer, M. & Esquerré-Tugayé, M.-T. Cloning and characterization of a cDNA encoding an elicitor of Phytophthora parasitica var. nicotianae that shows cellulose-binding and lectin-like activities. Molecular Plant-Microbe Interactions 10, 1045–1053 (1997).
Orsomando, G. et al. Phytotoxic protein PcF, purification, characterization, and cDNA sequencing of a novel hydroxyproline-containing factor secreted by the strawberry pathogen Phytophthora cactorum. Journal of Biological Chemistry 276, 21578–21584 (2001).
Baxter, L. et al. Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science 330, 1549–1551 (2010).
Huang, J., Yang, M. & Zhang, X. The function of small RNAs in plant biotic stress response. Journal of Integrative Plant Biology 58, 312–327 (2016).
Dussert, Y. et al. Draft genome sequence of Plasmopara viticola, the grapevine downy mildew pathogen. Genome Announcements 4, e00987–16 (2016).
Yin, L. et al. Genome sequence of Plasmopara viticola and insight into the pathogenic mechanism. Scientific Reports 7, 46553 (2017).
Sharma, R. et al. Genome analyses of the sunflower pathogen Plasmopara halstedii provide insights into effector evolution in downy mildews and Phytophthora. BMC Genomics 16, 741 (2015).
McCarthy, C. G. P. & Fitzpatrick, D. A. Phylogenomic reconstruction of the oomycete phylogeny derived from 37 genomes. mSphere 2, e00095–17 (2017).
Xiang, J. et al. Studying the mechanism of Plasmopara viticola RxLR effectors on suppressing plant immunity. Frontiers in Microbiology 7, 709 (2016).
Vleeshouwers, V. G. A. A. et al. Understanding and exploiting late blight resistance in the age of effectors. Annual Review of Phytopathology 49, 507–531 (2011).
Kemen, E. et al. Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana. PLoS Biology 9, e1001094 (2011).
Yin, L. et al. Characterization of the secretome of Plasmopara viticola by de novo transcriptome analysis. Physiological and Molecular Plant Pathology 91, 1–10 (2015).
Vergara, R., Parada, F., Rubio, S. & Pérez, F. J. Hypoxia induces H2O2 production and activates antioxidant defence system in grapevine buds through mediation of H2O2 and ethylene. Journal of Experimental Botany 63, 4123–4131 (2012).
Weiberg, A. et al. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 342, 118–123 (2013).
Wang, M. et al. Bidirectional cross-kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nature Plants 2, 16151 (2016).
Gobbin, D., Rumbou, A., Linde, C. C. & Gessler, C. Population genetic structure of Plasmopara viticola after 125 years of colonization in European vineyards. Molecular Plant Pathology 7, 519–531 (2006).
Nowara, D. et al. HIGS: Host-induced gene silencing in the obligate biotrophic fungal pathogen Blumeria graminis. The Plant Cell 22, 3130–3141 (2010).
Koch, A. et al. An RNAi-based control of Fusarium graminearum infections through spraying of long dsRNAs involves a plant passage and is controlled by the fungal silencing machinery. PLOS Pathogens 12, e1005901 (2016).
Lenzi, L., Caruso, C., Bianchedi, P. L., Pertot, I. & Perazzolli, M. Laser microdissection of grapevine leaves reveals site-specific regulation of transcriptional response to Plasmopara viticola. Plant and Cell Physiology 57, 69–81 (2015).
Si-Ammour, A., Mauch-Mani, B. & Mauch, F. Quantification of induced resistance against Phytophthora species expressing GFP as a vital marker: ß-aminobutyric acid but not BTH protects potato and Arabidopsis from infection. Mol.Plant Pathol. 4, 237–248 (2003).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Simpson, J. T. et al. ABySS: A parallel assembler for short read sequence data. Genome Research 19, 1117–1123 (2009).
Boisvert, S., Laviolette, F. & Corbeil, J. Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. Journal of Computational Biology 17, 1519–1533 (2010).
Haas, B. J. et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461, 393–398 (2009).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62 (2006).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Parra, G., Blanco, E. & Guigó, R. GeneID in Drosophila. Genome Research 10, 511–515 (2000).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research 25, 955–964 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research 35, 3100–3108 (2007).
Alexeyenko, A., Tamas, I., Liu, G. & Sonnhammer, E. L. L. Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics 22, e9–15 (2006).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30, 772–780 (2013).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology 56, 564–577 (2007).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology 59, 307–321 (2010).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Research 42, D222–D230 (2014).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protocols 7, 562–578 (2012).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Zheng, Y., Li, Y.-F., Sunkar, R. & Zhang, W. SeqTar: an effective method for identifying microRNA guided cleavage sites from degradome of polyadenylated transcripts in plants. Nucleic Acids Research 40, e28 (2011).
Šurbanovski, N., Brilli, M., Moser, M. & Si-Ammour, A. A highly specific microRNA-mediated mechanism silences LTR retrotransposons of strawberry. The Plant Journal 85, 70–82 (2016).
Acknowledgements
The authors wish to thank Claudia Kutter at Karolinska Institute (Stockholm, Sweden) for critical comments. We thank the Autonomous Province of Trento for financial support.
Author information
Authors and Affiliations
Contributions
M.B. and A.S.A. designed the experiments. M.B., E.A., P.B., M.P., M.M. and A.S.A. performed the experiments. M.B. and A.S.A. analyzed the results. M.B. and A.S.A. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brilli, M., Asquini, E., Moser, M. et al. A multi-omics study of the grapevine-downy mildew (Plasmopara viticola) pathosystem unveils a complex protein coding- and noncoding-based arms race during infection. Sci Rep 8, 757 (2018). https://doi.org/10.1038/s41598-018-19158-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19158-8
This article is cited by
-
A suitable alternative to antifungal agents for the control of early blight disease-Alternaria alternata of tomato
Australasian Plant Pathology (2024)
-
Unravelling molecular mechanisms involved in resistance priming against downy mildew (Plasmopara viticola) in grapevine (Vitis vinifera L.)
Scientific Reports (2023)
-
MicroRNA-mediated post-transcriptional regulation of Pinus pinaster response and resistance to pinewood nematode
Scientific Reports (2022)
-
Molecular characterization of the Rpv3 locus towards the development of KASP markers for downy mildew resistance in grapevine (Vitis spp.)
Euphytica (2022)
-
Metabolomics and transcriptomics to decipher molecular mechanisms underlying ectomycorrhizal root colonization of an oak tree
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
