
Abstract
The parasite Plasmodium falciparum causes the most severe form of malaria. Cell communication between parasites is an important mechanism to control population density and differentiation. The infected red blood cells (iRBCs) release small extracellular vesicles (EVs) that transfer cargoes between cells. The EVs synchronize the differentiation of the asexual parasites into gametocytes to initiate the transmission to the mosquito. Beside their role in parasite communication, EVs regulate vascular function. So far, the exact cargoes responsible for cellular communication remain unknown. We isolated EVs from cultured iRBCs to determine their small RNA content. We identified several types of human and plasmodial regulatory RNAs. While the miRNAs and tRNA-derived fragments were the most abundant human RNAs, we also found Y-RNAs, vault RNAs, snoRNAs and piRNAs. Interestingly, we found about 120 plasmodial RNAs, including mRNAs coding for exported proteins and proteins involved in drug resistance, as well as non-coding RNAs, such as rRNAs, small nuclear (snRNAs) and tRNAs. These data show, that iRBC-EVs carry small regulatory RNAs. A role in cellular communication is possible since the RNAs were transferred to endothelial cells. Furthermore, the presence of Plasmodium RNAs, in EVs suggests that they may be used as biomarker to track and detect disease.
Similar content being viewed by others


Introduction
Malaria affects over 400 million individuals worldwide every year, killing about 0.5 million1. The complications of the disease arise during the blood stage, which is characterized by symptoms such as cerebral malaria, severe anemia, metabolic acidosis and respiratory distress2. During the blood stage, the parasites release toxic factors that contribute to inflammation. In addition, iRBCs secrete small vesicles that contain human and parasite-derived biomolecules3. The EVs derived from iRBCs are elevated in patients suffering from malaria and are particularly elevated in severe disease4. Interestingly, the severity of the disease significantly correlates with the level of EVs in the plasma5. Therefore, EVs might be used as biomarker to track the progress of the disease and treatment. EVs of a size between 100–300 nm have been characterized to contain both human and parasite proteins3,6.
Surprisingly, EVs induce the conversion of parasites into gametocytes3. Thus, EVs act like a master regulator that orchestrate and synchronize the conversion to gametocytes to optimize the transmission of the parasites to mosquito. Although the molecules responsible for the commitment have not yet been identified, EVs transfer nucleic acids from parasite to parasite as demonstrated by the transfer of a plasmid DNA encoding for a gene conferring resistance to a particular drug6. Only the parasites that have acquired the selection marker can grow under the drug selection pressure. While these experiments demonstrated the potential of EVs to carry and transfer functional nucleic acids between parasites, the authors characterized for the first time a mechanism of communication between parasites. Interestingly malaria EVs contain RNAs, in particular small RNAs7. However so far, little is known about the nature of these RNAs.
During malaria, host derived miRNAs constitute essential mediators of cellular communication. In fact, functional miR451a, a miRNA expressed in RBCs is transferred from iRBCs to endothelial cells via EVs. Remarkably, miR451a is bound to Argonaute-2 and forms a functional silencing complex. Once transferred, miR451a destabilizes specific mRNA to regulate endothelial cell barrier function7.
Besides miRNAs, many other small RNA species have regulatory functions. However so far most of the analysis of EV RNAs have focused on miRNAs. High-throughput RNA-Seq has allowed the identification of new small RNAs with regulatory properties. Such as tRNA-derived fragments, piwi-RNAs (piRNAs), Vault RNAs (vtRNAs), Y RNAs and snoRNAs8,9. Furthermore, P. falciparum might use EVs to shuttle its own plasmodial RNAs. Currently no study has directly addressed the RNA composition of EVs.
Here we collected EVs derived from in vitro cultures of P. falciparum and performed RNA-Seq to characterize for the first time the small RNA content of iRBCs derived EVs. We found several species of host as well as plasmodial RNAs. Remarkably, EVs transferred Plasmodium RNAs to endothelial cells. While our data highlight a rich repository for infection-related biomarkers, we also identify an entirely new strategy by which Plasmodium likely manipulates host defensive barriers. Further exploration of EV functions may identify novel strategies for malaria control.
Results
Characterization and properties of extracellular vesicles isolated by differential centrifugation
To further investigate the potential regulatory RNAs present in EVs, we collected EVs from P. falciparum iRBCs cultures and purified the vesicles by ultracentrifugation. To verify the vesicular structure of the isolated EVs, we used transmission electron microscopy (TEM). Analysis by TEM revealed that EVs have a size of 100–250 nm and demonstrated an intact lipid bilayer (Fig. 1a). The morphology is consistent with EVs as previously reported measuring about 150 nm in size as observed by TEM and the nanoparticle tracking system (Fig. 1b). Larger EVs observed by the nanoparticle tracking system might be the result of aggregate formation3. The purity of the EV preparations was determined by western blotting with a panel of antibodies. EVs were enriched in the lipid raft protein stomatin as well as the parasite protein RESA, while Bip a plasmodium protein specifically expressed in endoplasmic reticulum was absent (Fig. 1c).

Extracellular vesicles isolated from P. falciparum carry RNA. (a) Transmission electron microscopy of EV preparation, purified EVs show individual EVs and a few clumps of varying sizes and intact lipid bilayers. The scale bar is 100 nm. (b) Representative particle size distribution from purified EVs. The concentration and size are determined by Nanoparticle Tracking Analysis. One representative experiment is shown. (c) Isolated EVs were confirmed by Western Blot for the enrichment of EV markers stomatin and RESA and negative for the endoplasmic reticulum Bip. (d) Total and Small bioanalyzer profiles of EV RNAs and total cell RNA show enrichment of small RNAs in EVs. One representative experiment is shown.
To further define the EV cargoes, we isolated total RNA from EVs and intact iRBC cells. The quantity and quality of isolated RNA were determined using an Agilent Bioanalyzer. In contrast to intact cells, total RNA bioanalyzer profiles indicated that iRBC EVs contain minor amounts of 18 S and 28 S rRNA species. Furthermore, the RNA composition is different in EVs in comparison with intact cells, with enrichment in small RNAs below 300 nt in length in the vesicles (Fig. 1d).
Extracellular vesicles derived from iRBCs contain small RNAs
Next to decipher the RNA content of EVs, we generated small RNA libraries of the samples for small RNA-Seq from three biological replicates of EVs derived from iRBCs. The same 3D7 parasite strain was cultured in RBCs of three different donors.
Small RNA sequencing of the EV libraries yielded a total of 83’319’134 raw reads that were pre-processed and trimmed to remove adapter sequences using sRNAbench to identify high quality reads that were considered for further analysis. After processing for adapter and unmapped sequences, the number of reads was reduced to 26’647’563, 8’126’655 and 11’925’918 respectively. The total number of reads mapping to the human genome was 43’689’670, whereas the total number of reads mapping to the P. falciparum genome was 3’010’466. Therefore, the proportion of P. falciparum reads was 6.4% versus 93.6% of human (Fig. 2a and Table 1).

Characterization of small RNAs by NGS. (a) Composition of reads mapping to human and P. falciparum genomes. (b) Distribution of iRBC EV small RNAs. Segments of the bar indicate the percent of reads attributed to each RNA biotype among all RNA reads that mapped perfectly to known sequences, averaged across 3 biological replicates is shown. (c) Reads aligning to coding genes were mapped back to the human chromosomes and represented as the % reads aligning to each chromosome. (d) Reads aligning to coding genes were mapped back to the human chromosomes and after normalization to the nucleotide size of the chromosome.
Since small RNAs have regulatory functions, we focused our analysis on the identification of small RNAs. As expected miRNAs and tRNAs were widely and abundantly expressed. Then we performed a detailed quantification of annotated RNAs to functionally categorize the human small RNAs. The average across all three samples was calculated to obtain a representative profile of biological diversity between donors. Of total reads, the miRNAs were the most abundant small RNA species with 42.3%, followed by 34.79% of tRNAs, 8% piRNA, 7% Y RNA, 3.60% Metazoa Signal Recognition Particle (SRP) RNA, 2.17% U-RNA, 1.17% Vault RNA, 0.48% snoRNA and 0.14% pre-mature RNA (Fig. 2b). The reads map to all the human chromosomes in particular chromosome 1 and 3 (Fig. 2c). Since the chromosomes do not have the same size, we then normalized by size to look for specific enrichment. Since there are several copies of mitochondrial DNAs per cell, we excluded them from the analysis. After normalization, there was an enrichment in chromosome 18 and 22 (Fig. 2d).
EVs contain a subset of human regulatory miRNAs
A total of 305 human miRNAs were identified in at least one sample. While we identified many miRNAs, only a few sets were highly expressed7 (Fig. 3a). A total of 61 miRNAs were present in all the 3 samples and among them, miR451a was the most abundant (515 RPM) accounting for 26.9% of the total miRNA reads, followed by miR486-5p (22.51%), miR92a-3p (9.37%) and miR103a-3p (6.34%). The most abundant miRNAs represent over 65% of the total reads (Fig. 3a). Because only a few miRNA species accounted for the majority of miRNA sequencing reads, we focused on the top 16 miRNAs for our analysis of potential regulatory targets. We performed Kyoto Encyclopedia of Genes and Genomes (KEGG)10 pathway enrichment analysis of genes targeted by miRNAs relative to all target genes in miRDB (http://www.genome.jp/kegg). We found that pathways involved in cell cycle, cell adhesion and associated to cancer were significantly enriched (Fig. 3b). In addition to mature RNAs, we found pre-mature miRNAs. However, the RPM for the pre-mature miRNAs were low and only 3 had an RPM above 1. In fact, pre-mature miR451a and miR137 were the highest expressed with 3.4 and 1.5 RPM respectively. Although the abundance was low, they were expressed in all three preparations (Fig. 3c).

Profiling of human miRNAs and tRNAs isolated from P. falciparum infected EVs. (a) Top 16 most highly expressed unique miRNAs in EVs. Left axis and bars: percent of total miRNA reads that mapped to each of the top 16 most abundant miRNAs. Error bars are standard deviation (n = 3). Right axis and line: cumulative percent of total miRNA reads. (b) Significantly (Benjamini Hochberg corrected P value < 0.05) enriched KEGG pathways for target genes. The number of miRNA involved in targeting at least one gene in a given pathway are shown in parentheses. (c) Distribution of human pre-mature miRNAs (n = 3). (d) Distribution of known tRNA sequences based on the total read counts. tRNA sequences are categorized by number of read counts corresponding to each individual tRNA (n = 3). (e) Based on the small RNA-seq results, only tRNA fragments were identified in EVs. Shown are examples of tRNA structures for each species depicting the boundaries of the identified tRNA fragment. The read density at each position is shown as a heatmap.
The human tRNAs represented an abundant small RNA category in our samples. We found that 60 tRNAs were expressed out of which 42 tRNAs were expressed in all the three biological replicates. However over 75% of the reads were derived from tRNA-GlyCCC, tRNA-LysTTT, tRNA-PheGAA and tRNA-LysCTT. With an average of 615 RPM, tRNA-GlyCCC represents about 44% and tRNA-LysTTT 16% of the total tRNA reads (Fig. 3d). All the tRNAs identified were tRNA fragments, we did not discover full length tRNAs. We found the 3′ and 5′ halves of tRNA –GlyCCC, with each half consisting of about 32 nt. The 5′half was slightly more abundant than the 3′. In addition, we found 5′tRNA-LysTTT of about 24 nt. Finally, tRNA-PheGAA, was composed of a 5′tRNA fragment and a 3′ fragment of about 24 nt each (Fig. 3e and Supplementary Figure 1).
Extracellular vesicles contain a subset of Y-RNA, Vault RNA, SRP-RNA, U RNA, piRNA and snoRNAs
Although miRNAs and tRNAs have essential biological functions, other small non-coding RNAs have in general not been characterized in EVs. Our sequencing strategy favors the identification and quantification of additional RNAs. Apart from the miRNAs and tRNA fragments, other RNA biotypes were abundantly present in iRBC-EVs, including Y-RNA, vault-RNA, SRP-RNA (7SL-RNA), U-RNA, Piwi-RNA and sno-RNA.
Y-RNAs, which range in size from 83 to 112 nt, are cytoplasmic non-coding RNAs that are conserved in the animal kingdom11,12,13. Most Y-RNAs are bound to the proteins Ro60 and La to form Ro ribonucleoproteins (RNPs), which might be targeted by the immune system in autoimmune disease14. There are four non-coding Y-RNAs in humans (hY1, hY3, hY4 and hY5 RNAs). We identified 2 Y-RNAs in our samples, RNY4 (319.37 RPM) and RNY1 (3.2 RPM), (Fig. 4a). Therefore, RNY4, which was identified in all 3 replicates accounted for 99% of the RPM for the total Y-RNAs15.

Functional characterization of small RNAs. (a) Distribution of Y-RNA (b) Vault-RNA. (c) Distribution of Metazoa-RNA. (d) Distribution of U-RNA. (e) Distribution of PIWI-RNAs. (f) Distribution of snoRNAs.
Vault RNAs are the RNA components of Vault ribonucleoprotein particles, which are located in the cytoplasm. The three human vRNA genes contain 88 (vRNA2 and vRNA3) or 98 nucleotides (vRNA1) single-exon polymerase III transcripts16. A fourth vRNA-related sequence, vRNA4, presumed to be silent pseudogene is located on chromosome X16. Only 1 Vault RNA was identified, VTRNA1-1 (51.31 RPM) having the highest level of expression and representing 99.5% of the vRNA reads (Fig. 4b).
Next, we identified 8 different SRP-RNAs, with 4 of them expressed in all three samples. RN7SL5P was the most abundant with an average RPM count of 107, which represents 68% (Fig. 4c) of all the SRP-RNAs. SnRNAs, also commonly referred to as U-RNA are highly conserved non-coding RNAs located in the nucleus and associated with Sm ribonucleoproteins as well as other specific proteins, to form small nuclear ribonucleoproteins. They participate in the splicing of precursors mRNAs. SnRNAs have an average size of 150 nt. We identified a total of 6 different U RNA (U1, U2, U3, U4, U5 and U6) with U1 being the most abundant, representing 70% of the total counts (Fig. 4d).
Piwi-associated RNAs, as compared with miRNAs, are a distinct class of 24–31 nucleotide-long RNAs produced by a Dicer-independent mechanism from single-stranded precursor transcripts expressed from intergenic regions termed piRNA clusters17. In total, we detected 44 piRNAs of which 21 were expressed in all three samples. The three most abundant piRNAs (PIR13807, PIR58593 and PIR57690) together represent approximately 33% of the total piRNA reads (Fig. 4e).
Small nucleolar RNAs (snoRNAs) are a class of small RNA molecules that primarily guide chemical modifications of other RNAs, mainly ribosomal RNAs and small nuclear RNAs18. We identified 7 snoRNAs with a total of 35 RPM (Fig. 4f). SNORD116-2 was the most abundant with 16 RPM and 46% of the total count.
P. falciparum iRBCs release plasmodial RNAs
Next, to investigate whether plasmodial RNA is released in the supernatant, we labelled newly transcribed RNAs in tightly synchronized iRBCs. Ring stage parasites were incubated with 5-ethynyl uridine (EU), which is incorporated into RNA during transcription. First, we monitored the cells for active transcription and, only iRBCs were positive for EU as measured by flow cytometry (Fig. 5a). Then, we collected the supernatants and freshly transcribed RNA was detected by flow cytometry in the supernatant with more than 80% of events being positive for freshly synthesized RNAs. Since RBCs are transcriptionally inactive, we assume that the labelled RNA is derived from the parasites (Fig. 5a). This experiment gave us strong confidence that EVs contain plasmodial RNA. In order to identify the plasmodial sequences, we aligned our RNA-Seq results generated previously to the P. falciparum genome (Plasmodium 6.1). We found that several plasmodial RNAs including tRNAs, rRNAs and mRNAs were present in EVs. In total, we identified 126 RNAs mapping to the Plasmodium genome of which 28 were identified in all the 3 samples (Supplementary Table 1). Most of the reads mapped to chromosome 1 (10.7%), chromosome 5 (33.1%), chromosome 7 (33.6%) and chromosome 13 (9.42%), only a minor amount mapped to mitochondrial and apicoplast DNAs (0.75 and 0.30%, respectively) (Fig. 5b). Then, we normalized by the length of the DNA, excluding mitochondrial and apicoplast DNAs since they are present in several copies per cell. Chromosome 1, 5 and 7 keep the highest number of mapped RNAs (Fig. 5c). Most of the RNA transcripts are derived from structural RNAs. There are a few putative novel ncRNAs, however their abundance is low. We have found mostly rRNA, tRNA and snoRNA in the three replicates from Plasmodium (Fig. 5d). In addition, a large amount of transcripts coding for proteins exported to the RBC cytosol such as RESA, ETRAMP and mRNAs from the PHIST family including Mal7P1.172 and PF0_0137 were present. Furthermore, EVs contain a large amount of apicoplast tRNAs19 and mitochondrial rRNAs (Fig. 5e), including tRNA-GLu-1, tRNA-Ser1, tRNA-Met1, tRNA-Pro1, tRNA-Arg1, tRNA-Trp1, tRNA-Arg3, tRNA-Cys1, pre-tRNA-Pr01, tRNA-SelCys1. Interestingly several RNAs were involved in drug resistance, such as PFE1150w (multidrug resistance protein 1), PFL1410c (ABC transporter, multidrug resistance-associated protein 2), PF13_0238 (Kelch-13).

EVs contain plasmodial RNAs. (a) Quantification of P. falciparum RNAs transferred to EVs. Infected RBCs were labeled with Click-It RNA Alexa Fluor 488. After 30 h. of incubation, EVs were collected and EU incorporation was analyzed by Flow Cytometry. (b) Distribution of the reads mapped to the P. falciparum genome according to the corresponding chromosomes. (c) Distribution of the reads mapped to the P. falciparum according to the size of the chromosome. (d) Types of Plasmodium genes in the EVs by RNA-Seq. (e) Extracellular vesicles contain a large number of Apicoplast tRNA and Mitochondrial rRNA. (f) RNA expression is different in EVs than mixed stage parasite expression pattern. (g) EV RNA composition is different that of iRBC. The expression levels shown by RNAseq of EV has no detectable correlation with that of steady state iRBC, showing that EV is not merely a smaller form of iRBC. Neither parametric (Pearson’s R) nor parametric correlations (Kendell’s tau) analysis detected correlation between EV and iRBC at any time points with correlation larger than 0.001. The X-axis shows the average steady state RNA expression levels.
We then compared the RNA EV expression profile with stage specific RNA profile of the blood stage and found that EV RNAs are mostly derived from genes with peak expression in merozoites and early rings. Very few RNAs are derived from other stages (Fig. 5f). Importantly, there was no direct correlation of the gene expression levels between iRBC and EV transcriptome (Fig. 5g).
Plasmodial extracellular RNAs are associated with EVs
The extracellular RNAs found in the plasma can be associated with proteo-lipid complexes and the complexes containing RNAs might be co-purified with EVs during ultracentrifugation. To exclude the possibility that plasmodial RNA molecules originate from protein–lipid complexes present in our EV preparation or in the conditioned medium derived from in vitro P. falciparum iRBCs, we treated EVs with proteinase K20. In fact, proteolytic digestion of the protein complex stabilizing the plasmodial RNAs would release the RNAs and render them sensitive to degradation. However, RNAs protected inside a vesicular structure would not be expected to show sensitivity to proteolysis. We treated EVs with or without proteinase K at 55 °C. The samples were incubated for 0, 15, 30 and 45 min followed by RNA extraction and quantification by qPCR. In fact, MAL5_18S, 28 S rRNA and PFA0110w were protected from degradation inside EVs (Fig. 6a–c).

Plasmodial RNAs are contained inside the EVs. (a) The RNAs are protected from proteinase K digestion, EVs were treated with proteinase K (5 mg/ml, close symbols) or untreated (open symbol) at 55 °C. At times indicated aliquots were removed and the RNA was isolated and the expression of (a) MAL5_18S, (b) 28 S rRNAs, (c) PFA0110w, was analyzed by qPCR. The mean + /− s.d. of one representative experiments of three is shown. (d) Elution profile of conditioned media collected from iRBC culture as measured by absorbance at 280 nm. Points represent the mean +/− s.d. of three experiments performed in triplicate. (e) Fractions from conditioned media were assayed for (e) PFA0110w, (f) MAL5_18S, (g) 28 S rRNA, (h) PF11_0381a and (i) miR451 using absolute quantification by qPCR. The mean + /− s.d. of one representative experiment is shown (n = 3).
To further confirm that the plasmodial RNAs are associated with EVs, we performed size exclusion chromatography, which allows efficient separation of vesicles from protein lipid complexes that are smaller and elute later than vesicles. For this purpose, we cultured iRBCs and collected the conditioned supernatant after 48 hours. We isolated 14 fractions by chromatography and most of the proteins were eluted in the latest fractions (9–12), only a minor portion was eluted in the fraction 5 that corresponds to the EV fractions (Fig. 6d). We can assume based on our standards that most of the proteins in the supernatant are albumin, gamma-globuline. We then isolated total RNA including small RNAs from each isolated fraction and performed qPCR. As miR451a, all the tested plasmodial RNAs were detected in fraction 5, which corresponds to the EV fraction (Fig. 6e–i). Altogether these experiments demonstrated that Plasmodium RNAs are located in EVs.
Plasmodium EVs deliver RNA cargo to human endothelial cells
To further understand the significance and potential function of the plasmodial RNAs in cellular communication, we tested their stability and potential transferability to endothelial cells. For this purpose, we incubated human bone marrow derived endothelial cells with (BMEC) EVs and noticed that the EVs were quickly taken up by the cells (Fig. 7a). We then monitored the transfer of RNAs by qPCR. No plasmodial RNA was detected in the absence of EVs. However, after a 12-hour EV-incubation the level of RNAs increased in endothelial cells. Therefore, the vesicles can act as a shuttle to transfer plasmodial RNAs to human endothelial cells. The inhibitor of the RNA Polymerase II and III (α-amanitin) was not able to inhibit the transfer, demonstrating that there was no active transcription of the Plasmodium genes in endothelial cells (Fig. 7b). To gain insight into the potential role of EV RNAs, we performed GO analysis. GO function analysis revealed that the RNA EVs were involved in RNA binding (Fig. 7c). Whereas GO cellular localization demonstrated that most of the RNAs are associated with the cytosol or vesicle membranes (Fig. 7d). Finally, the GO biological process showed that the RNAs are involved in translation regulation, regulation of gene expression and regulation of catalytic activity (Fig. 7e). Taken together these data further indicate a potential role of extracellular RNAs in regulation and cellular communication. Finally, we hypothesized that sequences that are identical between P. falciparum and human might have cross-regulatory function. From our RNA-Seq data, we identified several sequences that aligned to human and plasmodial genomes (Table 2).

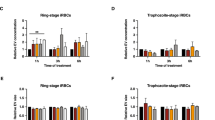
Transfer of plasmodial RNAs to endothelial cells via EVs. (a) Bone marrow endothelial cells (BMECs) were incubated with 100 μg of PKH67 fluorescently labelled EVs for 0 or 6 h. BMECs were stained for actin (phalloidin, red) and nuclei (DAPI, blue). Scale bar, 10 μm. (b) Endothelial cells were incubated with 100 μg of EVs for 12 hours. After isolation of their RNA, the transfer of plasmodial RNA was measured by qPCR. The average and S.D. of 3 experiments is shown. (mean ± s.e.m.; n = 3 experiments), ***P < 0.0001 versus control (Student’s t-test). (c) Functional classification of the identified Plasmodium RNAs in EVs by gene ontology (GO) analysis conducted using Cytoscape with the Cluego plugin. This highlights the regulatory function, which plasmodial RNAs may exert in the recipient cells. (c) GO molecular function (d) GO cellular localization (e) GO biological function.
Discussion
To date, a detailed analysis of the nucleic acid composition of malarial EVs has been lacking and only the host miRNA composition has been determined7. This study is the first systematic description of EV small-RNAs in P. falciparum iRBCs. With our approach, we found that EVs contain human, as well as plasmodial RNAs with potential regulatory functions. Interestingly the EVs were able to deliver plasmodial RNAs to endothelial cells suggesting a potential role in pathogen-host cellular communication. Furthermore, the full characterization of the EV RNA content offers a large number of promising new biomarkers for the diagnosis and the prediction of the progression of the disease.
Transfer of functional RNAs between cells via EVs is a universal mechanism of cellular communication21. Here, we identified several small RNAs with regulatory function. The host miRNAs were the most abundant RNA species detected in the EV samples. miRNAs are crucial in regulating posttranscriptional gene expression22. Recently miRNAs were shown to shuttle from cell to cell via EVs to mediate cellular communication23. In the context of malaria, we have previously shown that miR451a regulates cellular functions after transfer to endothelial cells7. Here, in addition to miR451a, we identified miR486-5p and miR92a-3p as the highest expressed human miRNAs. All the three miRNAs are expressed in RBCs and play an essential role in their maturation24,25. They regulate erythropoiesis by inhibiting proliferation or differentiation, protecting erythroid cells from oxidative stress or controlling iron transport and metabolism. However, these miRNAs have a function beside their role in RBC maturation26,27. There is only little overlap between the genes targeted by the EV miRNAs. However, the miRNAs target the same functional pathways. This raises the possibility that the transferred EV miRNAs (including mir451a, mir486-5p and mir92a-3p) might act synergistically to target multiple genes part of the same pathways involved in malaria infections and pathogenesis. In fact, our analysis of the KEGG pathways showed that iRBC-EV miRNAs targeted pathways involved in cell proliferation and adherence junction. Although little is known about the specific role of iRBC EVs in Cerebral Malaria pathogenesis, the concentration of EVs derived from RBCs, platelets, endothelial cells and leucocytes are markedly elevated in patients and there is a clear association between EV concentration and pathogenesis. More direct evidences for a role of EVs are coming from the rodent model of malaria. The reduction of EVs in transgenic mice or with drugs confers protection against severe disease and cerebral malaria. Furthermore, EVs isolated from mice with malaria are potently activating macrophages4,5,28,29,30,31,32,33. It remains to be demonstrated whether EV RNAs derived from different cellular sources can act synergistically to modulate the pathogenesis. In addition to miRNAs, we identified numerous host tRNA fragments. This is in agreement with other studies that found tRNAs in immune cells derived EVs8, in semen EVs9 and in EVs secreted by pathogens such as Trypanosoma cruzi34 and leishmania35. While tRNAs, which are 73–90 nt in length are components of the translational machinery, a functional role in gene regulation has been proposed for tRNA fragments. In our samples the most abundant tRNA (tRNA-Glyc) was derived from a 5′ and 3′ halves. In addition, we found 5′ end tRNA fragments, predominantly 30–34 nt tRNA halves derived from tRNA-Lys and tRNA-Phe. These derivatives of tRNAs are not degradation products but are specific cleavage products that function in pathophysiological conditions36. During oxidative stress, angiogenin cleaves the full-length mature tRNAs and produces 5′ and 3′ halves that are 30–40 nt in length37. The 5′ ends of these tRNA halves inhibit translation by displacing components of the translation initiation complex38. tRNA fragments are heterogeneous in size, nucleotide composition, biogenesis and function. Besides tRNA halves, shorter tRNA-derived fragments (tRFs) in the size range of 13–30 nt have been described in organisms spanning all domains of life. These RNAs can be both constitutively generated and produced in the context of stress. Although angiogenin is not detected in EVs3, it is expressed in RBCs39. The oxidative environment that triggers the parasite in the RBCs might favor the generation of tRFs that are then secreted via EVs. While, the role of tRNA in cellular communication is unknown, their biological function range from translation control, over RNA silencing, to regulating apoptosis40,41. Similar to earlier descriptions of exosomal and circulating tRNAs, we identified primarily 18–35 nt fragments in our analysis, supporting the potential processing of tRNA to miRNA-like molecules8,9.
While numerous piRNAs and snoRNAs are found in the circulation, their significance in biological processes or diseases is unknown42,43. In our samples, the piRNAs were almost as abundant as the miRNAs. Canonical piRNAs are defined as small RNAs bound to PIWI proteins44. Recently, functional roles for piRNA beyond transposon silencing have emerged including genome rearrangement45, epigenetic regulation46 regulation of mRNA47 and lncRNAs48. piRNAs target the 3′UTR of several gene products that have to be degraded by acting similarly to miRNAs. Therefore, piRNAs may have potentially an important role in gene regulation and cellular communication.
Moreover, snoRNAs can give origin to shorter miRNA-like RNAs capable of regulating translation due to complementary interactions with mRNA49. The importance of snoRNAs in biological processes is illustrated by the absence of SNORD116 (SNO1354) in the Prader-Willi syndrome (PWS), a severe hereditary disease associated with obesity, mental retardation, and some other symptoms19,50.
PIR13807, which was the most abundant piRNA in our study, as well as PIR58593 were identified in the plasma of a large cohort of individuals42. SNO1408 (SNORD10) and SNO1257 (SNORD114) were also identified in plasma of healthy individuals42.
In addition, in our samples we found Y RNAs. Y RNAs are components of ribonucleoproteins (RNPs) complexed with Ro60 and La proteins. Although full length Y RNAs have a size of approximately 100 nt, high-throughput RNA sequencing has revealed that small Y RNA fragments, 22–36 nt, are also highly abundant in cells, tissues and body fluids including in EVs of humans, as well as in a range of tumors9,51,52.
We found that RNY4 is the most abundant in EVs. Interestingly Y RNA-derived fragment is the most abundant fragment in plasma and serum in healthy patients52, as well as in EVs found in semen9.
Altogether, the function of the extracellular tRNAs, piRNAs, snoRNAs and Y-RNAs is currently unknown. Interestingly, only the processed forms of the RNAs were found in the EVs. Therefore, it can be speculated that the small RNAs are specifically targeted to the EVs. It is an intriguing possibility that small RNAs could mediate intercellular physiological signals to the hosts or between parasites.
While host small RNAs were the most abundant, we also found Plasmodium small RNAs such as tRNA, snoRNAs. In addition, we found Plasmodium mRNAs coding for proteins with gene regulatory properties such as the Alba proteins. The Alba proteins bind to RNA and fine tune the translation of RNAs linked to invasion53. We also identified Plasmodium RNAs with regulatory properties such as PFC0425w (Plant Homeodomain (PHD) finger protein) and PFF1185w (Smarca-related protein) which have chromatin-mediated gene regulation, as well as transcription factor with an AP2 domain (PF08_0074). It remains to be investigated whether the parasite mRNAs are translated in the recipient cells.
Parasite resistance to antimalarial drugs is an increasing problem in the fight against malaria54. Surprisingly, EVs carry host and Plasmodium RNAs involved in drug resistance. The vtRNAs are part of a large cellular ribonucleoparticles called vaults, which are implicated in chemotherapy resistance and have been implicated in the regulation of several cellular processes including transport mechanisms55, signal transmissions and immune responses56. vtRNAs are upregulated in several tumors where they mediate drug resistance, which can be conferred either by direct binding to chemotherapeutic compounds57,58 or by the regulation of expression of multidrug-associated proteins59. Whether vtRNA1-1 plays a role in acquiring drug resistance to malaria therapies is an intriguing possibility. It might act by binding to a drug to participate in the export of malaria therapeutics58.
Furthermore, we identified several parasite-derived mRNAs coding for proteins involved in drug resistance such as PFE1150w (multidrug resistance), PFL1410c (ABC transporter, multidrug resistance-associated protein 2). As well as PFKELCH13, which is involved in artemisinin resistance60.
After alignment of the RNAseq reads against the plasmodial genome, we found that EVs contain plasmodial RNAs. Most of the transcripts are derived from structural RNAs, including a few putative novel ncRNAs. We demonstrated the presence of PF11_0381a snoRNA U2 spliceosome in EVs and its EV-mediated transfer to endothelial cells. In addition, some plasmodial mRNAs were identified. Interestingly a large number of the plasmodial mRNAs are coding for exported proteins61, such as PHIST B and PHIST C, PFA0110w, PFD1170c, MAL7P1.171, MAL7P1.172, PF08_0137. Why and how plasmodial RNAs are exported to EVs is not clear. However, the large percentage of small RNAs with potential gene regulatory function suggests they might be involved in cellular communication. The recent discovery of pathogen derived regulatory RNAs in several EVs suggests that regulatory RNAs may function in interspecies regulation involving microbial RNAs and host genes. It is possible that P. falciparum transfer its own RNAs via EVs to human cells to regulate gene expression to its own advantage. Some outstanding questions remain such as how P. falciparum targets specifically these RNAs to EVs or are the transferred RNAs transcribed in the target cells, either host cells or parasites.
While our and other results suggested that iRBCs secrete about 15 more EVs than uninfected RBCs, our current results with the transfer of plasmodial RNA showed that our preparations may contain more than 80% EVs derived from iRBCs3,4.
Taken together our data identified for the first time the RNAs species contained in EVs. We found host as well as parasite derived RNAs. Most of the RNAs have gene regulatory functions and might therefore be involved in cellular communication. In addition, we found several RNAs involved in drug resistance and aspect that will require more investigations. Finally, we also found Plasmodium derived RNAs. Therefore, our study offers a wide range of new biomarkers as well as new potential pathways that can be targeted by drugs.
Materials and Methods
Cell culture of parasites
The P. falciparum strain 3D7 was used for this study. Parasites were kept in fresh type 0 + human red blood cells, suspended at 4% hematocrit in HEPES-buffered RPMI 1640 containing 10% (w/v) heat inactivated human serum, 0.5 ml Gentamycin, 2.01 g sodium bicarbonate and 0.05 g Hypoxanthine at pH 6.74. Prior to culture, the complete medium was depleted from extracellular vesicles and debris by ultracentrifugation at 100000 g for 1 hour. The parasite cultures were maintained in a controlled environment at 37 °C in a gassed chamber at 5% CO2 and 1% O2.
Synchronization of parasites
In order to obtain highly synchronized parasite cultures we performed a combination of Percoll and sorbitol. Parasites at 42–45 hours post-invasion were purified using a 70% Percoll. Fresh blood was added to the isolated schizonts. After 8 hours, 5% sorbitol was used to eliminate the remaining schizonts to yield highly synchronized rings.
Purification of EVs
EVs from iRBCs were isolated from cell culture supernatants as described3. In brief, cell culture supernatants of Plasmodium falciparum-infected RBCs were collected. Cells and cellular debris were removed from the supernatant by centrifugation at 600 g, 1600 g, 3600 g and finally 10000 g for 15 min. To further concentrate the EVs, the supernatant was filtered through a Vivacell 100 filter (100 kDa molecular weight cut off; Sartorius). Then, the concentrated supernatant was pelleted at 100000 g, the pellet resuspended in PBS and layered on top of a 60% sucrose cushion and spun at 100000 g for 16 hours. The interphase was collected and washed with PBS twice at 100000 g for 1 hour to yield EVs.
Isolation of RNA and gene expression studies in BMEC-1
Total RNA was isolated from EVs and BMEC-1 using miRNeasy kit (Qiagen, Hilden, Germany). The concentration and integrity of total RNA was measured using a NanoDrop-1000 spectrophotometer (Thermo Scientific, Wilmington, DE). Size distribution was determined by Agilent 2100 Bioanalyzer with the Agilent Small RNA Chip (Agilent Technologies, Santa Clara, CA).
For Plasmodial RNAs, reverse transcription reactions were performed with 1 μg total RNA using M-MLV Reverse Transcriptase kit (Promega) after DNase I treatment (Invitrogen).
Mature miR-451a and selected miRNAs were detected by quantitative RT-PCR (qRT-PCR) using TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). Small nuclear RNA U6 (RNU6) (for miR-451a) and 18 S rRNA and EF1 (for mRNAs) were used as reference genes for relative quantitation using the 2^− Ct method.
Metabolic labeling of RNA and imaging
Ring stage parasites at a parasitemia of about 5% (3D7), (15 hours post-invasion) were incubated at a final concentration of 0.5 mM 5-ethynyl uridine (EU) for 30 h. EVs were isolated from the conditioned medium as described above with exception of the sucrose gradient. The cells and EVs were subsequently fixed with 4% para-formaldehyde and permeabilized with 0.1% Triton-X-100. EU incorporated EV RNA was detected using Click chemistry according to the manufacturer’s protocol (Invitrogen, C10329) and nuclei were counterstained using Hoechst 33342 and analyzed with a MACSQuant VYB flow cytometer. Flow cytometry data acquisition was performed using FlowJo X (Tree Star, Ashland, OR).
Endothelial cell culture
A semi-immortalized human bone marrow derived endothelial cell line (BMEC-1)62 was cultured in Endothelial Cell Growth Medium MV (Promocell) supplemented with 10% fetal bovine serum (Cedarlane, Burlington, Canada) and maintained at 37 °C under 5% CO2.
EV internalization assays with BMEC-1. Purified EVs were labelled with PKH67 red fluorescent labelling kit (Sigma-Aldrich) and incubated with BMECs for 12 h before washing cells three times to remove unbound EVs. For uptake inhibition experiments endothelial cells were co-incubated with different compounds, as described in Fig. 5. Cells were fixed, permeabilized and stained for F-actin with CF594 (Biotium) and with the nuclear dye DAPI (Sigma-Aldrich). Leica TCS SP5. Epifluorescence was performed on a Axiovert 200 M microscope (Carl Zeiss. Germany) equipped with a camera (Orca charge-coupled device; Hamamatsu Photonics, Japan) using a 40 Plan-Neofluar (NA 1.3) oil immersion objective. Intensity analysis was performed with the Axiovision 4.6.3 software. Background subtraction was performed for each field. Confocal microscopy was performed on a Zeiss LSM 510 Laser Scanning Microscope using a 63 Plan-Neofluar water-immersion objective.
Size exclusion chromatography
Experiments to determine possible presence of serum-derived Ago2-miRNA complexes were performed essentially as described7.
Briefly, sephacryl S-500 resin (GE Healthcare) was packed in a chromatography column (0.9 Å~ 30 cm, 19.1 mL bed volume). Before injection, the column was equilibrated with 25 mL of PBS solution at 0.5 mL/min at room temperature. The column was injected with 0.5 mL of undiluted fresh serum or purified EVs and eluted at 4 °C for approximately 1 h with PBS solution (pH 7.4) at a flow rate of 0.5 mL/min. A total of 25 fractions of 1 mL each were collected. Fractions were stored at 4 °C before use. Protein molecular weight standards included BSA (67 kDa; GE Healthcare) and tyrosine (0.181 kDa; Sigma-Aldrich).
Gene ontology analysis
ClueGO was used to find over-represented GO terms in the categories “biological process”, “cellular component” and molecular function”. Benjamini-Hochberg correction was performed for multiple testing-controlled P values. The GO analysis was conducted using Cytoscape 3.2.1 software63 with the ClueGO64 plugin. The GO analysis was conducted using a two-sided hypergeometric test with Bonferroni correction. The GO term levels were from five to ten. The minimum number of genes to form a cluster was set at three, while the minimum percentage of genes covered by our data set against the database was set at 7%. The rest of the settings were left as defaults.
RNA-Seq
Each sequencing library was constructed from 2 ng of isolated and treated plasma RNA. All libraries were uniquely bar-coded with index primer for multiplexing into sequencing lanes. The small RNA libraries were prepared and amplified using the NEBNext small RNA Library Prep Set (New England BioLabs, Ipswitch, MA, USA) following manufacturer instruction. The amplified libraries were resolved on a 10% Novex TBE gel (Life technologies) for size selection and the 140 to 160 nucleotide bands that correspond to adapter-ligated constructs derived from the 21 to 40 nucleotide RNA fragments were excise and recovered in DNA elution buffer. The average size distribution of each library was determined using Agilent Bioanalyzer with High Sensitivity Chip Kit (Agilent, Santa Clara, CA, USA) and quantified on ABI 7900HT Fast RT-PCR instrument using the KAPA Library Quantification kit according to the manufacture’s protocol (Kapa Biosystems, Woburn, MA, USA). Each library was adjusted to final concentration of 2 nM, pooled, and sequenced on an Illumina HiSeq. 2000 or MiSeq sequencer for single read 50 cycles at the Center for Cancer Computational Biology at Dana-Farber Cancer Institute.
Sequence Analysis
The BCL files were de-multiplexed using CASAVA v1.82, and the adaptor sequences within the read sequences were trimmed by FastX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit). The processed sequences were filtered for small RNAs greater than 16 nucleotides in length. The sequences were then aligned, quantified and annotated using sRNABench 1.0 pipeline65. Briefly, the pipeline implemented hierarchical sequence mapping strategy that first mapped and remove spike-in library, contaminants, and rRNA before sequentially mapped to known mature miRNA, tRNA, snoRNA and piRNA onto the human genome sequence (hg19) using Bowtie266 with parameters that allow for 1 mismatch in seed alignment (-N 1), try two set of seeds (-R 2), and set the length of seed substrings to be 16 (-L 16). Mapped small RNA species was quantified to read counts and normalized to RPM as described in sRNABench. Detected species were mapped to mature miRNA only and not precursor miRNA. Reads derived from microRNA with multiple copies in the genome were summed together, and read counts from sample duplicates were aggregated by mean for where it is applicable. All statistical analysis was performed using R version 3.2.
References
WHO. WHO Malaria Report 2012. (2012).
Miller, L. H., Baruch, D. I., Marsh, K. & Doumbo, O. K. The pathogenic basis of malaria. Nature 415, 673–679, https://doi.org/10.1038/415673a (2002).
Mantel, P. Y. et al. Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell host & microbe 13, 521–534, https://doi.org/10.1016/j.chom.2013.04.009 (2013).
Nantakomol, D. et al. Circulating red cell-derived microparticles in human malaria. J Infect Dis 203, 700–706, https://doi.org/10.1093/infdis/jiq104 (2011).
Pankoui Mfonkeu, J. B. et al. Elevated cell-specific microparticles are a biological marker for cerebral dysfunctions in human severe malaria. PloS one 5, e13415, https://doi.org/10.1371/journal.pone.0013415 (2010).
Regev-Rudzki, N. et al. Cell-cell communication between malaria-infected red blood cells via exosome-like vesicles. Cell 153, 1120–1133, https://doi.org/10.1016/j.cell.2013.04.029 (2013).
Mantel, P. Y. et al. Infected erythrocyte-derived extracellular vesicles alter vascular function via regulatory Ago2-miRNA complexes in malaria. Nature communications 7, 12727, https://doi.org/10.1038/ncomms12727 (2016).
Nolte-‘t Hoen, E. N. et al. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic acids research 40, 9272–9285, https://doi.org/10.1093/nar/gks658 (2012).
Vojtech, L. et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic acids research 42, 7290–7304, https://doi.org/10.1093/nar/gku347 (2014).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic acids research 45, D353–D361, https://doi.org/10.1093/nar/gkw1092 (2017).
Hendrick, J. P., Wolin, S. L., Rinke, J., Lerner, M. R. & Steitz, J. A. Ro small cytoplasmic ribonucleoproteins are a subclass of La ribonucleoproteins: further characterization of the Ro and La small ribonucleoproteins from uninfected mammalian cells. Molecular and cellular biology 1, 1138–1149 (1981).
Wolin, S. L. & Steitz, J. A. Genes for two small cytoplasmic Ro RNAs are adjacent and appear to be single-copy in the human genome. Cell 32, 735–744 (1983).
O’Brien, C. A. & Harley, J. B. A subset of hY RNAs is associated with erythrocyte Ro ribonucleoproteins. The EMBO journal 9, 3683–3689 (1990).
Lerner, M. R., Boyle, J. A., Hardin, J. A. & Steitz, J. A. Two novel classes of small ribonucleoproteins detected by antibodies associated with lupus erythematosus. Science 211, 400–402 (1981).
Kowalski, M. P. & Krude, T. Functional roles of non-coding Y RNAs. Int J Biochem Cell Biol 66, 20–29, https://doi.org/10.1016/j.biocel.2015.07.003 (2015).
van Zon, A. et al. Multiple human vault RNAs. Expression and association with the vault complex. The Journal of biological chemistry 276, 37715–37721, https://doi.org/10.1074/jbc.M106055200 (2001).
Vagin, V. V. et al. A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313, 320–324, https://doi.org/10.1126/science.1129333 (2006).
Kishore, S. & Stamm, S. Regulation of alternative splicing by snoRNAs. Cold Spring Harb Symp Quant Biol 71, 329–334, https://doi.org/10.1101/sqb.2006.71.024 (2006).
Cassidy, S. B., Schwartz, S., Miller, J. L. & Driscoll, D. J. Prader-Willi syndrome. Genet Med 14, 10–26, https://doi.org/10.1038/gim.0b013e31822bead0 (2012).
Arroyo, J. D. et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proceedings of the National Academy of Sciences of the United States of America 108, 5003–5008, https://doi.org/10.1073/pnas.1019055108 (2011).
Valadi, H. et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature cell biology 9, 654–659, https://doi.org/10.1038/ncb1596 (2007).
Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004).
Mittelbrunn, M. et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nature communications 2, 282, https://doi.org/10.1038/ncomms1285 (2011).
Patrick, D. M. et al. Defective erythroid differentiation in miR-451 mutant mice mediated by 14-3-3zeta. Genes Dev 24, 1614–1619, https://doi.org/10.1101/gad.1942810 (2010).
Wang, L. S. et al. MicroRNA-486 regulates normal erythropoiesis and enhances growth and modulates drug response in CML progenitors. Blood 125, 1302–1313, https://doi.org/10.1182/blood-2014-06-581926 (2015).
Mees, S. T. et al. Analyzing miRNAs in ductal adenocarcinomas of the pancreas. J Surg Res 169, 241–246, https://doi.org/10.1016/j.jss.2009.10.005 (2011).
Narducci, M. G. et al. MicroRNA profiling reveals that miR-21, miR486 and miR-214 are upregulated and involved in cell survival in Sezary syndrome. Cell Death Dis 2, e151, https://doi.org/10.1038/cddis.2011.32 (2011).
Combes, V., Coltel, N., Faille, D., Wassmer, S. C. & Grau, G. E. Cerebral malaria: role of microparticles and platelets in alterations of the blood-brain barrier. Int J Parasitol 36, 541–546, https://doi.org/10.1016/j.ijpara.2006.02.005 (2006).
Combes, V. et al. Circulating endothelial microparticles in malawian children with severe falciparum malaria complicated with coma. JAMA 291, 2542–2544, https://doi.org/10.1001/jama.291.21.2542-b (2004).
Couper, K. N. et al. Parasite-derived plasma microparticles contribute significantly to malaria infection-induced inflammation through potent macrophage stimulation. PLoS Pathog 6, e1000744, https://doi.org/10.1371/journal.ppat.1000744 (2010).
El-Assaad, F., Wheway, J., Hunt, N. H., Grau, G. E. & Combes, V. Production, fate and pathogenicity of plasma microparticles in murine cerebral malaria. PLoS Pathog 10, e1003839, https://doi.org/10.1371/journal.ppat.1003839 (2014).
Tiberti, N. et al. Exploring experimental cerebral malaria pathogenesis through the characterisation of host-derived plasma microparticle protein content. Sci Rep 6, 37871, https://doi.org/10.1038/srep37871 (2016).
Penet, M. F. et al. Protection against cerebral malaria by the low-molecular-weight thiol pantethine. Proceedings of the National Academy of Sciences of the United States of America 105, 1321–1326, https://doi.org/10.1073/pnas.0706867105 (2008).
Garcia-Silva, M. R. et al. Extracellular vesicles shed by Trypanosoma cruzi are linked to small RNA pathways, life cycle regulation, and susceptibility to infection of mammalian cells. Parasitol Res 113, 285–304, https://doi.org/10.1007/s00436-013-3655-1 (2014).
Lambertz, U. et al. Small RNAs derived from tRNAs and rRNAs are highly enriched in exosomes from both old and new world Leishmania providing evidence for conserved exosomal RNA Packaging. BMC Genomics 16, 151, https://doi.org/10.1186/s12864-015-1260-7 (2015).
Lee, Y. S., Shibata, Y., Malhotra, A. & Dutta, A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 23, 2639–2649, https://doi.org/10.1101/gad.1837609 (2009).
Yamasaki, S., Ivanov, P., Hu, G. F. & Anderson, P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol 185, 35–42, https://doi.org/10.1083/jcb.200811106 (2009).
Ivanov, P., Emara, M. M., Villen, J., Gygi, S. P. & Anderson, P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell 43, 613–623, https://doi.org/10.1016/j.molcel.2011.06.022 (2011).
Pavlov, N. et al. Angiogenin distribution in human term placenta, and expression by cultured trophoblastic cells. Angiogenesis 6, 317–330, https://doi.org/10.1023/B:AGEN.0000029412.95244.81 (2003).
Maute, R. L. et al. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America 110, 1404–1409, https://doi.org/10.1073/pnas.1206761110 (2013).
Goodarzi, H. et al. Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell 161, 790–802, https://doi.org/10.1016/j.cell.2015.02.053 (2015).
Freedman, J. E. et al. Diverse human extracellular RNAs are widely detected in human plasma. Nature communications 7, 11106, https://doi.org/10.1038/ncomms11106 (2016).
Huang, X. et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics 14, 319, https://doi.org/10.1186/1471-2164-14-319 (2013).
Sarkar, A., Volff, J. N. & Vaury, C. piRNAs and their diverse roles: a transposable element-driven tactic for gene regulation? FASEB J, https://doi.org/10.1096/fj.201600637RR (2016).
Mochizuki, K. & Gorovsky, M. A. A Dicer-like protein in Tetrahymena has distinct functions in genome rearrangement, chromosome segregation, and meiotic prophase. Genes Dev 19, 77–89, https://doi.org/10.1101/gad.1265105 (2005).
Lee, H. C. et al. C. elegans piRNAs mediate the genome-wide surveillance of germline transcripts. Cell 150, 78–87, https://doi.org/10.1016/j.cell.2012.06.016 (2012).
Rouget, C. et al. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature 467, 1128–1132, https://doi.org/10.1038/nature09465 (2010).
Watanabe, T., Cheng, E. C., Zhong, M. & Lin, H. Retrotransposons and pseudogenes regulate mRNAs and lncRNAs via the piRNA pathway in the germline. Genome Res 25, 368–380, https://doi.org/10.1101/gr.180802.114 (2015).
Ender, C. et al. A human snoRNA with microRNA-like functions. Mol Cell 32, 519–528, https://doi.org/10.1016/j.molcel.2008.10.017 (2008).
Sahoo, T. et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet 40, 719–721, https://doi.org/10.1038/ng.158 (2008).
Meiri, E. et al. Discovery of microRNAs and other small RNAs in solid tumors. Nucleic acids research 38, 6234–6246, https://doi.org/10.1093/nar/gkq376 (2010).
Dhahbi, J. M. et al. 5′-YRNA fragments derived by processing of transcripts from specific YRNA genes and pseudogenes are abundant in human serum and plasma. Physiological genomics 45, 990–998, https://doi.org/10.1152/physiolgenomics.00129.2013 (2013).
Vembar, S. S., Macpherson, C. R., Sismeiro, O., Coppee, J. Y. & Scherf, A. The PfAlba1 RNA-binding protein is an important regulator of translational timing in Plasmodium falciparum blood stages. Genome Biol 16, 212, https://doi.org/10.1186/s13059-015-0771-5 (2015).
White, N. J. Does antimalarial mass drug administration increase or decrease the risk of resistance? Lancet Infect Dis 17, e15–e20, https://doi.org/10.1016/S1473-3099(16)30269-9 (2017).
van Zon, A. et al. Vault mobility depends in part on microtubules and vaults can be recruited to the nuclear envelope. Exp Cell Res 312, 245–255, https://doi.org/10.1016/j.yexcr.2005.10.016 (2006).
Kowalski, M. P. et al. Host resistance to lung infection mediated by major vault protein in epithelial cells. Science 317, 130–132, https://doi.org/10.1126/science.1142311 (2007).
Mashima, T. et al. Interactions between antitumor drugs and vault RNA. Nucleic Acids Symp Ser (Oxf), 217–218, https://doi.org/10.1093/nass/nrn110 (2008).
Gopinath, S. C., Matsugami, A., Katahira, M. & Kumar, P. K. Human vault-associated non-coding RNAs bind to mitoxantrone, a chemotherapeutic compound. Nucleic acids research 33, 4874–4881, https://doi.org/10.1093/nar/gki809 (2005).
Persson, H. et al. The non-coding RNA of the multidrug resistance-linked vault particle encodes multiple regulatory small RNAs. Nature cell biology 11, 1268–1271, https://doi.org/10.1038/ncb1972 (2009).
Mbengue, A. et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520, 683–687, https://doi.org/10.1038/nature14412 (2015).
Marti, M., Good, R. T., Rug, M., Knuepfer, E. & Cowman, A. F. Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science 306, 1930–1933, https://doi.org/10.1126/science.1102452 (2004).
Schweitzer, K. M. et al. Characterization of a newly established human bone marrow endothelial cell line: distinct adhesive properties for hematopoietic progenitors compared with human umbilical vein endothelial cells. Lab Invest 76, 25–36 (1997).
Cline, M. S. et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2, 2366–2382, https://doi.org/10.1038/nprot.2007.324 (2007).
Bindea, G. et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093, https://doi.org/10.1093/bioinformatics/btp101 (2009).
Barturen, G. et al. sRNAbench: profiling of small RNAs and its sequence variants in single or multi-species high-throughput experiments. Methods in Next Generation Sequencing: Methods in Next Generation Sequencing (2014).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25, https://doi.org/10.1186/gb-2009-10-3-r25 (2009).
Acknowledgements
The authors wish to thank Alex Holman and Yaoyu E. Wang of The Center for Cancer Computational Biology, Dana-Farber Cancer Institute, Boston, MA for assistance with RNA-sequencing and data analysis. This study was financially supported in part by the Novartis foundation for medical- biological research (PYM), the Gottfried and Julia Bangerter-Rhyner-Stiftung (to MW and PYM) and the research pool of the University of Fribourg (PYM). Additional grants include the Swiss Excellence Scholarships for Foreign Scholars (to KAB and SM). The funding bodies had no role in the design of the study, in collection, analysis, and interpretation of data, or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
P.Y.M. designed the project; P.Y.M., K.A.B., S.M., M.A.H.C. and M.W. performed experiments; P.Y.M., K.A.D., S.M. and M.A.H.C. performed data analysis; S.R.A., R.H.Y.J. and L.F. performed bioinformatics analysis. I.G. supported the RNA-Seq. P.Y.M., K.A.B., S.M., M.A.H.C., I.G. and L.F. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Babatunde, K.A., Mbagwu, S., Hernández-Castañeda, M.A. et al. Malaria infected red blood cells release small regulatory RNAs through extracellular vesicles. Sci Rep 8, 884 (2018). https://doi.org/10.1038/s41598-018-19149-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19149-9
This article is cited by
-
Extracellular vesicles could be a putative posttranscriptional regulatory mechanism that shapes intracellular RNA levels in Plasmodium falciparum
Nature Communications (2023)
-
Starvation induces changes in abundance and small RNA cargo of extracellular vesicles released from Plasmodium falciparum infected red blood cells
Scientific Reports (2023)
-
Surface proteome of plasma extracellular vesicles as mechanistic and clinical biomarkers for malaria
Infection (2023)
-
Extracellular vesicles in malaria: an agglomeration of two decades of research
Malaria Journal (2021)
-
Prediction of gene expression regulation by human microRNAs in Plasmodium falciparum
Genes and Environment (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
