
Abstract
Exposure to lipopolysaccharides (LPS) causes extensive neutrophilic inflammation in the airways followed by mucous cell hyperplasia (MCH) that is sustained by the anti-apoptotic protein, Bcl-2. To identify inflammatory factor(s) that are responsible for Bcl-2 expression, we established an organ culture system consisting of airway epithelial tissue from the rat nasal midseptum. The highest Muc5AC and Bcl-2 expression was observed when organ cultures were treated with brochoalveolar lavage (BAL) fluid harvested from rats 10 h post LPS instillation. Further, because BAL harvested from rats depleted of polymorphonuclear cells compared to controls showed increased Bcl-2 expression, analyses of cytokine levels in lavages identified IL-13 as an inducer of Bcl-2 expression. Ectopic IL-13 treatment of differentiated airway epithelial cells increased Bcl-2 and MUC5AC expression in the basal and apical regions of the cells, respectively. When Bcl-2 was blocked using shRNA or a small molecule inhibitor, ABT-263, mucous cell numbers were reduced due to increased apoptosis that disrupted the interaction of Bcl-2 with the pro-apoptotic protein, Bik. Furthermore, intranasal instillation of ABT-263 reduced the LPS-induced MCH in bik +/+ but not bik −/− mice, suggesting that Bik mediated apoptosis in hyperplastic mucous cells. Therefore, blocking Bcl-2 function could be useful in reducing IL-13 induced mucous hypersecretion.
Similar content being viewed by others


Introduction
Bcl-2 is a founding member of a family of proteins that maintain cellular homeostasis by regulating apoptosis. Bcl-2 protects cells against a wide range of cell death stimuli1,2 by stabilizing the mitochondrial membrane and preventing permeabilization and release of death mediators3. Bcl-2 is inserted in the outer mitochondrial membrane and can be inactivated by binding to pro-apoptotic members of the family. While Bcl-2 is classified as an oncogene because it causes the onset of many cancers including lymphoma, it also sustains the function of thymocyte subpopulations during development4.
Because of its importance in various biological processes and diseases, understanding the regulation of Bcl-2 expression is very critical. Bcl-2 levels are regulated by various cytokines, including IL-1β and IGF-1 in airway epithelial cells5,6, IL-6 in lymphoblast cells7, IL-7 and IL-21 in T lymphoid cells8,9, IL-10 in tumor-associated macrophages10, andIL-22 in renal cortex tissue11. Many of these cytokines converge into the NF-κB pathway12,13 and other signaling molecules like janus kinase/signal transducer and activator of transcription (JAK/STAT)14,15 and phosphatidylinositol 3-kinase (PI3K)/PKB (protein kinase B)16,17 to increase Bcl-2 expression. However, the LPS-induced inflammatory mediators that affect Bcl-2 expression in non-hematopoietic and primary non-cancerous cells have not been extensively studied.
The airway epithelium modulates pulmonary immune responses and is a key player in the pathogenesis of chronic lung diseases18,19. As part of the innate immunity, airway epithelial cells (AECs) produce mucins to trap and clear inhaled particulates by mucociliary action20,21. In healthy subjects, few mucous cells are present in the conducting airways, but in subjects with asthma, cystic fibrosis (CF), and chronic obstructive pulmonary disease (COPD), the number of hyperplastic mucous cells increases leading to mucous hypersecretion and airway plugging21. MUC5AC is one of the major secretory polymeric mucins upregulated in hyperplastic mucous cells and contributes to airway reactivity22,23. Surprisingly, therapeutics to reduce the debilitating mucous hypersecretion are limited with only few potential drugs currently in preclinical or clinical trials24,25.
Following inflammatory responses to LPS or allergen exposure, Bcl-2 expression is upregulated in airway epithelial cells of animal models of mucous hypersecretion and in patients with cystic fibrosis, asthma, and chronic bronchitis5,26,27. Bcl-2 expression is upregulated in airway mucous cells to sustain hyperplastic mucous cells in animal models with acute and chronic inflammatory settings26,28,29. Therefore, the goal of the present study was to understand the pathways responsible for the coordinated induction of Bcl-2 and MUC5AC in AECs and help to identify novel intervention strategies to control mucous cell hyperplasia.
In the present study, we identified IL-13 as an important inflammatory factor that induces Bcl-2 and MUC5AC expression in response to a neutrophilic inflammation induced by LPS. When Bcl-2 function was suppressed, the role of IL-13 was switched from causing proliferation30 to inducing cell death in AECs in differentiated airway cultures in vitro and in hyperplastic mucous cells in vivo in a Bik-dependent manner. The small molecule BH3 domain mimetic compounds targeting the hydrophobic groove of Bcl-2 has been very successful strategy against cancer using ABT-73731 and it’s orally bioavailable derivative ABT-263 or navitoclax32. We further found that ABT-263 at very low doses alleviated LPS-induced mucous cell hyperplasia (MCH).
Results
LPS-induced BAL potentiates mucous cell hyperplasia and Bcl-2 expression
To identify inflammatory factors that induce Bcl-2 in hyperplastic mucous cells, we established a nasal epithelial explant organ culture system. We used the nasal explant culture to identify the inflammatory factors regulating Bcl-2 expression in mucous cells, because we previously have shown that nasal epithelium undergoes mucous cell hyperplasia in response to LPS injury with concomitant epithelial expression of Bcl-233. The nasal explant culture avoids any alteration to the cells present in vivo. In addition, several studies have shown that the mucociliary epithelium of the nose has many properties that resemble the respiratory epithelium of the lung34. Similar to what is observed in the lung epithelium33, intranasal instillation of LPS caused MCH in the rat nasal distal midseptal epithelium (Fig. 1A) and the mucous cells showed increased Bcl-2 expression (Fig. 1B). Rat midseptal nasal explants when cultured on an air-liquid interface and treated with 1, 10, or 100 µg/ml LPS for 24 h and analyzed after an additional 48 h in LPS-free medium showed a dose-dependent increase in the Muc5AC mRNA (Fig. 1C) and in the amount of stored mucosubstances or Vs (Fig. 1D). However, because the quantity of stored mucosubstances was much lower than that observed in vivo (Fig. 1A) we postulated that inflammatory factors in the bronchoalveolar lavage (BAL) may potentiate the extent of MCH. Therefore, in addition to the 100 µg/ml LPS, explant cultures were treated with BAL fluid harvested at 24 h post LPS instillation, which results in amount of stored mucosubstances similar to that observed in vivo (Fig. 1E). At 24 h post LPS instillation, LPS activity in the BAL fluid was reduced drastically to 1% of the instilled amount, suggesting little contribution of the initially instilled LPS in inducing mucosubstances (Supplemental Fig. S1).

LPS exposure increases inflammatory factors in the BAL that augment Muc5AC and Bcl-2 expression. (A) LPS induced mucous cell metaplasia in rat nasal epithelium. Representative micrographs of nasal epithelia from non-treated (NT) and LPS-instilled rats stained with AB-PAS. Quantification of mucous cells and volume density of intraepithelial stored mucosubstances (Vs) at 3 d post LPS instillation. Data shown as mean ± SEM (n = 7/group) (B) LPS-induced Bcl-2 expression in mucous cells. A representative nasal epithelial section from LPS-treated rat showing Bcl-2-immunopositivity (red) among Muc5AC-positive (green) mucous cells (MCs) and the nuclei are stained with DAPI (blue). (C) MUC5AC mRNA levels in LPS-treated organ cultures quantified by q-PCR. The fold-change over non-treated controls is shown. (D) Quantity of the intraepithelial stored mucosubstances (Vs) in LPS-treated organ cultures stained with AB-PAS. (E) Representative photomicrographs of nasal explants treated with BALF from LPS-instilled rats or with BALF and 100 µg/ml LPS (BALF+LPS), and the quantity of Vs in explants at 24 h following each treatment. Data shown as mean ± SEM (n = 3/group); *p < 0.05; **p < 0.01; ***p < 0.001.
Identification of BAL inflammatory factors that augment Bcl-2 positivity in mucous cells
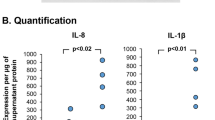
To identify the inflammatory factor(s) responsible for induction of Bcl-2 in mucous cells, BAL was harvested at 3, 10, and 24 h post LPS instillation and fractionated into cell-free supernatant and cell membrane-bound fractions. Our previously published studies have extensively characterized the mucous cell hyperplasia and Bcl-2 expression when rats are instilled with 1000 µg/ml LPS5,26,35. In addition, the detailed kinetics of the inflammatory response and mucous cell hyperplasia was followed over 90 days post instillation of 1000 µg LPS and reported36. Therefore, we used 1000 µg LPS to identify the inflammatory cytokines that induce Bcl-2 expression. Treatment of organ cultures with the supernatant fraction of BAL harvested at 10 h post LPS instillation compared to the media-treated controls induced maximal Bcl-2-positivity and mucous cells (MCs)/mm basal lamina (BL) (Fig. 2A), while the BAL collected at 3 h post LPS instillation 15% of the LPS remaining (Supplemental Fig. S1) showed no effect (data not shown). Because we have previously observed that depletion of polymorphonuclear cells (PMNs) leads to increased Bcl-2 positivity in hyperplastic mucous cells in vivo 37, we treated the explants with BAL harvested at 10 h post LPS instillation from PMN-depleted and non-depleted controls injected with normal rabbit serum (NRS) (Fig. 2B). Consistent with Bcl-2 expression in rats depleted of PMNs37, explant cultures treated with BAL from PMN-depleted rats compared with controls showed increased Bcl-2 positivity (Fig. 2C). Multiplex analysis of cytokines showed increased levels of IL-1β, IL-6, TNFα and IL-13 in BAL samples from LPS- compared to saline-instilled rats, but only IL-1β and IL-13 were significantly higher in the BAL from PMN-depleted compared to NRS-injected rats (Fig. 2D). Levels of MCP-2 and GRO-KC were not affected by PMN depletion (Suppl. Fig. S2). Together, these findings suggested that IL-1β and IL-13 were the main inducers of Bcl-2 expression in hyperplastic mucous cells.

Identification of LPS-induced inflammatory factors that are required for Muc5AC and Bcl-2 expression. (A) Representative photomicrographs of nasal explants treated with media only or with the BAL supernatant harvested 10 h after LPS instillation showing Muc5AC- (green) and Bcl-2- (red) positivity with DAPI-stained nuclei (blue). The number of Bcl-2-positive mucous cells per mm basal lamina in the organ cultures treated with LPS-induced BAL collected at 10 h post instillation. BAL was used as Whole (BAL fluid and lavaged cells), Sup (the cell-free supernatant), or Memb (the membrane fraction prepared by lysing lavaged cells). Data shown as mean ± SEM (n = 3/group) (B) Rats were injected IP with anti-PMN or NRS 24 h before LPS-instillation and BAL was collected 10 h post LPS challenge. The numbers of neutrophils (PMNs), lymphocytes (Lymphs) and macrophages (Mac) in the BAL was quantified from cytospins stained with Wright-Giemsa. (C) Quantitation of Bcl-2-positive mucous cells in the nasal organ cultures treated with BAL supernatant from NRS- or anti-PMN treated rats collected at 10 h post LPS-instillation. Data shown as mean ± SEM (n = 5/group for NRS and n = 7/group for anti-PMN) (D) Inflammatory factors measured in the BAL supernatant at 10 h post LPS challenge from rats treated with NRS or anti-PMN. Data shown as mean ± SEM (n = n = 5/group for NRS and n = 7/group for anti-PMN /group); *p < 0.05.
IL-13 induces Bcl-2 and MUC5AC in human airway epithelial cells (HAECs)
Our previous study had demonstrated the role of IL-1β in inducing Bcl-2 expression as discovered by microarray analyses5. Therefore, the present study focused on investigating the physiological importance of IL-13 in mediating Bcl-2 and mucin (MUC5AC) expression. In primary HAECs, IL-13 treatment increased the number of cells that immunostained positive for Bcl-2 and MUC5AC expression (Fig. 3A). In differentiated HAEC, Bcl-2 was localized around perinuclear and basal regions while MUC5AC was localized in the apical regions, as analyzed by 3-D imaging (Fig. 3B, middle panels) and lateral 2-D image algorithms (Fig. 3B, right panels).

IL-13 induces MUC5AC and Bcl-2 expression in human airway epithelial cells (HAECs). (A) Cytometric analysis of Bcl-2 (red) and MUC5AC (green) positivity in HAECs treated with IL-13 (10 ng/ml) or media for 48 h as shown in representative micrographs in top panels with DAPI-stained nuclei (blue). Approximately 300 cells from each treatment were analyzed to calculate the percentage of Bcl-2-positive (Bcl-2-pos) and MUC5AC-positive (MUC5AC-pos) cells. Data shown as mean ± SEM (n = 10/group)***p < 0.001. (B) Representative micrographs of differentiated cells treated with IL-13 or left untreated (NT). Differentiated HAECs were treated with IL-13 or left untreated and were co-immunostained for Bcl-2 (red) and MUC5AC (green) and analyzed by laser-scanning confocal microscopy. A 2-D image rendering of lateral views of differentiated cells treated with IL-13 showing using transparent-rendering (left-panel) and surface-rendering (right panel) algorithms. A 3-D image rendering of differentiated cells treated with IL-13 showing apical and basal views of a rotated image of the differentiated culture mount. (C) Suppression of Bcl-2 expression and the effect on MUC5AC expression levels in cells transfected with shBcl-2 or shCTRL, and treated with IL-13. Representative micrographs of HAECs transfected with shBcl-2 or shCTRL, and treated with IL-13 showing Bcl-2 (red) and MUC5AC (green) immunostaining and DAPI-stained nuclei (blue). Bcl-2- and MUC5AC-positive cells were quantified. Data shown as mean ± SEM with n = 3 per treatment group. ***p < 0.001. (D) Analysis of apoptotic cells recovered after IL-13 treatment of untransfected (UT) or shCTRL- or shBcl-2-transfected cells. The cells were stained with Annexin V (AnnV) and propidium iodide (PI) to analyze apoptotic cells by FACS analysis. Early (AnnV) and late (AnnV+PI) apoptotic cells were increased by 4–5-fold. Data shown are representative of 3 experiments.
Suppression of Bcl-2 reduces MUC5AC expression by inducing apoptosis
To investigate the role of Bcl-2 in mucin expression, we blocked Bcl-2 expression using retroviral shRNA expression vector (shBcl-2). Both Bcl-2- and MUC5AC-positivity were significantly reduced in shBcl-2 compared with shCTRL cells (Fig. 3C). This reduction was due to apoptotic death of cells in shBcl-2-transfected cells at 24 h post treatment reduction as shown by Annexin V staining (Fig. 3D).
ABT-263 treatment reduces MCH in the mouse model of LPS exposure
We further investigated the therapeutic effect of blocking Bcl-2 using the small molecule BH3 mimetic, ABT-263, in the mouse model of LPS-induced inflammation and MCH6,37. The number of MCs/mm BL was significantly reduced in mice intranasally treated with ABT-263 (2 mg/kg body weight) for 2 consecutive days starting 6 d post instillation of mice with 50 µg of LPS (as illustrated in Fig. 4A) compared with mice instilled with vehicle (Fig. 4B). Because we failed to detect dying MUC5AC-positive mucous cells, we used Scgb1A1, a secretoglobulin detected in secretory Club cells38, as an alternative to identify dying secretory epithelial cells. The number of cells positive for active (cleaved) caspase 3 (Ac-Casp3) (Fig. 4C) and TUNEL (Fig. 4D) were increased among Scgb1A1-positive secretory cells in ABT-263- compared to vehicle-treated mice.

ABT-263 reduces endotoxin-induced mucous cell hyperplasia in vivo in a Bik-dependent manner. (A) Experimental outline for testing therapeutic efficacy of ABT-263 in LPS-induced MCH in mice. (B) Representative micrographs of lung tissue sections stained with Alcian-Blue (AB) and H&E from LPS-challenged mice treated with vehicle or ABT-263 (2 mg/Kg) are shown. Quantification of mucous cell numbers per mm BL. (C) Representative micrographs showing activated (cleaved) caspase 3 or Ac-Casp3 (green) among Scgb1a1-positive (red) secretory cells in mouse axial airways. The relative fold-change in the number of ac-Casp3+ secretory cells in LPS-challenged mice treated with vehicle or ABT-263. (D) Representative micrographs showing TUNEL-positivity (green) in Scgb1a1+ (red) secretory cells in mouse axial airways treated with ABT-263 and DAPI-stained nuclei (blue). The relative fold-change in the number of TUNEL+ secretory cells in mice challenged with LPS and treated with vehicle or ABT-263. (E) STAT-1 phosphorylation in HAECs following 0, 15, and 60 minutes of IL-13 treatment. Cropped Western blots are displayed. (F) Bik and Bcl-2 mRNA levels in IL-13 treated STAT1 −/− and STAT1 +/+ MEFs compared with the respective non-treated cells. (G) Immunoprecipitation with anti-Bcl-2 antibodies of proteins extracted from HAECs treated with nothing or ABT-263. Bik levels are reduced in the pull-down while increased in the flow-through of HAECs treated with ABT-263 compared with non-treated controls. Cropped Western blots are displayed. (H) Representative micrographs of lung tissue sections from LPS-challenged mice stained with AB-H&E and quantification of mucous cell numbers per mm BL in mice treated with vehicle or ABT-263 (2 mg/Kg) following LPS challenge. Scale = 20 µM; Data shown as mean ± SEM (n = 5–10 mice/group); *p < 0.05; **p < 0.01; ***p < 0.001.
Our previous studies have established that Bik expression causes cell death in airway epithelial cells, and cells from bik −/− mice are resistant to IFNγ-induced cell death39. Therefore, we investigated whether IL-13 increases Bik expression. IL-13 activates signal transduction and transactivator 1 (STAT1) in several cell types40, a transcription factor responsible for Bik induction39. We found that IL-13 in HAECs phosphorylated STAT1 at 15 and 60 min of treatment (Fig. 4E) and thereby increased Bik expression, because STAT1 −/− compared with STAT1 +/+ mouse embryonic fibroblasts (MEFs) when treated with murine IL-13 showed significantly lower Bik mRNA levels, while Bcl-2 levels remained unaffected (Fig. 4F). In addition, the amount of Bik protein that immunoprecipitated by Bcl-2 antibodies was significantly reduced by ABT-263 and remained in the input of ABT-263-treated cell extracts (Fig. 4G), suggesting that ABT-263 disrupted Bik/Bcl-2 interaction. The importance of Bik in the resolution of LPS-induced MCH was assessed by exposing bik +/+ and bik −/− mice to ABT-263 or vehicle following LPS challenge. ABT-263 treatment suppressed the MCH in bik +/+ but not in bik −/− mice (Fig. 4H) confirming the role of Bik in causing cell death in hyperplastic mucous cells when Bcl-2 is blocked with ABT-263.
Discussion
Our previous studies showed that exposure of rodent lungs to LPS causes extensive neutrophilic inflammation and mucous cell hyperplasia that is sustained by Bcl-2 expression in epithelial mucous cells26,27,33,35,36,41. By using BAL fluid from rats depleted of PMNs, the current studies identify IL-13 as the factor that induce MUC5AC and Bcl-2 expression in airway cells. Because Bcl-2 sustains hyperplastic mucous cells, targeted inactivation of Bcl-2 using the BH3 mimetic, ABT-263, suppresses LPS-induced mucous cell hyperplasia in a Bik-dependent cell death pathway.
LPS-induced inflammation is characterized by an initial influx of large number of PMNs over the first 3–8 h followed by macrophages and lymphocytes over 6–12 h post LPS instillation36. The inflammatory factors derived from these cells affect the airway epithelium to establish mucous cell hyperplasia over 48–72 h post instillation37,41,42. We were able to replicate the extent of MCH observed in the nasal midseptum in vivo, when treating the organ culture system with BAL fluid harvested from 10 h post LPS instillation. However, the whole BAL that included lavaged cells did not induce Bcl-2 expression, suggesting that the inflammatory cells released contents that inhibit Bcl-2 expression. The effect of additional LPS from the BAL fluid is minimal, given that we had already added 100 μg/ml LPS to the explant culture. If higher LPS levels was causing Bcl-2 positivity, BAL fluid collected at 3 h post LPS-instillation that had even higher levels of LPS would be expected to induce Bcl-2 expression, but showed no significant change in Bcl-2 expression. These findings suggest that the Bcl-2-inducing IL-13 and IL-1β reach maximum levels at 10 h post instillation, because BAL fluid harvest at 3 or 24 h was not effective in inducing Bcl-2 expression. Significant neutrophilic inflammation sets in within 3 hour of LPS exposure in rodents as well as in humans36,43. However, the effect of neutrophils on other cell types is highly complex, given that the lung consists of many cell types. Our previous study demonstrated that depletion of PMNs caused an increase in Bcl-2-positive mucous cells in vivo 37. The ex vivo midseptum culture also showed increased Bcl-2 positivity when treated with BAL from rats depleted of PMNs, suggesting that the organ culture system reliably replicated the in vivo findings. Analyses of cytokines that are differentially increased in BAL fluid from PMN-depleted compared to non-depleted controls allowed us to identify IL-13 as one of the inflammatory factors responsible for Bcl-2 expression in mucous cells. While LPS-induced inflammation is primarily known to increase the cytokines IL-1β, IL-6, TNF-α37 and IL-1836, macrophages from various rat strains, when stimulated with anti-CD8 antibody in vitro, also produce IL-1344. Many studies have shown that IL-13 is expressed by several inflammatory cells including T helper 2 (TH2) cells, type 2 innate lymphoid cells (ILC2s), invariant natural-killer T (iNKT) cells, eosinophils, or alternatively activated macrophages45. However, while T cells are found in the BAL of LPS-instilled rats36, these T cells do not produce TH2 cytokines. Rather other cell types, including endothelial cells and epithelial cells produce IL-13 when rats are challenged with LPS30,46. Further, LPS at low concentrations induces IL-13 production from mast cells by activating TLR2 and TLR4 receptors47,48,49. Therefore, depletion of neutrophils may have enriched for the IL-13 detected in the BAL of LPS-instilled rats and facilitated increased expression of Bcl-2 in hyperplastic mucous cells.
IL-13 plays an important role in proliferation and repair of airway epithelial cells and promotes mucous cell hyperplasia50,51,52 by inducing expression of the Sam pointed domain-containing ETS transcription factor (SPDEF) in Club cells through a STAT6-dependent mechanism. Because Bcl-2 and MUC5AC expression have repeatedly been observed to occur within the same cells26,29,33, it is likely that IL-13 may activate the same pathways to induce expression of these two genes. Similar to MUC5AC53, induction of Bcl-2 expression is mediated through transactivation of EGFR pathway5, and EGFR activation is necessary for IL-13–mediated MCH30,54,55. IL-13 indirectly activates EGFR via production of TGF-α56, HB-EGF30, or epigen57. IL-13–induced TACE and release of TGF-α is also directly implicated in the airway epithelial hyperproliferation56. Therefore, airway IL-13 levels may be critical for normal cellular homeostasis in the setting of airway epithelial injury because it coordinates the proliferative and cytoprotective activity. Similarly, IL-13 when co-incubated with IL-9 is protective against spontaneous or corticosteroid-induced apoptosis by upregulating Bcl-251,58. Unfortunately, therapeutic targeting of IL-13 results in adverse events because this cytokine also elicits immunoregulatory functions59. For instance, IL-13 suppresses Th17 cytokine production in an IL-10-dependent manner60 and thereby may play an important role in Th17-associated autoimmune diseases like multiple sclerosis, rheumatoid arthritis, and colitis61. Consequently, IL-13–targeted therapeutics result in significant adverse events involving the musculoskeletal diseases that are associated with Th17 cytokines62. Therefore, targeting of Bcl-2 function to reduce MCH by a Bik-mediated cell death is an approach that is more specific to mucous cells and likely to have less side effects.
Bcl-2 has been found in the nucleus, as well as associated with the ER63 or mitochondria64. However, in differentiated HAECs of Bcl-2 was localized in the basal and peri-nculear areas while MUC5AC was enriched in the epical region as previously reported65,66. Although we have reported that suppression of Bcl-2 causes reduction of mucous cells26, the inducer of cell death when the anti-apoptotic Bcl-2 was downregulated, had not been identified. The present study demonstrates that IL-13 increased expression of Bcl-2 and Bik to alter the airway epithelial cell fate and that the pro-apoptotic Bik is required for ABT-263 mediated suppression of LPS-induced MCH. Our previous studies have established that Bik expression causes cell death in airway epithelial cells as bik +/+ but not bik −/− mice resolve MCH during prolonged exposure to allergen, and bik −/− cells are resistant to IFNγ-induced cell death39. In addition, clinical findings show that Bik mRNA levels are significantly reduced in airway cells of asthmatics39 and chronic bronchitics67 compared to non-diseased controls. Most importantly, Bik expression causes cell death only in proliferating airway epithelial cells67 but not in other cell types, such as hematopoietic and endothelial cells68. ABT-263 has been successfully used in Phase II clinical trials for cancer treatment32,69. The dose used for cancer patients is 250 mg/d over 21 days. In contrast, we found efficacy in reducing LPS-induced MCH at doses that are a 100-fold lower. Therefore, the small molecule Bcl-2 inhibitors, when delivered directly to the lung, may provide better treatment options against mucous hypersecretion.
The utility of rat nasal organ culture to identify the regulatory mechanisms underlying MCH and Bcl-2 expression in the lower airways supports the recent findings that cells of the upper airways strongly resemble the airway cells lining the lung airways70,34. The upper and lower airway diseases may display different manifestations of the same inflammatory process71. Therefore, suppression of Bcl-2 expression or blocking Bcl-2 function as shown in this study may also have beneficial effects in rhinosinusitis in the context of chronic mucus hypersecretion.
The present studies provide a novel paradigm to primarily target hyperplastic mucous cells by suppressing Bcl-2 function and thereby switching the proliferative function of IL-13 into an efficient suppressor of MCH. These findings support the development and use of the small molecule Bcl-2 inhibitors as a novel treatment modality for patients with cystic fibrosis and chronic bronchitis when delivered directly to the lung. ABT-263 is currently being tested for cancer therapy, specifically in lymphomas and leukemia32, and for senescent stem cells72. Together with the previous findings in a mouse model of asthma73, the findings suggest that this compound may also represent an effective treatment for targeting hyperplastic mucous cells. Whether blocking Bcl-2 may have therapeutic effects in chronic diseases other than LPS- and allergen-induced mucus hypersecretion that are mediated by IL-13 needs to be explored in the future.
Methods
Laboratory Animals
Specific pathogen-free F344/NCrR male rats, 6–8 wk of age, were obtained from NCI (Frederick, MD) and were housed until 8–10 weeks of age. Rats were housed in pairs and were provided food and water ad libitum, a 12-h light/dark cycle at 22.2 °C, and 30–40% humidity. The bik −/− mice on C57BL/6 were made available by Dr. Andreas Strasser (Walter and Eliza Hall Institute) and bred at the Lovelace Respiratory Research Institute (LRRI) and genotyped as described68. Pathogen-free wild-type C57BL/6J mice were purchased from The Jackson Laboratory or from in-house breeding. Rodents were housed in isolated cages under specific pathogen-free conditions. All experiments were approved by the LRRI Institutional Animal Care and Use Committee and were performed in accordance with relevant guidelines and regulations at LRRI. LRRI is a facility approved by the Association for the Assessment and Accreditation for Laboratory Animal Care International.
LPS Challenge
F344/NCrR male rats (NCI, Frederick, MD) of 6–8 wk of age were briefly anesthetized with 5% isoflurane in oxygen and instilled intratracheally with 1000 μg of LPS (Pseudomonas aeruginosa serotype 10, Sigma, St. Louis, MO) in 0.5 ml of 0.9% pyrogen-free saline. Control rats were instilled with 0.5 ml of 0.9% pyrogen-free saline. Rats were sacrificed at 72 h post LPS-instillation for lung tissue analysis and BAL was collected at various time-points post LPS-instillation as described below. Similarly, C57BL/6 mice (both male and female) at 6–8 wks of age were briefly anesthetized with 5% isoflurane in oxygen and instilled intranasally with 50 μg of LPS (P. aeruginosa serotype 10, lot 31K4122, 3,000,000 LPS units (EU)/mg, Sigma, St. Louis, MO) in 0.05 ml of 0.9% pyrogen-free saline. One group of mice received ABT-263 (0.05 mg/Kg in 0.05 ml of 0.9% pyrogen-free saline) intranasally on day 5 and 6 after LPS challenge. Control mice were instilled with 0.05 ml of 0.9% pyrogen-free saline. Mice were sacrificed 24 h post last exposure and lung tissues were processed and analyzed.
Preparation and treatment of rat nasal epithelial organ cultures
The preparation and LPS treatment of explant cultures from rat nasal midsepta was essentially as described previously74. Briefly, following exsanguination, after removing the lower jaw the head was split in half longitudinally and the nasal midseptum and maxilloturbinates removed by microdissection using a Leica MZ 7.5 stereo zoom microscope (Leica Microsystems, Inc., Bannockburn, IL). The septum was cut into three sections, one proximal and two distal. The explants were cultured for 72 h by placing them epithelium side-up in transwell dishes (Corning, Incorporated Life Sciences., Acton, MA), and cultured in supplemented Ham’s/F-12 media (HyClone, Logan, UT). The nasal explants were treated with LPS for 24 h and replenished with fresh media for maintaining cultures over 48 h. We selected this time-point based on our previous studies showing that Bcl-2 expressing hyperplastic mucous cells by LPS in vivo peaks at 48 h post instillation26. Following treatments, explant cultures were fixed in zinc formalin for a minimum of 24 h, embedded in paraffin, and processed for microscopy.
Neutrophil depletion and bronchoalveolar lavage fractionation
Rats were intraperitoneally (i.p.) injected with 1 ml of rabbit anti-rat polymorphonuclear neutrophil (PMN) antiserum or with normal rabbit serum (NRS) as control (#AIAD11540, Accurate Chemical Corp., Westbury, NY) 24 h before LPS-instillation. Rats were sacrificed at 3, 10, and 24 h post-instillation with an injection of pentobarbital sodium and exsanguinated through the renal artery. Lungs were removed with cannulated trachea, placed in ice-cold saline for 3 min, and then lavaged three times with 5 ml of Ham’s F-12 media. BAL for treatment of organ cultures was used immediately after collection and fractionation, or was stored in 0.2-ml aliquots at −80 °C until further use. For fractionation, BAL was centrifuged at 1000 × g for 10 min at 4 °C, the supernatant was separated from the cell pellets and kept at −80 °C until needed. To isolate the cell membrane fraction, cells were lysed in cold (4 °C) sterile milli-Q-filtered water, centrifuged, and the supernatant from the lysed cells was discarded. The lysis was repeated twice to achieve complete lysis and culture medium was added to the membrane fraction to obtain the original volume of the BAL.
LPS quantification
The amount of LPS recovered in the BAL fluid at 3, 10, and 24 h post instillation was assayed using the Cambrex LAL Limulus Amoebocyte Assay (Walkersville, MD) according to package directions. Values are expressed as percentage of 1000 μg initially instilled intratracheally.
Histological Analysis
Histochemical staining for Alcian Blue and periodic acid Schiff (AB-PAS) was carried out as previously described35. Airway epithelial cell and mucous cell numbers per mm basal lamina (BL) were measured by counting the number of nuclei and mucous cells, respectively, and dividing by the length of the BL. Images were taken using a light microscope (Eclipse E600W; Nikon) with a Plan Fluor 60× NA 0.85 objective and a digital camera (DXM1200F; Nikon) with ACT-1 acquisition software (version 2.62 l Nikon). In all cases, the VisioMorph system (Visiopham A/S, Horsholm, Denmark) was used for morphometry by a person unaware of slide identity.
Luminex Analysis
The levels of cytokines and chemokines (IL-1β, IL-6, IL-13, TNFα, Gro/KC, and MCP-1) in BAL fluid were determined using a multiplex assay kit (Lincoplex panel, Linco Research, Inc., St. Charles, MO) according to the manufacturer’s instructions. Briefly, the BALF was filtered to remove cells and debris, then beads were incubated with diluted standards, or BALF overnight followed by a detector antibody cocktail for 60 min each at room temperature. After two washes in PBS supplemented with 0.02% Tween 20, 0.1% BSA, and 0.02% NaN3, the beads were incubated for 30 min with fluorescent dye-conjugated streptavidin. Cytokine levels were measured using a flow cytometer and were analyzed with Flowmetrix software (Luminex, Ausitn, TX). Standard curves for each cytokine and chemokine were generated on a log-log plot for each assay, and the concentrations in each sample were calculated from the corresponding curve-fitting equations.
Immunofluorescence analysis
Tissue sections were deparaffinized, hydrated in graded ethanol and deionized water, then washed in 0.05% v Brij-35 in Dulbecco’s PBS (pH 7.4). The antigens were unmasked by treating with Digest-All kit (Zymed Laboratories, San Francisco, CA) at a 1:3 dilution of trypsin to diluent at 37 °C for 10 min. Sections were then blocked using 0.2% Triton X-100 with 0.2% Saponin in a blocking solution containing 3% IgG-free BSA, 1% Gelatin and 2% normal donkey serum followed by incubation with anti-Bcl-2 (#sc-492, Santa Cruz Biotech, CA), anti-MUC5AC (#MAB2011, Millipore Inc.), anti-active Caspase 3 (#9661, Cell Signaling Technologies, CA) or isotype controls (Cell Signaling Technologies, CA). The immunolabeled cells were detected using F(ab)2-fragments of respective secondary antibodies conjugated to either DylightTM-549 or DylightTM-649 (Jackson Immunoresearch, West Grove, PA) and mounted with 4′,6-diamidino-2-phenylindole (DAPI) containing Fluormount-GTM (SouthernBiotech, Birmingham, AL) for nuclear staining.
For cytometry, cells were grown on Lab-Tek-II 8-chamber slides (Nalgene Nunc International, Rochester, NY) and treated with 10 ng/ml of IL-13 or were left untreated and were fixed using 3% paraformaldehyde with 3% sucrose in PBS and processed for immunostaining as described above. Micrographs were captured using either Zeiss LSM 510 Meta confocal microscope (Carl Zeiss MicroImaging, Inc, Thornwood, NY) or using the Axioplan 2 fluorescent imaging system (Carl Zeiss, Thornwood, NY) equipped with a charge-coupled device camera (ORCA-ER; Hamamatsu Photonics, Iwata City, Japan) and SlideBook 6 acquisition software (Intelligent Imaging Innovations, Denver, CO). Quantification of Bcl-2-positive and MUC5AC-positve cells per mm of basal lamina was performed using the VisioMorph system (Visiopham A/S, Horsholm, Denmark) or NIH ImageJ (http://imagej.nih.gov/ij/) software. Cells cultured in air-liquid interface were also immunostained for Bcl-2 and MUC5AC similarly.
TUNEL Assay
For detection of apoptotic cells, deparaffinized lung sections were stained using TACS® 2 TdT Fluorescein Kit (Trevigen Inc., Gaithersburg, MD) and fluorescent TUNEL-positive cells were detected as described earlier for fluorescent staining. In all cases, quantification of TUNEL-positivity was carried out by a person unaware of slide identity.
Cell culture
The human airway epithelial cells (HAECs) were maintained in bronchial epithelial growth medium (BEGM, Lonza, Walkersville, MD). Primary HAECs were purchased from Clontech (Walkersville, HD). For air-liquid interface culture primary HAECs were seeded on Transwell membranes and differentiated for 14 days. Following treatments, the membrane quarters were used for qRT-PCR and membrane halves embedded in paraffin for immunostaining. Cell viability was determined by trypan blue exclusion.
Immunoprecipitation and Western blot analysis
Protein was extracted from cells or tissues by homogenization in RIPA buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% deoxycholate, 0.1% SDS, and 5 mM EDTA) supplemented with a protease inhibitor cocktail (Sigma Chemical Co., St. Louis, MO). Protein concentration was determined using the BCA kit (Pierce, Thermo Fisher Scientific, Rockford, IL) and 50–100 μg of protein lysate was analyzed by Western blotting. For immunoprecipitation using the Pierce Crosslink IP Kit (# 26147, Thermo Fisher Scientific), cells were rinsed twice with cold PBS, scraped into cold PBS plus protease inhibitors and analyzed per manufacturer’s instructions. The Bcl-2 (#sc-7382) antibody was from Santa Cruz Biotechnology Inc., CA) and antibodies to Bik (#4592), p-STAT1 (#9167) and STAT1 (#9172) were from Cell Signaling Technologies (Boston, MA). The proteins were detected using appropriate peroxidase-conjugated secondary antibodies and visualized by chemiluminescence (Perkin Elmer, Waltham, MA) using the FujiFilm Image Reader LAS-4000 (Valhalla, NY).
Quantitative RT-PCR
RNA was isolated from the snap-frozen right lungs of animals using TRIzol as described previously39 whereas RNA from cultured cells was extracted using the RNeasy kit (Qiagen, Valencia, CA) and concentration was determined using the Thermo Scientific Nanodrop 1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). The primer/probe sets for MUC5AC and CDKN1B were obtained from Life Technologies (Carlsbad, CA) and were amplified by quantitative real-time PCR using RT-PCR Master Mix (Life Technologies (Carlsbad, CA)) in the ABIPRISM 7900HT Real-Time PCR System. Relative quantities were calculated by normalizing averaged CT values to CDKN1B to obtain ΔCt, and the relative standard curve method was used for determining the fold change as described previously75.
Blocking Bcl-2 with retroviral transfection and ABT-263
HAECs were transfected with Bcl-2 shRNA containing retroviral vectors or control vectors (Origene Technologies, Inc., Rockville, MD) as per manufacturer’s instructions. After infection with Bcl-2 or control shRNAs, cells were treated with 10 ng/ml human IL-13, and 48 h later cells were assessed for Bcl-2 expression by immunofluorescence or Western blotting.
Statistical analysis
Grouped results were expressed as means ± SEM. Data were analyzed using GraphPad Prism Software (GraphPad Software, Inc., San Diego, CA). Grouped results were analyzed using analysis of variance. We performed the Kruskal–Wallis 2-sample non-parametric H test to compare the NRS and anti-PMN groups for the sample comparisons of unequal sample size. When significant main effects were detected (P < 0.05), Fishers least significant difference test was used to determine differences between groups. In addition, data were log transformed to correct for possible heteroscedasticity and reanalyzed for statistical differences. For all analyses the results remained unchanged.
References
Czabotar, P. E., Lessene, G., Strasser, A. & Adams, J. M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nature reviews. Molecular cell biology 15, 49–63, https://doi.org/10.1038/nrm3722 (2014).
Delbridge, A. R., Grabow, S., Strasser, A. & Vaux, D. L. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nature reviews. Cancer 16, 99–109 (2016).
Moldoveanu, T., Follis, A. V., Kriwacki, R. W. & Green, D. R. Many players in BCL-2 family affairs. Trends Biochem Sci 39, 101–111 (2014).
Tani-ichi, S. et al. Interleukin-7 receptor controls development and maturation of late stages of thymocyte subpopulations. Proceedings of the National Academy of Sciences of the United States of America 110, 612–617, https://doi.org/10.1073/pnas.1219242110 (2013).
Chand, H. S. et al. Intracellular Insulin-like Growth Factor-1 Induces Bcl-2 Expression in Airway Epithelial Cells. J Immunol 188, 4581–4589, https://doi.org/10.4049/jimmunol.1102673 (2012).
Chand, H. S., Woldegiorgis, Z., Schwalm, K., McDonald, J. & Tesfaigzi, Y. Acute Inflammation Induces Insulin-like Growth Factor-1 to Mediate Bcl-2 and Muc5ac Expression in Airway Epithelial Cells. American journal of respiratory cell and molecular biology 47, 784–791, https://doi.org/10.1165/rcmb.2012-0079OC (2012).
Qin, B., Zhou, Z., He, J., Yan, C. & Ding, S. IL-6 Inhibits Starvation-induced Autophagy via the STAT3/Bcl-2 Signaling Pathway. Scientific reports 5, 15701 (2015).
Akashi, K., Kondo, M., von Freeden-Jeffry, U., Murray, R. & Weissman, I. L. Bcl-2 rescues T lymphopoiesis in interleukin-7 receptor-deficient mice. Cell 89, 1033–1041 (1997).
Ostiguy, V., Allard, E. L., Marquis, M., Leignadier, J. & Labrecque, N. IL-21 promotes T lymphocyte survival by activating the phosphatidylinositol-3 kinase signaling cascade. Journal of leukocyte biology 82, 645–656 (2007).
Yang, C. et al. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med Oncol 32, 352, https://doi.org/10.1007/s12032-014-0352-6 (2015).
Xu, M. J. et al. IL-22 ameliorates renal ischemia-reperfusion injury by targeting proximal tubule epithelium. Journal of the American Society of Nephrology: JASN 25, 967–977 (2014).
Kurland, J. F. et al. NF-kappaB1 (p50) homodimers contribute to transcription of the bcl-2 oncogene. The Journal of biological chemistry 276, 45380–45386 (2001).
Catz, S. D. & Johnson, J. L. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 20, 7342–7351 (2001).
Sepulveda, P., Encabo, A., Carbonell-Uberos, F. & Minana, M. D. BCL-2 expression is mainly regulated by JAK/STAT3 pathway in human CD34+ hematopoietic cells. Cell death and differentiation 14, 378–380 (2007).
Chetoui, N., Boisvert, M., Gendron, S. & Aoudjit, F. Interleukin-7 promotes the survival of human CD4+ effector/memory T cells by up-regulating Bcl-2 proteins and activating the JAK/STAT signalling pathway. Immunology 130, 418–426 (2010).
Minshall, C. et al. IL-4 and insulin-like growth factor-I inhibit the decline in Bcl-2 and promote the survival of IL-3-deprived myeloid progenitors. J Immunol 159, 1225–1232 (1997).
Barata, J. T. et al. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. The Journal of experimental medicine 200, 659–669 (2004).
Whitsett, J. A. & Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nature immunology 16, 27–35, https://doi.org/10.1038/ni.3045 (2015).
Hammad, H. & Lambrecht, B. N. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity 43, 29–40, https://doi.org/10.1016/j.immuni.2015.07.007 (2015).
Gao, W. et al. Bronchial epithelial cells: The key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology 20, 722–729, https://doi.org/10.1111/resp.12542 (2015).
Fahy, J. V. & Dickey, B. F. Airway mucus function and dysfunction. The New England journal of medicine 363, 2233–2247 (2010).
Roy, M. G. et al. Muc5b is required for airway defence. Nature 16, 412–416, https://doi.org/10.1038/nature12807 (2014).
Young, H. W. et al. Central role of Muc5ac expression in mucous metaplasia and its regulation by conserved 5′ elements. American journal of respiratory cell and molecular biology 37, 273–290 (2007).
Barnes, P. J. Chronic obstructive pulmonary disease. The New England journal of medicine 343, 269–280 (2000).
Ha, E. V. & Rogers, D. F. Novel Therapies to Inhibit Mucus Synthesis and Secretion in Airway Hypersecretory Diseases. Pharmacology 97, 84–100 (2016).
Harris, J. F. et al. Bcl-2 sustains increased mucous and epithelial cell numbers in metaplastic airway epithelium. American journal of respiratory and critical care medicine 171, 764–772 (2005).
Chand, H. S. et al. A Variant of p53 Restricts the Mucus Secretory Phenotype by Regulating SPDEF and Bcl-2 Expression. Nat Commun 5, 5667 (2014).
Tesfaigzi, Y., Harris, J. F., Hotchkiss, J. A. & Harkema, J. R. DNA synthesis and Bcl-2 expression during the development of mucous cell metaplasia in airway epithelium of rats exposed to LPS. American journal of physiology. Lung cellular and molecular physiology. 286, L268–274 (2004).
Bartner, L. R., Robinson, N. E., Kiupel, M. & Tesfaigzi, Y. Persistent mucus accumulation: a consequence of delayed bronchial mucous cell apoptosis in RAO-affected horses. American journal of physiology. Lung cellular and molecular physiology. 291, L602–609 (2006).
Allahverdian, S., Harada, N., Singhera, G. K., Knight, D. A. & Dorscheid, D. R. Secretion of IL-13 by airway epithelial cells enhances epithelial repair via HB-EGF. American journal of respiratory cell and molecular biology 38, 153–160, https://doi.org/10.1165/rcmb.2007-0173OC (2008).
Leverson, J. D. et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med 7, 279ra240 (2015).
Tse, C. et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer research 68, 3421–3428 (2008).
Tesfaigzi, J., Hotchkiss, J. A. & Harkema, J. R. Expression of the Bcl-2 protein in nasal epithelia of F344/N rats during mucous cell metaplasia and remodeling. American journal of respiratory cell and molecular biology 18, 794–799 (1998).
Zhang, X. et al. Similarities and differences between smoking-related gene expression in nasal and bronchial epithelium. Physiol Genomics 41, 1–8, https://doi.org/10.1152/physiolgenomics.00167.2009 (2010).
Tesfaigzi, Y., Harris, J. F., Hotchkiss, J. A. & Harkema, J. R. DNA synthesis and Bcl-2 expression during development of mucous cell metaplasia in airway epithelium of rats exposed to LPS. American journal of physiology. Lung cellular and molecular physiology. 286, L268–274 (2004).
Harris, J. F., Aden, J., Lyons, C. R. & Tesfaigzi, Y. Resolution of LPS-induced airway inflammation and goblet cell hyperplasia is independent of IL-18. Respiratory research 8, 24 (2007).
Foster, J. E., Gott, K., Schuyler, M. R., Kozak, W. & Tesfaigzi, Y. LPS-induced neutrophilic inflammation and Bcl-2 expression in metaplastic mucous cells. American journal of physiology. Lung cellular and molecular physiology. 285, L405–414 (2003).
Laucho-Contreras, M. E. et al. Club Cell Protein 16 (CC16) Augmentation: A Potential Disease-modifying Approach for Chronic Obstructive Pulmonary Disease (COPD). Expert Opin Ther Targets 20, 869–883, https://doi.org/10.1517/14728222.2016.1139084 (2016).
Mebratu, Y. A., Dickey, B. F., Evans, C. & Tesfaigzi, Y. The BH3-only protein Bik/Blk/Nbk inhibits nuclear translocation of activated ERK1/2 to mediate IFNgamma-induced cell death. The Journal of cell biology 183, 429–439, https://doi.org/10.1083/jcb.200801186 (2008).
Ingram, J. L. et al. Opposing actions of Stat1 and Stat6 on IL-13-induced up-regulation of early growth response-1 and platelet-derived growth factor ligands in pulmonary fibroblasts. J Immunol 177, 4141–4148 (2006).
Tesfaigzi, Y., Fischer, M. J., Martin, A. J. & Seagrave, J. Bcl-2 in LPS- and allergen-induced hyperplastic mucous cells in airway epithelia of Brown Norway rats. American journal of physiology. Lung cellular and molecular physiology. 279, L1210–1217 (2000).
Snyder, J. C. et al. Clara cells attenuate the inflammatory response through regulation of macrophage behavior. American journal of respiratory cell and molecular biology 42, 161–171 (2010).
Sandstrom, T., Bjermer, L. & Rylander, R. Lipopolysaccharide (LPS) inhalation in healthy subjects increases neutrophils, lymphocytes and fibronectin levels in bronchoalveolar lavage fluid. The European respiratory journal 5, 992–996 (1992).
Sirois, J. & Bissonnette, E. Y. Alveolar macrophages of allergic resistant and susceptible strains of rats show distinct cytokine profiles. Clinical and experimental immunology 126, 9–15 (2001).
Gour, N. & Wills-Karp, M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 75, 68–78 (2015).
Hocke, A. C. et al. Regulation of interleukin IL-4, IL-13, IL-10, and their downstream components in lipopolysaccharide-exposed rat lungs. Comparison of the constitutive expression between rats and humans. Cytokine 33, 199–211, https://doi.org/10.1016/j.cyto.2006.01.007 (2006).
Chiba, N., Masuda, A., Yoshikai, Y. & Matsuguchi, T. Ceramide inhibits LPS-induced production of IL-5, IL-10, and IL-13 from mast cells. Journal of cellular physiology 213, 126–136, https://doi.org/10.1002/jcp.21101 (2007).
Supajatura, V. et al. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. The Journal of clinical investigation 109, 1351–1359, https://doi.org/10.1172/JCI14704 (2002).
Masuda, A., Yoshikai, Y., Aiba, K. & Matsuguchi, T. Th2 cytokine production from mast cells is directly induced by lipopolysaccharide and distinctly regulated by c-Jun N-terminal kinase and p38 pathways. J Immunol 169, 3801–3810 (2002).
Kuperman, D. A. et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nature medicine 8, 885–889 (2002).
Xiang, J., Rir-Sim-Ah, J. & Tesfaigzi, Y. IL-9 and IL-13 Induce Mucous Cell Metaplasia That Is Reduced by IFN{gamma} in a Bax-mediated Pathway. American journal of respiratory cell and molecular biology 38, 310–317 (2008).
Yu, H., Li, Q., Kolosov, V. P., Perelman, J. M. & Zhou, X. Interleukin-13 induces mucin 5AC production involving STAT6/SPDEF in human airway epithelial cells. Cell communication & adhesion 17, 83–92, https://doi.org/10.3109/15419061.2010.551682 (2010).
Atherton, H. C., Jones, G. & Danahay, H. IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. American journal of physiology. Lung cellular and molecular physiology. 285, L730–739 (2003).
Tyner, J. W. et al. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. The Journal of clinical investigation 116, 309–321 (2006).
Shim, J. J. et al. IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. American journal of physiology. Lung cellular and molecular physiology. 280, L134–140 (2001).
Booth, B. W., Adler, K. B., Bonner, J. C., Tournier, F. & Martin, L. D. Interleukin-13 induces proliferation of human airway epithelial cells in vitro via a mechanism mediated by transforming growth factor-alpha. American journal of respiratory cell and molecular biology 25, 739–743 (2001).
Taniguchi, K. et al. Epigen is induced during the interleukin-13-stimulated cell proliferation in murine primary airway epithelial cells. Experimental lung research 37, 461–470, https://doi.org/10.3109/01902148.2011.596894 (2011).
Singhera, G. K., MacRedmond, R. & Dorscheid, D. R. Interleukin-9 and -13 inhibit spontaneous and corticosteroid induced apoptosis of normal airway epithelial cells. Experimental lung research 34, 579–598 (2008).
Wynn, T. A. Type 2 cytokines: mechanisms and therapeutic strategies. Nature reviews. Immunology 15, 271–282, https://doi.org/10.1038/nri3831 (2015).
Newcomb, D. C. et al. IL-13 Regulates Th17 Secretion of IL-17A in an IL-10-Dependent Manner. J Immunol 188, 1027–1035 (2012).
Miossec, P., Korn, T. & Kuchroo, V. K. Interleukin-17 and type 17 helper T cells. The New England journal of medicine 361, 888–898 (2009).
Corren, J. et al. Lebrikizumab treatment in adults with asthma. The New England journal of medicine 365, 1088–1098, https://doi.org/10.1056/NEJMoa1106469 (2011).
Thomenius, M. J., Wang, N. S., Reineks, E. Z., Wang, Z. & Distelhorst, C. W. Bcl-2 on the endoplasmic reticulum regulates Bax activity by binding to BH3-only proteins. The Journal of biological chemistry 278, 6243–6250 (2003).
Zutter, M., Hockenbery, D., Silverman, G. A. & Korsmeyer, S. J. Immunolocalization of the Bcl-2 protein within hematopoietic neoplasms. Blood 78, 1062–1068 (1991).
Vaughan, M. B., Ramirez, R. D., Wright, W. E., Minna, J. D. & Shay, J. W. A three-dimensional model of differentiation of immortalized human bronchial epithelial cells. Differentiation 74, 141–148 (2006).
Ehre, C. et al. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proceedings of the National Academy of Sciences of the United States of America 109, 16528–16533, https://doi.org/10.1073/pnas.1206552109 (2012).
Mebratu, Y. A., Schwalm, K., Smith, K. R., Schuyler, M. & Tesfaigzi, Y. Cigarette smoke suppresses Bik to cause epithelial cell hyperplasia and mucous cell metaplasia. American journal of respiratory and critical care medicine 183, 1531–1538 (2011).
Coultas, L. et al. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Molecular and cellular biology 24, 1570–1581 (2004).
Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine 19, 202–208, https://doi.org/10.1038/nm.3048 (2013).
Gower, A. C., Steiling, K., Brothers, J. F. 2nd, Lenburg, M. E. & Spira, A. Transcriptomic studies of the airway field of injury associated with smoking-related lung disease. Proceedings of the American Thoracic Society 8, 173–179, https://doi.org/10.1513/pats.201011-066MS (2011).
Compalati, E. et al. The link between allergic rhinitis and asthma: the united airways disease. Expert review of clinical immunology 6, 413–423, https://doi.org/10.1586/eci.10.15 (2010).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nature medicine 22, 78–83 (2016).
Chand, H. S., Mebratu, Y. A., Kuehl, P. J. & Tesfaigzi, Y. Blocking Bcl-2 resolves IL-13-mediated mucous cell hyperplasia in a Bik-dependent manner. The Journal of allergy and clinical immunology 140, 1456–1459, https://doi.org/10.1016/j.jaci.2017.05.038 (2017). e1459.
Fanucchi, M. V., Harkema, J. R., Plopper, C. G. & Hotchkiss, J. A. In vitro culture of microdissected rat nasal airway tissues. American journal of respiratory cell and molecular biology 20, 1274–1285 (1999).
Schwalm, K. et al. Expression of the pro-apoptotic protein bax is reduced in bronchial mucous cells of asthmatics. American journal of physiology. Lung cellular and molecular physiology. 294, L1102–1109 (2008).
Acknowledgements
The authors thank Lois Herrera for assistance on the selected experiments and Mr. Hans Petersen for help with statistical analysis after log transformation of data. NIH R01HL068111 and NIH R01ES015482 (Y.T.)
Author information
Authors and Affiliations
Contributions
Y.T. designed and conceived the studies; H.S.C. and J.F.H. conducted the experiments; Y.T., H.S.C., and J.F.H. analyzed the data and H.S.C. and Y.T. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chand, H.S., Harris, J.F. & Tesfaigzi, Y. IL-13 in LPS-Induced Inflammation Causes Bcl-2 Expression to Sustain Hyperplastic Mucous cells. Sci Rep 8, 436 (2018). https://doi.org/10.1038/s41598-017-18884-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18884-9
This article is cited by
-
Indoor-air purification by photoelectrochemical oxidation mitigates allergic airway responses to aerosolized cat dander in a murine model
Scientific Reports (2023)
-
A long noncoding RNA antisense to ICAM-1 is involved in allergic asthma associated hyperreactive response of airway epithelial cells
Mucosal Immunology (2021)
-
Effects of N-acetyl cysteine on mitochondrial ROS, mitochondrial dynamics, and inflammation on lipopolysaccharide-treated human apical papilla cells
Clinical Oral Investigations (2021)
-
GFI-1 Protects Against Lipopolysaccharide-Induced Inflammatory Responses and Apoptosis by Inhibition of the NF-κB/TNF-α Pathway in H9c2 Cells
Inflammation (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
